
Abstract
Keywords
Introduction
MS is a chronic disease of the CNS that ultimately results in demyelination and axonal degeneration. Although the pathophysiology of MS is not well understood, it is known to involve localized inflammation and breakdown of the blood–brain barrier (BBB) with infiltration and activation of immune cells. 1 More recently, oxidative stress, potentially resulting from inflammation, has been shown to play a role in the pathogenesis of the disease. 2 Oxidative stress can contribute to mitochondrial dysfunction and apoptosis, 3 cytotoxicity,4,5 increased BBB permeability and leukocyte migration. 6
BG-12 is an oral formulation of dimethyl fumarate, the first drug in development for MS that has been shown to activate the Nrf2 pathway in vitro. 7 Activation of this pathway may potentially reduce the cytotoxic effects of oxidative stress. 8 In a randomized, placebo-controlled phase 2b study in patients with relapsing–remitting MS (RRMS), BG-12 240 mg three times daily reduced the total number of gadolinium-enhanced (Gd+) lesions over four brain MRI scans at weeks 12, 16, 20 and 24 by 69% compared with placebo (p < 0.0001; primary end point). In addition, BG-12 demonstrated a favourable safety profile over 24 weeks of treatment compared with placebo. Common adverse events (AEs) associated with BG-12 included headache, gastrointestinal symptoms and flushing; the severity of gastrointestinal AEs was mild to moderate in most cases. The incidence of serious AEs and infection were low, and rates in the BG-12 and placebo groups were similar. 9
Previous studies of disease modifying treatments for MS considered age, gender, lesion activity and disability at baseline as potentially important indicators of clinical outcomes for patients with RRMS on therapy. The objective of the present analyses was to explore the effect of BG-12 240 mg three times daily on total Gd+ lesion development from weeks 12 to 24 seen in the phase 2b study in subgroups of the overall study population. The subgroups analysed were based on disease activity (Expanded Disability Status Scale [EDSS] score [≤2.5, >2.5], Gd+ lesions [absent, present]) or demographics (age [< 40 years, ≥ 40 years], gender [male, female] and median disease duration [≤ 6 years, > 6 years]) at baseline. The results of all subgroup analyses are presented here.
Patients and methods
Phase 2b study description
Detailed methods for the primary phase 2b study of BG-12 have been published previously. 9 Briefly, 257 male and female patients aged 18 to 55 years (inclusive) diagnosed with RRMS according to McDonald criteria (nos. 1–4) 10 were included. Patients eligible had a baseline EDSS 11 score between 0.0 and 5.0 (inclusive) and at least one relapse within 12 months before randomization, with a previous brain MRI scan consistent with MS or Gd+ lesions on MRI scans performed within six weeks before randomization. In this multicentre, randomized, double-blind, lacebo-controlled, parallel-group, dose-ranging study patients initially were randomized 1:1:1:1 to receive oral BG-12 120 mg once daily, 120 mg three times daily (360 mg/day), 240 mg three times daily (720 mg/day) or placebo during an initial 24-week treatment period (weeks 1–24). All patients then received BG-12 during a 24-week, dose-blinded safety-extension period (weeks 25–48). The primary end point was the sum of the total number of new Gd+ lesions over four MRI scans at weeks 12, 16, 20 and 24. This time frame was selected based on the time to MRI effect reported in a pilot study in patients with RRMS who received BG-12 240 mg three times daily. 12 A list of investigators for the BG-12 phase 2b study group can be found in the Appendix.
Patients
Subgroup analyses were based on the patients from the efficacy-evaluable (EE) population who received placebo or BG-12 240 mg three times daily. The EE population was prespecified as part of the primary end point analysis and consisted of patients who had no missing data from four MRI scans at weeks 12, 16, 20 and 24.
