
Abstract
Background:
Macrophages are dynamic participants in destruction of white matter in active multiple sclerosis (MS) plaques. Regulation of phagocytosis and myelin degradation along endosomal pathways in macrophages is highly-orchestrated and critically-dependent upon acidification of endosomal lumena. Evidence from in vitro studies with macrophages and THP-1 cells suggests that sodium channel Nav1.5 is present in the limiting membrane of maturing endosomes where it plays a prominent role in the accumulation of protons. However, a contribution of the Nav1.5 channel to macrophage-mediated events in vivo has not been demonstrated.
Method:
We examined macrophages within active MS lesions by immunohistochemistry to determine whether Nav1.5 is expressed in these cells in situ and, if expressed, whether it is localized to specific compartments along the endocytic pathway.
Results:
Our results demonstrate that Nav1.5 is expressed within macrophages in active MS lesions, and that it is preferentially expressed in late endosomes and phagolysosomes (Rab7+, LAMP-1+), and sparsely expressed in early (EEA-1+) endosomes. Triple-immunolabeling studies showed localization of Nav1.5 within Rab7+ endosomes containing proteolipid protein, a myelin marker, in macrophages within active MS plaques.
Conclusions:
These observations support the suggestion that Nav1.5 contributes to the phagocytic pathway of myelin degradation in macrophages in vivo within MS lesions.
Introduction
Macrophage-mediated destruction of white matter, including phagocytosis of myelin fragments by infiltrating macrophages, is a pathological hallmark of active lesions in multiple sclerosis (MS).1–4 Phagocytosis of axonal components by macrophages may also contribute to white matter damage in MS. 5 While some stages of the highly-orchestrated process of engulfment and subsequent degradation of extracellular material through the endosomal pathway have been elucidated, 6 an understanding of phagocytotic and degradative processes by macrophages is incomplete. Maturation of endosomes along the endocytic pathway, including transition from macropinocytotic vesicles to early endosomes to late endosomes and phagolysosomes, is a complex process involving multiple protein–protein interactions and increasing acidification of endosomal lumena. Recent work demonstrates that endosomal acidification via proton accumulation is critically dependent upon ion fluxes across endosomal and lysosomal membranes. 7 These ionic fluxes include significant alterations in [Cl-], [Na+], [K+], and [Ca2+] that accompany acidification of endosomes. Carrithers et al. 8 recently described the expression of the tetrodotoxin-resistant (TTX-R) cardiac sodium channel, Nav1.5, in late endosomes of cultured primary monocyte-derived macrophages. Nav1.5 expression was shown to be confined to endosomal limiting membrane, and was not seen at the plasma membrane. This study also showed that activity of Nav1.5 contributes to regulation of macrophage phagocytosis and endosomal pH during lipopolysaccharide (LPS)-stimulated endosomal acidification. More recently, knockdown of Nav1.5 was shown to attenuate the initial calcium response to bacterial challenge in cultured macrophages, and to prevent calcium oscillations in maturing endosomes. 9
While observations on cultured macrophages suggest a role for Nav1.5 in pathways that regulate macrophage phagocytosis and endosomal/phagolysosomal maturation in vitro, the possible presence of Nav1.5 and its subcellular localization have not been studied in macrophages in situ, and no information is currently available about the expression of Nav1.5 in macrophages in the human central nervous system (CNS). Systemic delivery of sodium channel blocking agents (i.e. phenytoin, flecainide, carbamazepine, and lamotrigine), which block both TTX-sensitive (TTX-S) and TTX-R sodium channels, 10 to rodents with experimental autoimmune encephalomyelitis (EAE) attenuates axonal loss, improves clinical status, and reduces immune cell infiltrates.11–13 Sodium channel blockade may protect axons in rodents with EAE by two converging pathways: 13 1) blockade of sodium influx into axons via axonal sodium channels, preventing inappropriate reverse operation of the sodium-calcium exchanger, which can lead to import of damaging levels of Ca2+ that initiate degradative pathways;14,15 and 2) direct effect on effector functions of immune cells, including macrophages, microglia; and T-lymphocytes.16–18 Importantly, recent work demonstrates that phenytoin attenuates LPS-induced phagocytosis in microglia in vitro, 17 but the detailed mode of action of sodium channel blockade on phagocytic pathways is not known.
