
Abstract
Keywords
Introduction
Multiple sclerosis (MS) is one of the most common neurologic diseases in young adults. It is characterized by myelin loss, various degrees of axonal pathology, and progressive neurological dysfunction 1 . Although the pathogenesis of MS is poorly understood, evidence suggests that both genetic and environmental components play important roles in disease development, both independently and interactively 2 . The role of genetics in MS and its interaction with environmental triggers are currently being extensively studied. MS is a disease with clear geographic variability in both prevalence and incidence. The role of environmental factors has historically been thought to be important. The geographical distribution and familial aggregation of MS have often been ascribed to the role of infectious agents, but there is no consensus regarding this theory 3 . The frequency of MS among first-degree, non-biological relatives living with the index case was no greater than the expected frequency from the general population prevalence data and was significantly less than the frequency for biological relatives. Which supports the hypothesis that the familial aggregation of MS is genetically determined rather than environmentally determined 3,4 .
The contribution of genetics to MS is supported by many reports showing familial aggregation of the disease, high concordance rates among twins, and an increased risk among relatives of patients with MS. People with MS have a 5–26% chance of having one or more affected relatives 5 –7 . A major contributor to the genetic risk is the major histocompatibility complex (MHC) antigen 8,9 . Several alleles have been identified as heritable risk factors for MS. Genetic complexity, especially related to human leukocyte antigens (HLA) of the MHC and, to a lesser extent, non-MHC-related genes, plays a major role in influencing disease susceptibility, phenotypic expression and clinical outcome through genotypic and epistatic interactions with MHC 10 .
Genome-wide association studies (GWASs) have helped identifying genetic regions of interest for many common complex diseases and traits 11 . Multiple GWASs that included thousands of MS patients with large numbers of normal controls have been reported in recent years. Most of these studies were reasonably powered and have identified over 50 risk loci that exert modest individual effects on MS susceptibility 12 –20 . The most recent study, a large multinational GWAS, confirmed 23 previously reported genetic variants that were associated with MS and implicated 29 new genetic variants in the susceptibility to MS 20 . The most important SNP predictors of MS was rs9271366, which has been previously shown to tag DRB1*1501 12 .
Studying large families with more than one affected family member have been useful in identifying the causative genetic sites in single gene disorders. Examining the distribution of DNA polymorphisms in families with a history of MS (FMS) and in the appropriate controls may help to identify putative causal variants. We have shown that parent’s consanguinity (PC) is associated with a higher probability of having FMS and an increased likelihood of the disease aggregating in families 21 . Having said that; studying populations with a high prevalence of consanguinity and, possibly, an increased risk of MS as a result of PC could be helpful.
Subjects/Materials and Methods
Participants for this study were identified via the MS registry at our center. This database contains prospectively collected demographic, clinical and radiological data from all patients. All patients are of Saudi origin. MS was diagnosed based on the revised McDonald criteria 22 . The local Institutional Review Board approved the study. All participants consented to participate in this study and to have their DNA banked.
Four subject groups were included in the study. The first group consisted of patients diagnosed with MS who did not have a family history of MS (sporadic MS). The second group included patients diagnosed with MS but had at least one family member known to have MS (FMS). The third group included individuals who were relatives of FMS patients but appeared to be free of MS symptoms (related controls). The last group included healthy volunteers (independent controls). The FMS patients were recruited from 14 different families who are currently registered and being followed in the authors MS registry. The independent control patients were recruited from regular blood donors who are usually young males. All patients diagnosed with MS were contacted, and if they agreed to participate, a blood DNA sample was collected and their information from the database was included. The collected data included the patient demographics, the date of disease onset and the information regarding parental consanguinity. Genomic DNA was extracted from the EDTA treated peripheral blood of both the patients and controls using standard Qiagen methodology.
Subjects were genotyped for 15 SNPs using an ABI 3010 DNA sequencer with a custom set of primers designed for the SNPlex Genotyping System (Applied Biosystems Inc., Foster City, USA). The selected SNPs were selected from previously identified ones in MS genome-wide association studies (GWAS). These 15 SNPs were selected based on the data available when this study was planned. The 15 SNPs included were rs10735781, rs10984447, rs10975200, rs12044852, rs11164838, rs2104286, rs12487066, rs1321172, rs3135388, rs4763655, rs6680578, rs6897932, rs7536563, rs757736, and rs6498169.