Study assessments
Analyses were performed on the following subgroups according to their baseline status: EDSS score ≤ 2.5, EDSS score > 2.5, 0 Gd+ lesions, ≥ 1 Gd+ lesions, age < 40 years, age ≥ 40 years, female patients, male patients, disease duration ≤ 6 years and disease duration > 6 years. These factors were selected based on previous studies suggesting that they may be important predictors of clinical outcomes in patients with RRMS on therapy.13–16
Statistical analyses
The effect of treatment on the sum of the total number of new Gd+ lesions over weeks 12, 16, 20 and 24 was analysed using the non-parametric Wilcoxon rank-sum test. In all subgroups, analyses were based on the EE population. Two-sided statistical tests with an alpha value of 0.05 were used in all cases. Because of the exploratory nature of these analyses, alpha adjustment for multiple comparisons was not applied.
Results
Patients
In the primary study population, 257 patients were randomized to receive BG-12 120 mg once daily (n = 64), 120 mg three times daily (n = 64), 240 mg three times daily (n = 64) or placebo (n = 65). One patient who received 240 mg three times daily withdrew from the study before the first treatment. 9 Overall, 235 patients completed the placebo-controlled portion (weeks 1–24) and 219 completed the safety-extension portion (weeks 25–48). 9
Subgroup analyses were conducted on 108 patients (placebo, n = 54; BG-12 240 mg three times daily, n = 54) from the EE population. Baseline characteristics of the placebo and BG-12 240-mg three-times-daily groups of the EE population were similar to those of the intent-to-treat (ITT) population (Table 1). 9 The mean age of patients was 35.2 years in patients who received placebo and 36.5 years in patients who received BG-12 240 mg three times daily. Approximately one-third of patients in each treatment group were female. At baseline, the mean EDSS score was 2.58 for patients randomized to placebo and 2.87 for patients randomized to BG-12 240 mg three times daily. The mean number of Gd+ lesions at baseline was 0.8 and 1.2 for placebo and BG-12 240 mg three times daily, respectively.
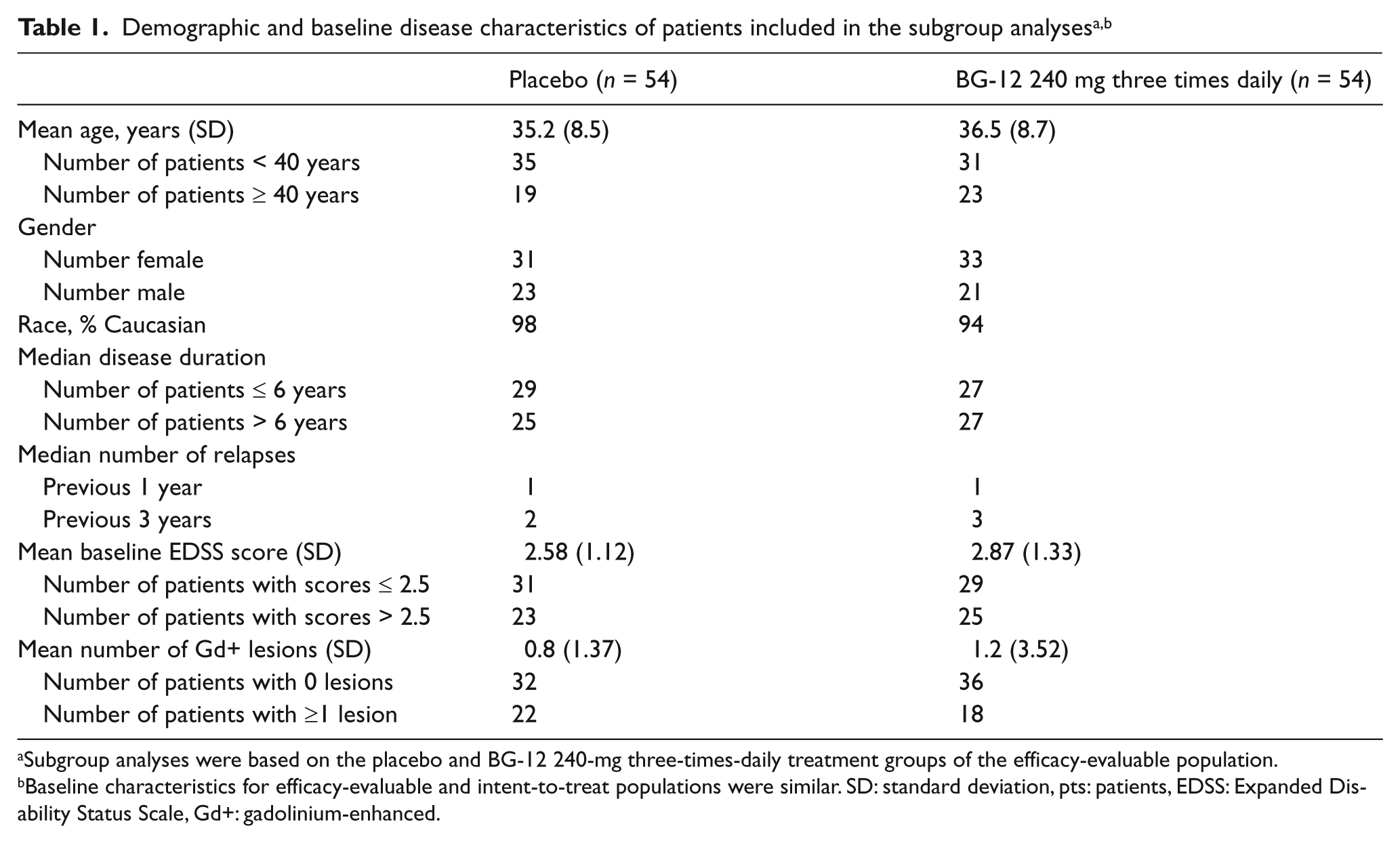
Subgroup analyses were based on the placebo and BG-12 240-mg three-times-daily treatment groups of the efficacy-evaluable population.
Baseline characteristics for efficacy-evaluable and intent-to-treat populations were similar. SD: standard deviation, pts: patients, EDSS: Expanded Disability Status Scale, Gd+: gadolinium-enhanced.
Primary end point analysis: overall population
BG-12 240 mg three times daily significantly reduced the total number of new Gd+ lesions from weeks 12 to 24 by 69% compared with placebo (1.4 vs. 4.5; p < 0.0001; Figure 1A). 9 A sensitivity analysis using the ITT population confirmed that patients receiving BG-12 240 mg three times daily had fewer total new Gd+ lesions compared with those receiving placebo (1.3 vs. 4.8; p < 0.0001). 