The experiments described above, carried out in vitro and in animal models, are consistent with the expression of sodium channels in immune cells, including macrophages, in vivo. The possible presence of sodium channels within these cells assumes special relevance in the context of recent clinical studies 19 of sodium channel blockers in patients with MS. In the present study, we examined macrophages within active lesions of MS subjects to determine whether these immune cells express Nav1.5 and, if so, whether it is expressed in endosomal compartments along the phagocytic pathway.
Methods
Multiple sclerosis tissue
Post-mortem CNS tissue, acquired via a rapid autopsy protocol from subjects with disabling secondary progressive MS (Table 1; n=12 active lesion samples from seven subjects; age 49.2
Basic clinical data.

SP: secondary progressive; MSQ: active; NC: normal control; CB: cerebellum; F: frontal; O: occipital; P: parietal; pons: structure in the brainstem; SC: spinal cord; Sv: subventricular; V: ventricle wall.
ORO macrophages and haematoxylin-stained perivenular lymphocyte cuffing were scored on a scale of 0 to 5, as described by Newcombe and Cuzner. 20 The first number is the score for ORO-positive macrophages and the second number is the score for perivenular cuffing obtained with haematoxylin staining.
Immunocytochemistry
A total of 10 µm cryosections were processed as described previously. 21 Sections were fixed for 5 minutes in 4% paraformaldehyde and rinsed in phosphate-buffered saline (PBS). Sections were sequentially incubated in: 1) blocking solution (PBS with 3% fish skin gelatin, 3% bovine serum albumin, 0.3% Triton X-100, 0.02% sodium azide, and 0.1 mg/ml human IgG); 2) primary antibodies (rabbit Nav1.5, 1:100, Alomone, Jerusalem, Israel; mouse EEA-1, 1:100, mouse LAMP-1, 1:100, BD Transduction Laboratories, San Jose, CA; goat EEA-1, 1:200, goat Rab7, 1:100, goat LAMP-1, 1:100, Santa Cruz Biotechnology, Santa Cruz, CA; mouse Rab7, 1:200, mouse PLP, 1:1000, Abcam, Cambridge, MA; mouse CD68, 1:100, Dako, Carpenteria, CA; RCA I-biotin, 1:500, Vector Lab, Burlingame, CA) for 24–48 hours at 4oC; 3) PBS; 4) appropriate secondary antibodies (donkey anti-mouse or goat IgG-Alexa Fluor 488 or 546, 1:1000, donkey anti-rabbit IgG-Alexa Fluor 546, 1:1000, Invitrogen, Carlsbad, CA; donkey anti-mouse IgG DyLight-649, 1:200, Jackson ImmunoResearch, West Grove, PA; donkey anti-chicken-Alexa Fluor 488 or Cy5, 1:200, Chemicon, Temecula, CA; and, StreptAvidin-Alexa Fluor 633, 1:200, Invitrogen) for 12–24 hours at 4oC; 5) PBS; and 6) 4’,6’ diamino-2-phenylindole-2HCl (DAPI, 300 nM, Sigma-Aldrich, St. Louis, MO) and mounted with Aqua Poly mount (Polysciences, Warrington, PA).
Control experiments were performed with the omission of primary antibodies, and, for the Nav1.5 antibody, pre-adsorption with the cognate peptide (~48 M peptide to antibody molar concentration ratio) for 3 hours at room temperature. In both control experiments, only background labeling was observed.
Tissue analysis
Images of fluorescent-labeled tissue sections were accrued with Nikon C1si (Nikon USA, Melville, NY) and Zeiss LSM 510META confocal microscopes (Zeiss Microsystems, Thornwood, NY) operating with frame lambda (sequential) mode and saturation indicator to prevent possible bleed-through between channels. Images were composed and processed to enhance contrast with Photoshop (Adobe, San Jose, CA).
Quantitative co-localization of Nav1.5 and markers of endosomal compartments (EEA-1, Rab7, and LAMP-1) was performed with MetaMorph software (Molecular Devices, Sunnyvale, CA). The number of macrophages analyzed was: EEA-1, 24 cells from 4 subjects; Rab7, 18 cells from 4 subjects; LAMP-1, 21 cells from 3 subjects. A one-way ANOVA with post hoc Bonferroni test was performed with Origin 8.5 software (OriginLab, Northhampton, MA) for statistical comparisons.