Variations in the genotype distribution of each of the 15 SNPs across the study groups were analyzed by using logistic regression. To control for patients clustering within families, conditional logistic regression was utilized. A total of 15 models were developed; each model included the genotype of the SNP of interest as the dependent variable and the other study groups as the independent variables. Results were summarized and reported in terms of odds ratio with the corresponding 95% confidence limits. All analyses were performed by using SAS 9.2 (SAS institute, Inc.).
Results
A total of 342 subjects were included in the study, of which 99 were sporadic MS patients, 22 were FMS patients, 89 were related controls, and 132 were independent controls. The average age and the proportion of female patients in the sporadic MS, FMS, and relative control groups were 33.7, 66%; 30.7, 77%; and 35.0, 55%, respectively. The demographic data of the MS subjects are listed in table 1.
Demographics and general characteristics.
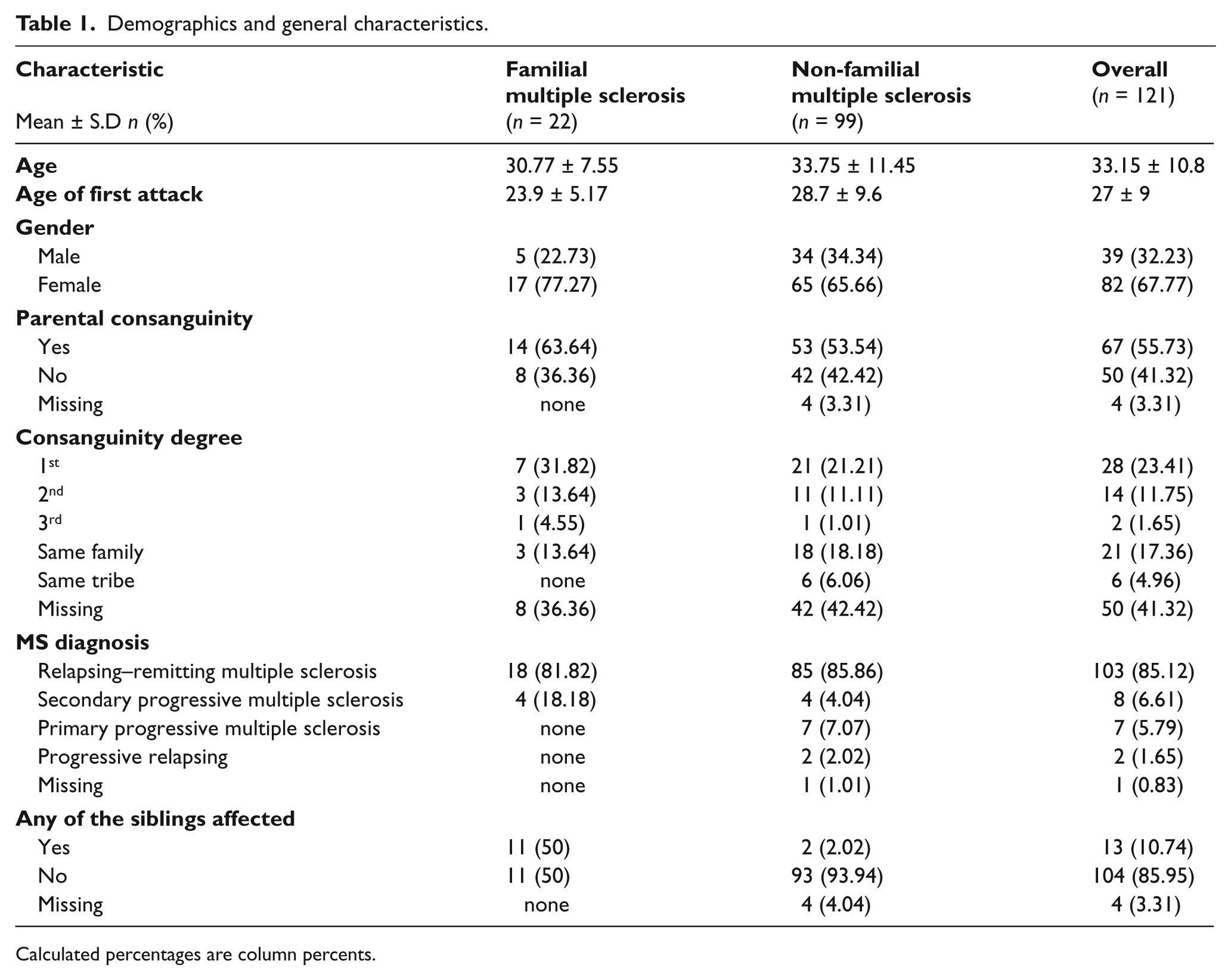
Calculated percentages are column percents.