9 The effect on new Gd+ lesions was apparent as early as eight weeks after initiating treatment (BG-12, 0.7; placebo, 1.3; p = 0.160; Figure 1B) and became significant at 12 weeks (BG-12, 0.5; placebo, 1.7; p = 0.007).
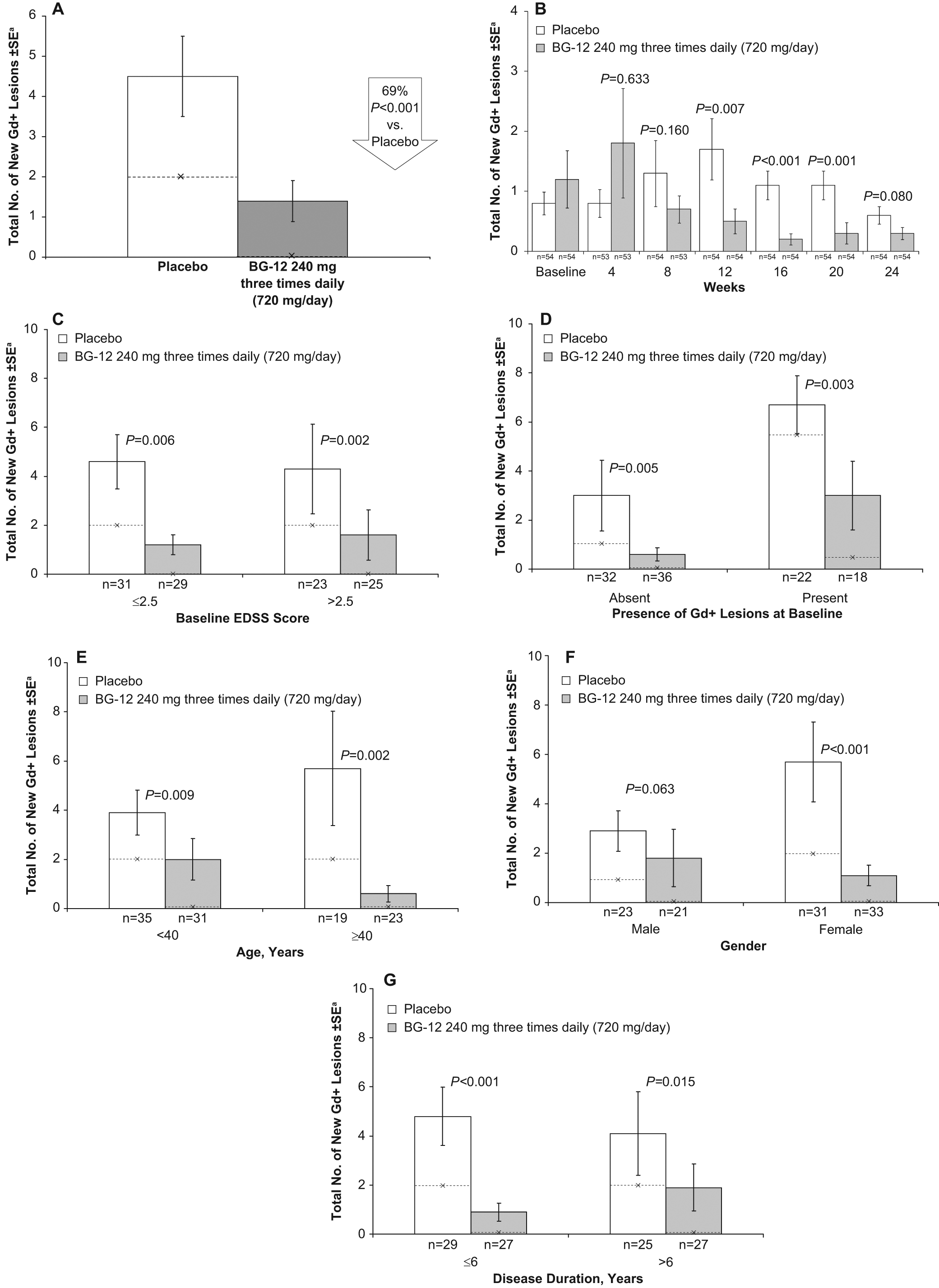
Effect of BG-12 on gadolinium-enhanced (Gd+) lesion accumulation from weeks 12 to 24 (A), total number of new Gd+ lesions on monthly MRI scans from baseline to 24 weeks (B). Effect of BG-12 on total number of new gadolinium-enhancing (Gd+) lesions by age (C), gender (D), disease duration (E), baseline Expanded Disability Status Scale (EDSS) score (F), and baseline Gd+ lesion load (G) in the efficacy-evaluable population. Bars represent mean values; --×-- represents median values
Subgroup analyses
Treatment with BG-12 reduced the total number of new Gd+ lesions from weeks 12 to 24 in subgroups of patients based on baseline disease characteristics. BG-12 240 mg three times daily reduced the total number of new Gd+ lesions by 74% (1.2 vs. 4.6; p = 0.006) in patients with baseline EDSS scores of 2.5 or lower (n = 60), by 63% (1.6 vs. 4.3; p = 0.002) in patients with baseline EDSS scores higher than 2.5 (n = 48) (Figure 1C), by 80% (0.6 vs. 3.0; p = 0.005) in patients with no Gd+ lesions at baseline (n = 68) and by 55% (3.0 vs. 6.7; p = 0.003) in patients with Gd+ lesions at baseline (n = 40) compared with placebo (Figure 1D).