Results
Sodium channel Nav1.5 has been previously shown to be expressed on the limiting membranes of late endosomes of interferon-γ primed and LPS-challenged THP-1 cells (a monocytic cell line) and peripherally-obtained, monocyte-derived macrophages studied in vitro. 8 To determine whether macrophages in situ express Nav1.5 in MS lesions, we examined active MS plaques to assess whether these cells express Nav1.5, and, if so, the subcellular localization of these channels.
We used triple-immunocytochemical labeling to detect the expression of Nav1.5 and endosomal markers (EEA-1, Rab7, and LAMP-1) in CD68+ cells within active MS lesions, in addition to the labeling of macrophage nuclei with DAPI. For clarity of co-localization of Nav1.5 with endosomal markers, Nav1.7 (red) and endosomal labels (green) signals were merged separately from merged DAPI and CD68 signals (Figures 1–3).
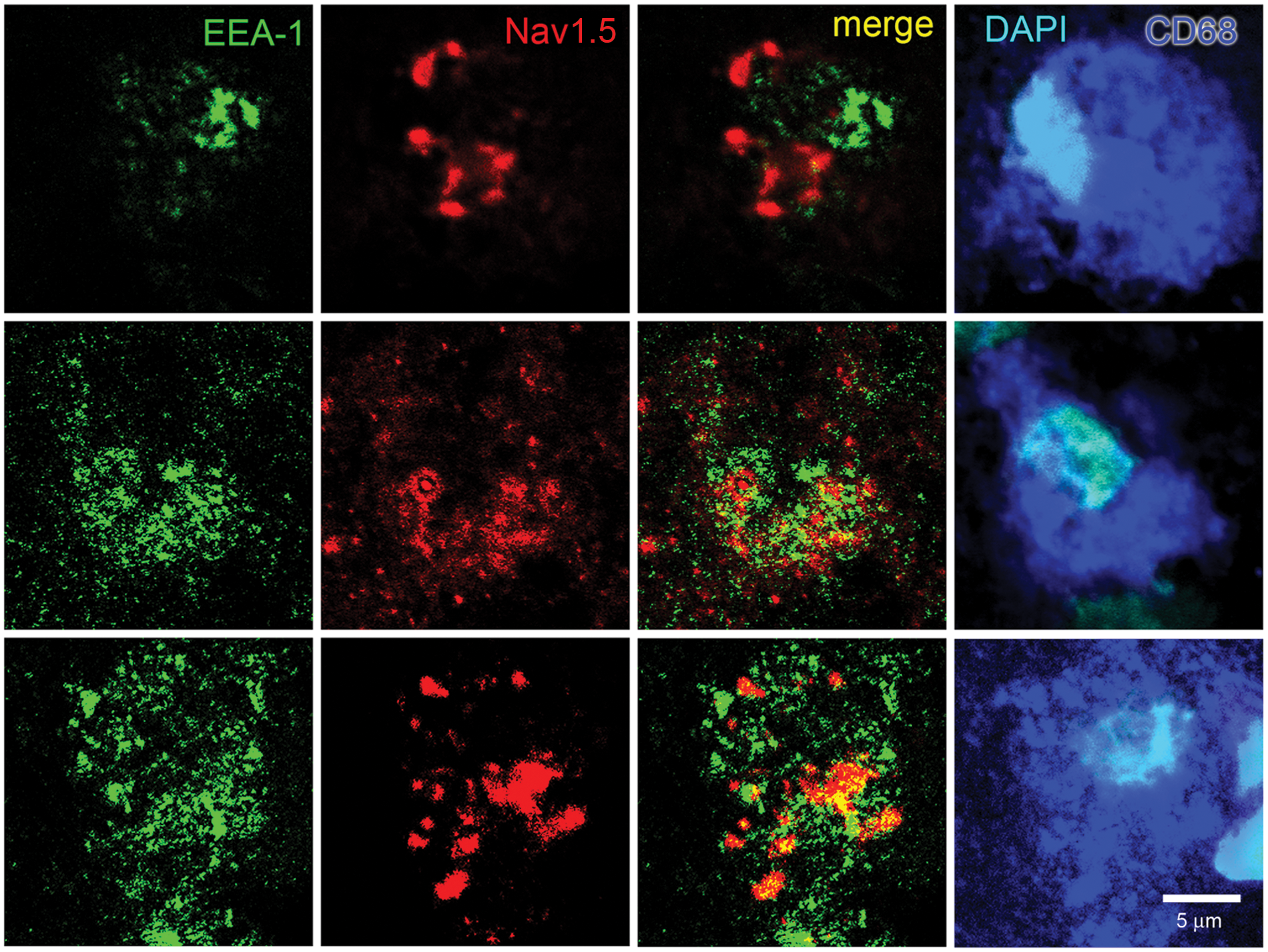
Co-immunocytochemistry for early endosome marker EEA-1 and Nav1.5 in macrophages within MS lesions. Individual macrophages from active lesions of 3 subjects are labeled with CD68 (blue) and nuclei stained with DAPI (cyan). The macrophages exhibit vesicular labeling for EEA-1 (green) and Nav1.5 (red), but only limited co-localization (yellow) of EEA-1 and Nav1.5.