When sporadic (without family history) MS patients were compared to the independent control subjects, multiple SNPs were showed tendency for association with MS but without statistical significance (figure 1). Only rs3135388 was found to be statistically significant [OR 2.22, CI (1.10, 4.48)]. Rs 3135388 SNP is located in chromosome 6 and correlate to MHC complex, specifically to HLA-DRB1 *1501 as reported previously 9 When the FMS patients were compared to the independent control subjects, almost all of the previously identified SNPs were found with a stronger association tendency (figure 2).Finally, when the patients and the controls were selected from a much more homogenous genetic pool, namely; the FMS group and the related controls, where only those SNPs that are highly linked to the disease would be expected to persist (figure 3), only two SNPs, rs6498169[OR 4.26, CI (1.17 – 15.51)], and rs10984447 [OR 13.63, CI (1.54, 120.83)] were associated with MS.
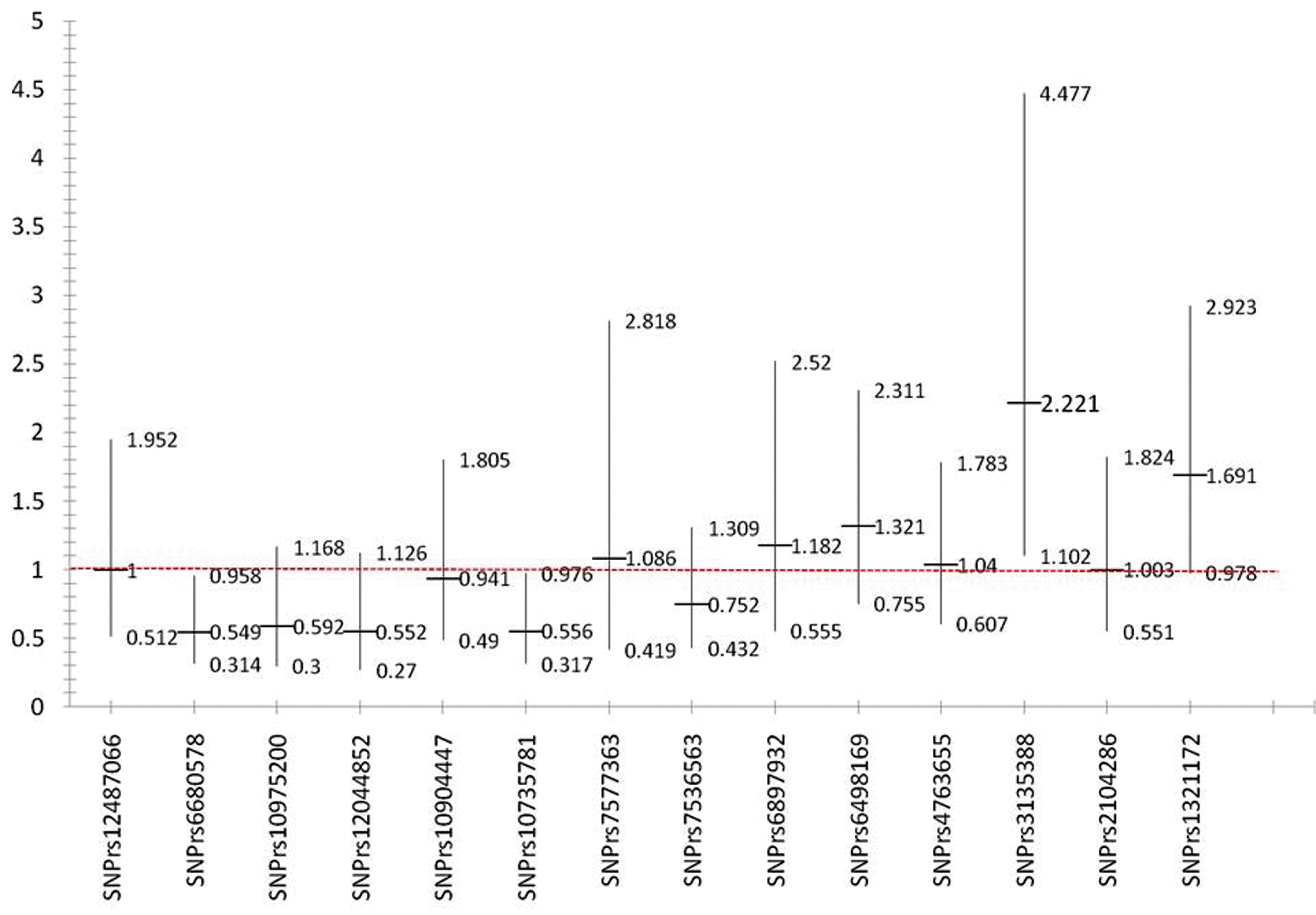
Odds ratios and confidence intervals for sporadic MS and independent control.