The efficacy of BG-12 240 mg three times daily on reducing development of new Gd+ lesions was demonstrated in all but one subgroup of patients based on demographics. BG-12 reduced the total number of new Gd+ lesions from weeks 12 to 24 by 49% (2.0 vs. 3.9; p = 0.009) in patients younger than 40 years of age (n = 66), by 89% (0.6 vs. 5.7; p = 0.002) in patients 40 years of age or older (n = 42) (Figure 1E), by 81% (1.1 vs. 5.7; p < 0.001) in female patients (n = 64) and by 38% (1.8 vs. 2.9; p = 0.063) in male patients (n = 44) compared with placebo (Figure 1F). Treatment with BG-12 240 mg three times daily reduced the total number of new Gd+ lesions by 81% (0.9 vs. 4.8; p < 0.001) in patients with a disease duration of six years or less (n = 56) and by 54% (1.9 vs. 4.1; p = 0.015) in patients with a disease duration longer than six years (n = 52) compared with placebo (Figure 1G). Median values for the total number of new Gd+ lesions were lower for BG-12 240 mg three times daily compared with placebo in all analysed subgroups.
Discussion
Consistent with the efficacy results for the overall population in the phase 2b study, the effect of BG-12 240 mg three times daily on the number of Gd+ lesions was qualitatively similar if not quantitatively identical across all of the baseline disease characteristics and demographic subgroups investigated. The reduction in the number of new Gd+ lesions remained significant (p < 0.05) in all subgroups except for male patients (p = 0.063). Male patients in the RRMS cohort treated with placebo had low disease activity in this study, a factor that – together with the relatively low numbers – may have contributed to not reaching a higher grade of significance.
Our results suggest that BG-12 prevents the development of Gd+ lesions by counteracting inflammatory activity and stabilizing the BBB. In addition, further CNS tissue damage and loss may be inhibited by the potential cytoprotective ability of BG-12 to prevent the transformation of Gd+ lesions into persistent T1-hypointense lesions. Another post hoc analysis of patients who had developed Gd+ lesions during the primary phase 2b study showed that treatment with BG-12 240 mg three times daily compared with placebo significantly reduced the percentage of Gd+ lesions that evolved into T1-non-enhanced hypointense lesions. 17
Because this phase 2b study was powered for an MRI outcome as the primary end point rather than relapse-related outcomes 9 and the reduction in annualized relapse rate was not significant in the primary phase 2b population (BG-12 240 mg three times daily, 0.44; placebo, 0.65 [+32%]; p = 0.524), 9 we did not analyse the effect on annualized relapse rates in the subgroups. Relapse end points are key outcomes in two multicentre, randomized, placebo-controlled, dose-comparison phase 3 studies being conducted to further investigate the efficacy of BG-12 in patients with RRMS: Determination of the Efficacy and Safety of Oral Fumarate in Relapsing–Remitting Multiple Sclerosis (DEFINE; primary end point proportion of patients with relapses at two years) and Comparator and an Oral Fumarate in Relapsing–Remitting Multiple Sclerosis (CONFIRM; primary end point annualized relapse rate at two years).
In addition to effectively reducing the number of new Gd+ lesions in the overall 9 and subgroup populations in the phase 2b study, BG-12 has demonstrated a favourable safety profile. 9 The rate of serious AEs and infections was low across all treatment groups, with flushing and MS relapses being the most common AEs. 9 If the ongoing phase 3 programme provides data that further support the effects shown in this phase 2b study, BG-12 has the potential to become a valuable treatment option for patients with relapsing MS. The proposed dual anti-inflammatory and neuroprotective mechanism of action is not yet completely understood. However, it is different from current MS treatment strategies and may serve as a rationale to combine BG-12 with other drugs currently licensed or being developed for the treatment of patients with MS.
Footnotes
Appendix: BG-12 Phase 2b Study Group
Acknowledgements
The authors acknowledge Sarita Shaevitz, Hema Gowda, Jill Licata and Matthew Hasson of Scientific Connexions (Newtown, PA, USA) for technical and editorial assistance with preparing the manuscript. The data presented within this manuscript have been presented previously as abstracts for the 60th Annual Meeting of the American Academy of Neurology and 22nd Annual Meeting of the Consortium of Multiple Sclerosis Centers.18,19
This study was supported by Biogen Idec, Inc., the Swiss MS Society (to LK), the Czech Ministry of Education (Research Programme MŠM 0021620849) (to EH) and the UCLH/UCL Comprehensive Biomedical Research Centre (CBRC).