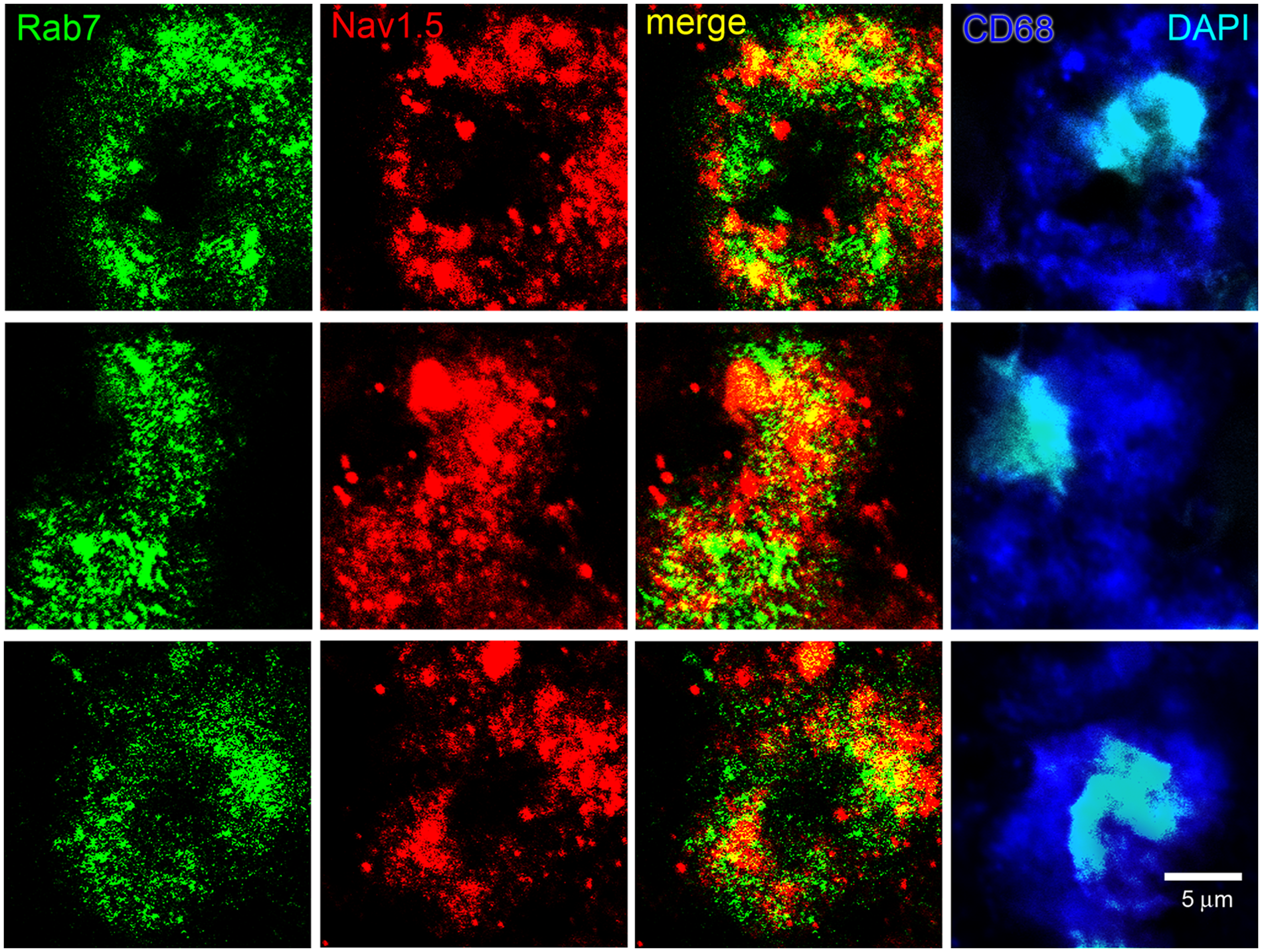
Co-localization of late endosome marker Rab7 and Nav1.5 in macrophages within MS lesions. Individual macrophages from active lesions of three subjects are labeled with CD68 (blue) and nuclei stained with DAPI (cyan). Late endosomes of activated macrophages display a vesicular pattern of Rab7 immunolabeling (green). Endosomes exhibit distinct Nav1.5 immunoreactivity (red), with some endosomes displaying intense labeling while others have limited immunoreactivity. Note the extensive co-localization (yellow) of Rab7 and Nav1.5 within macrophages.