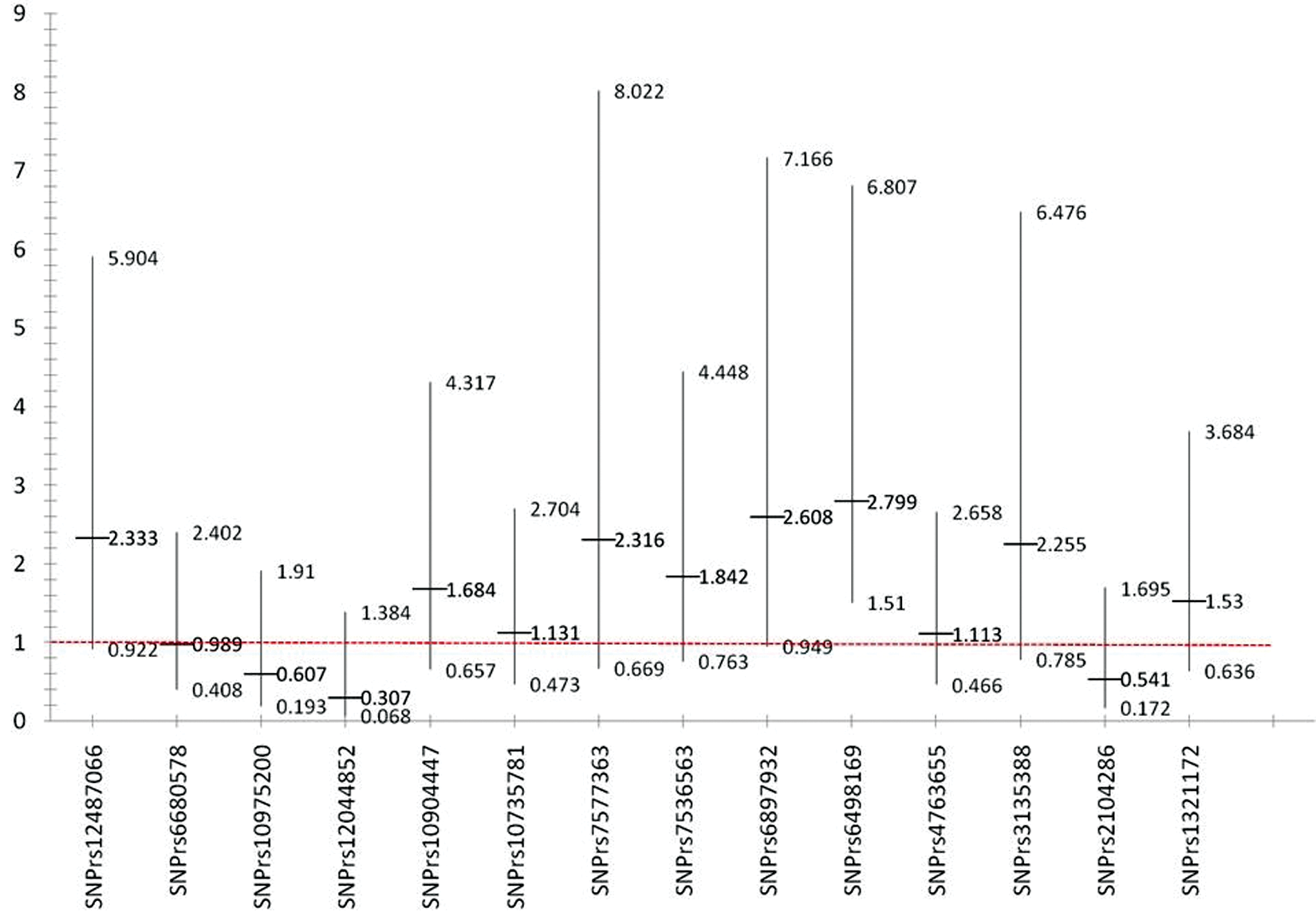
Odds ratios and confidence intervals for familial MS and independent control.