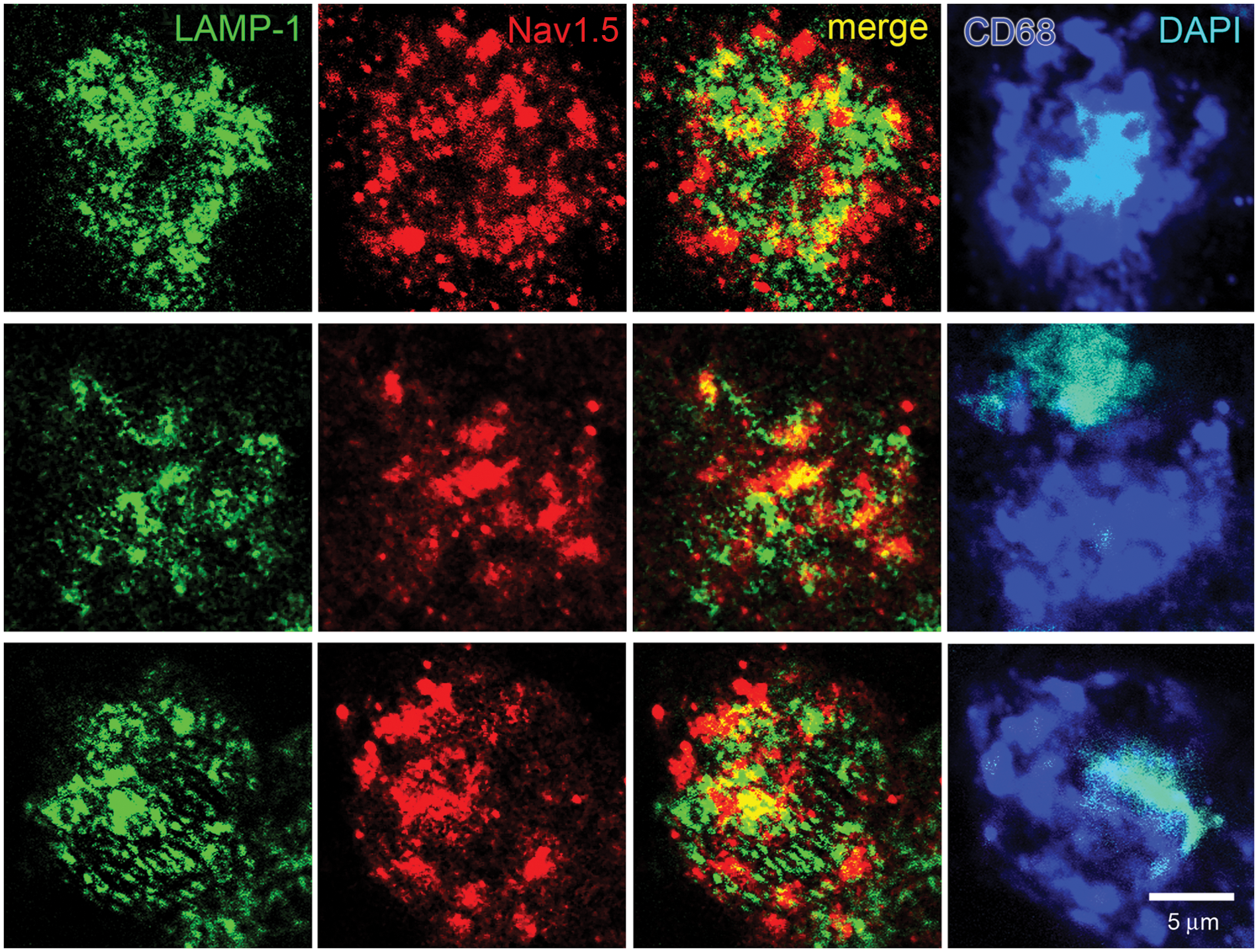
Co-localization of late endosome and phagolysosome marker LAMP-1 and Nav1.5 in macrophages within MS lesions. Individual macrophages from active lesions of two subjects are labeled with CD68 (blue) and nuclei stained with DAPI (cyan). Endosomal/phagolysosomal compartments in activated macrophages exhibit LAMP-1 labeling. LAMP-1+ vesicles exhibit expression of Nav1.5 that is not as extensive as that of Rab7 and Nav1.5.
Nearly all macrophages in active MS plaques exhibited vesicles that were labeled for EEA-1 (Figure 1). The macrophages also displayed vesicular Nav1.5 labeling (red), but the Nav1.5 labeling was generally not co-localized with EEA-1+ early endosomes. There was a tendency for the Nav1.5-positive vesicles to be of a larger size than the EEA-1+ vesicles, with a morphology similar to that of late endosomes which, as shown below, express this sodium channel isoform. Most macrophages in active plaques displayed clear labeling for the late endosomal marker Rab7, with the Rab7+ vesicles ranging in size from submicron to several microns (Figure 2). In contrast to the sparse expression of Nav1.5 in EEA-1+ early endosomes, there was substantial co-localization of Nav1.5+ and Rab7+ vesicles within macrophages in MS plaques. The late endosomal/phagolysosomal marker LAMP-1 was also detected within macrophages (Figure 3). Nav1.5 was clearly detectable in many LAMP-1+ endosomal vesicles, although the level of co-localization did not appear to be as extensive as that of Rab7 and Nav1.5. Carrithers et al. 8 utilized immunocryoelectron microscopy to demonstrate that Nav1.5 was localized within limiting membranes of late endosomes and phagolysosomes in macrophages in vitro. The light microscopic methods utilized in our study permitted us to demonstrate expression of Nav1.5 within late endosomes/phagolysosomes, but immunoultrastructural techniques, which would be required to localize Nav1.5 to endosomal membranes in the macrophages, were not feasible in this post-mortem tissue.
Quantification of the co-localization of Nav1.5 with the early (EEA-1) and late endosomal/phagolysosomal (Rab7 and LAMP-1) markers is shown in Figure 4. Nav1.5 immunolabeling was detectable in approximately 5% of the area labeled with EEA-1. In contrast, Nav1.5 labeling was present over nearly 20% of the area labeled with Rab7. The co-localization of Nav1.5 and LAMP-1 was intermediate between EEA-1 and Rab7, with Nav1.5 labeling present in approximately 12% of the LAMP-1 positive area. The expression of Nav1.5 in EEA-1+ endosomes was significantly (p<0.05) reduced compared to the expression of Nav1.5 in Rab7+ and LAMP-1+ endosomes.