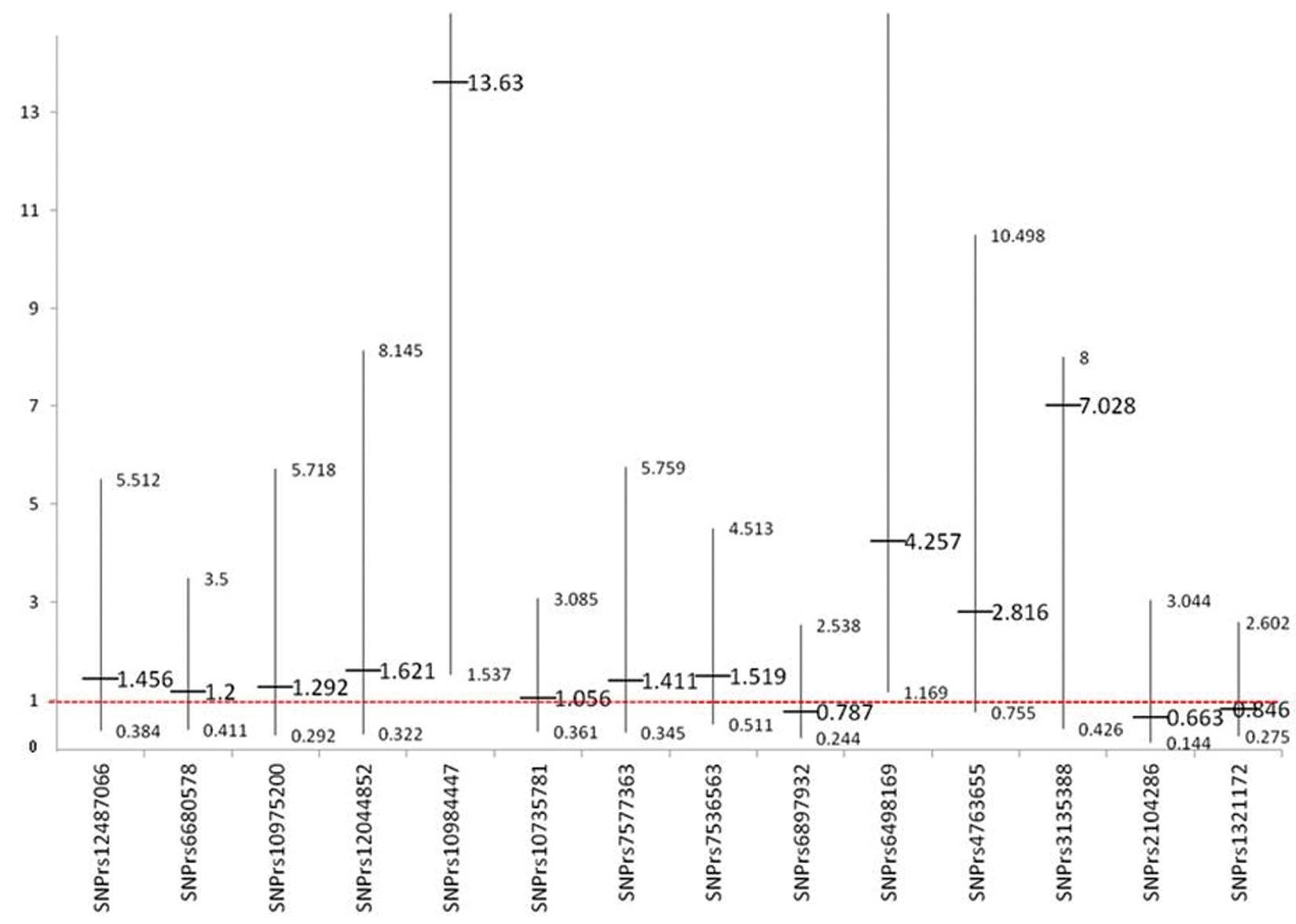
Odds ratios and confidence intervals for familial MS and relative control.