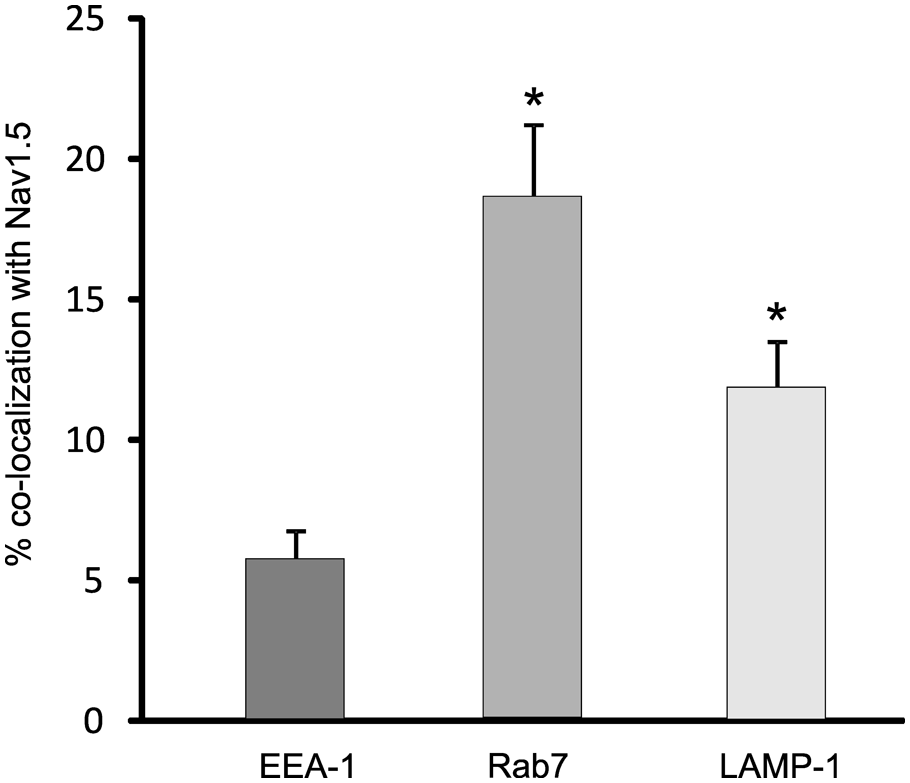
Quantification of co-localization of endosomal markers and Nav1.5. Approximately 5% of vesicles labeled with the early endosomal marker, EEA-1, show labeling for Nav1.5. In contrast, Nav1.5 immunoreactivity is present in ~20% of vesicles labeled with the late endosomal marker, Rab7, and in ~12% of vesicles labeled with late endosomal/phagolysosomal marker, LAMP-1.
We examined whether Nav1.5 was expressed only in macrophages within active MS lesions or was also expressed by microglia in peri-lesion areas or in control tissue. Shown in the left-hand panel of Figure 5 is a montage of images of subventricular tissue that extends from an active plaque (lower region) to adjacent normal-appearing white matter (NAWM; upper region). The active lesion exhibits an abundance of RCA-1+ macrophages, which include peripherally-recruited and resident cells, and a loss of PLP immunolabeling, while NAWM displays robust PLP immunolabeling and profiles of RCA-1+ endothelium. The three right-hand boxes of Figure 3 demonstrate, at increased magnification, examples of a macrophage within the active lesion (bottom box) and microglia at ~500 µm (middle box) and 1500 µm (upper box) from the lesion boundary. The macrophage within the plaque exhibits Nav1.5 and Rab7 immunolabeling, with many of the Rab7+ late endosomes displaying co-localization with Nav1.5. In contrast, Nav1.5 and Rab7 are not detectable in RCA-1+ microglia approximately 1500 µm from the lesion boundary. Microglia that are closer to the plaque boundary (~500 µm), and which have transformed to a more amoeboid morphology, also display a lack of Nav1.5 and Rab7 immunolabeling. Similar to the absence of detectable Nav1.5 labeling in microglia within NAWM distant from an active plaque border, Nav1.5 was not detectable in microglia in normal control brain tissue (data not shown).