Discussion
Studying families affected with certain complex, multi-factorial, diseases such as multiple sclerosis may help identifying the role of genetics in the pathogenesis of such diseases. Co-affected siblings can be used as a resource to identify susceptibility genes and have been the basis for genomic screens of various diseases through linkage analysis, including insulin-dependent diabetes 23 , bronchial asthma 24 , and MS 25 –28 . The data from the linkage analysis and the GWASs are helpful in identifying the genetic basis of non mendelian genetic diseases and the possible effect of genetics on the patient’s response to treatment and disease progression.
Genetic studies are usually more informative when performed on large families. The role of consanguinity is evident in inherited diseases. However, it is expected that consanguinity would not have any effect on the incidence or prevalence of complex, non-inherited diseases and may therefore be useful in genetic studies because of the concentration of the genetic pool, especially in populations with a high prevalence of consanguineous marriages. It is expected that a smaller sample size in such populations may be sufficient to identify the genetic associations of the disease since only most causal genetic variants would be expected to stand out. In this study, small sample size was able to show significant association of SNP; rs3135388 with sporadic MS patient [OR 2.22, CI (1.10, 4.48)]. Rs 3135388 is highly correlated with the HLA-DRB1*1501 allele; multiple studies have estimated the risk allele (T) is associated with a 3 to 6 fold higher risk for developing multiple sclerosis 13 . When patients and their controls were selected from a much more homogenous genetic pool, one would expect that only SNPs that are highly linked to the disease would show association. As shown in figures 1 and 2, most of the selected SNPs in our replicate studies showed a tendency for association with the disease when either sporadic or familial MS patients were compared to healthy volunteers (independent controls). However, when only familial MS patients were compared to the related controls, only two SNPs, rs6498169, and rs10984447 which is located on chromosome 16 and 9 respectively, showed statistically significant association with MS in our population. SNP rs6498169 showed higher association [OR 4.26, CI (1.17 – 15.51)] compared to published data with an odds ratio associated with this allele of 1.14 (CI 1.08-1.21) in North American, European and Australian patients13,29. As well SNP rs10984447 yielded high association with [OR 13.63, CI (1.54, 120.83)] compared to an odds ratio associated with this allele of 1.17 (CI 1.09-1.25) in North American and European patients 13 .
Our investigation (unpublished work) using whole genome, high-resolution array comparative genomic hybridization (array CGH) of variations in genomic dosage compared to the normal reference dosage within FMS cases, by using seven affected individuals from two large families, revealed the presence of potential genes flanking SNP rs6498169 and SNP rs10984447 within cytogenetic band 16p13.13 and 9q32 respectively. The genomic area on 16p13.13 contains genes such as SEPT12 and ABAT which have implicated roles in cytokinesis, and neurotransmitter 30,31 , where the 9q32 area contains genes such as TLR4 and TRAF1 involving role in the
innate immune system and regulation of cell survival and apoptosis 32,33. This result supports the linkage of the SNPs identified in our population to multiple sclerosis pathogenesis. Further validation of this predicted area to evaluate the substantial contribution of those SNPs to MS pathogenesis is being performed with the help of a large family and powerful molecular techniques.
Our findings are consistent with many other GWAS analyses in multiple studies from different countries that have shown the association of chromosome 16p13, mainly the KIAA 0350 gene (SNP rs6498169), with MS 29,34. It has also been shown that the CLEC16A gene, which is in close proximity to our identified SNP, is linked to several autoimmune diseases, including multiple sclerosis. This region includes the CLEC16A/KIAA0350 gene and the adjacent gene MHC2TA (MHC class II transactivator) 35,36.
Conclusions
SNPs rs6498169 and 10984447 are the two most significant multiple sclerosis associated SNP in our Saudi MS population out side the MHC region. Our results suggest that it is possible to use a more homogenous genetic pool to identify the most significant MS-associated SNPs in the Saudi population, even with a very small sample size. Our finding is in agreement with many other studies that used much larger sample sizes and were performed in many ethnic groups.
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest.
Funding
This research is supported by grant from the King Abdullah International Medical Research Center, grant number RC08/030.