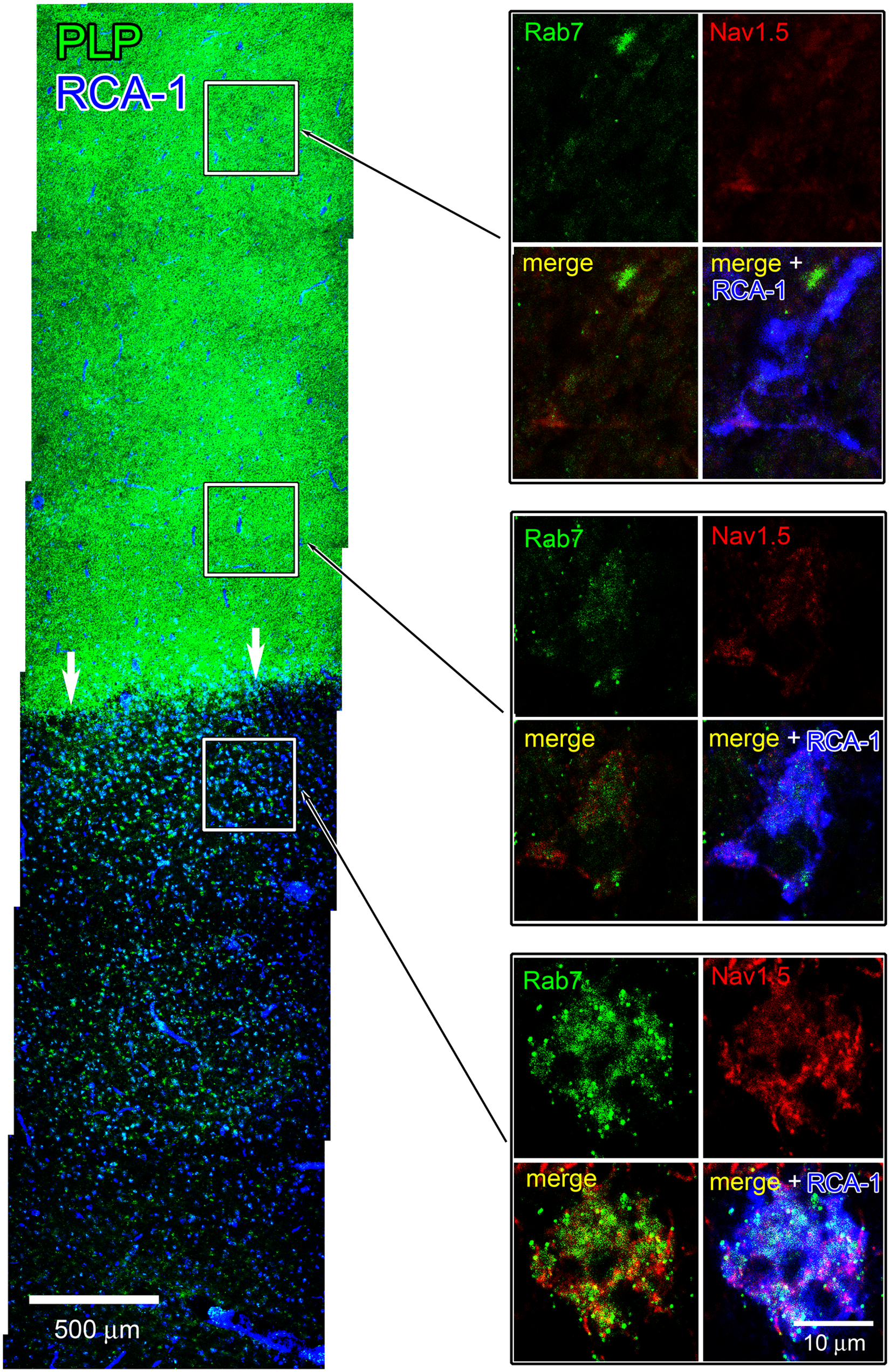
Nav1.5 expression in active lesion and NAWM. Left-hand panel: a montage of images is shown of subventricular tissue extending from an active lesion (arrows) through adjacent NAWM and reacted with RCA-1 and PLP. The active lesion exhibits an abundance of RCA-1+ macrophages (blue) and a lack of PLP staining (green). NAWM displays robust PLP labeling (green); note prominent labeling of endothelium with RCA-1 (blue) in NAWM. Right-hand panels: a section serial to that shown in the left-hand panel was immunoreacted with RCA-1, Nav1.5, and Rab7. Boxes illustrate RCA-1+ microglia (blue) ~1500 µm (top) and 500 µm (middle) outside of the lesion border. Note the lack of Nav1.5 (red) and Rab7 (green) immunolabeling in these microglia. Macrophage within the active lesion exhibits robust Nav1.5 and Rab7 immunolabeling, with substantial co-localization of Nav1.5+ and Rab7+ (lower box).
Phagocytosis of myelin by macrophages in MS lesions has been demonstrated in several studies,2–4 but has not been previously localized to specific endosomal compartments. As demonstrated in Figure 6(a) and (b), RCA-1+ macrophages within active MS plaques exhibit engulfed myelin fragments that are labeled with PLP antibody. Not all macrophages within active MS lesions exhibited PLP+ fragments of myelin; however, in the subpopulations of macrophages that displayed PLP immunoreactivity, ~30% (10/33) of the PLP+ myelin fragments were co-localized with LAMP-1. Additional experiments were performed to determine whether Nav1.5 was co-localized with the intra-cellular PLP+ fragments. As shown in Figure 7(a) and (b), there was co-localization of phagocytosed PLP+ myelin fragments and Nav1.5+ vesicles in RCA-1+ macrophages.
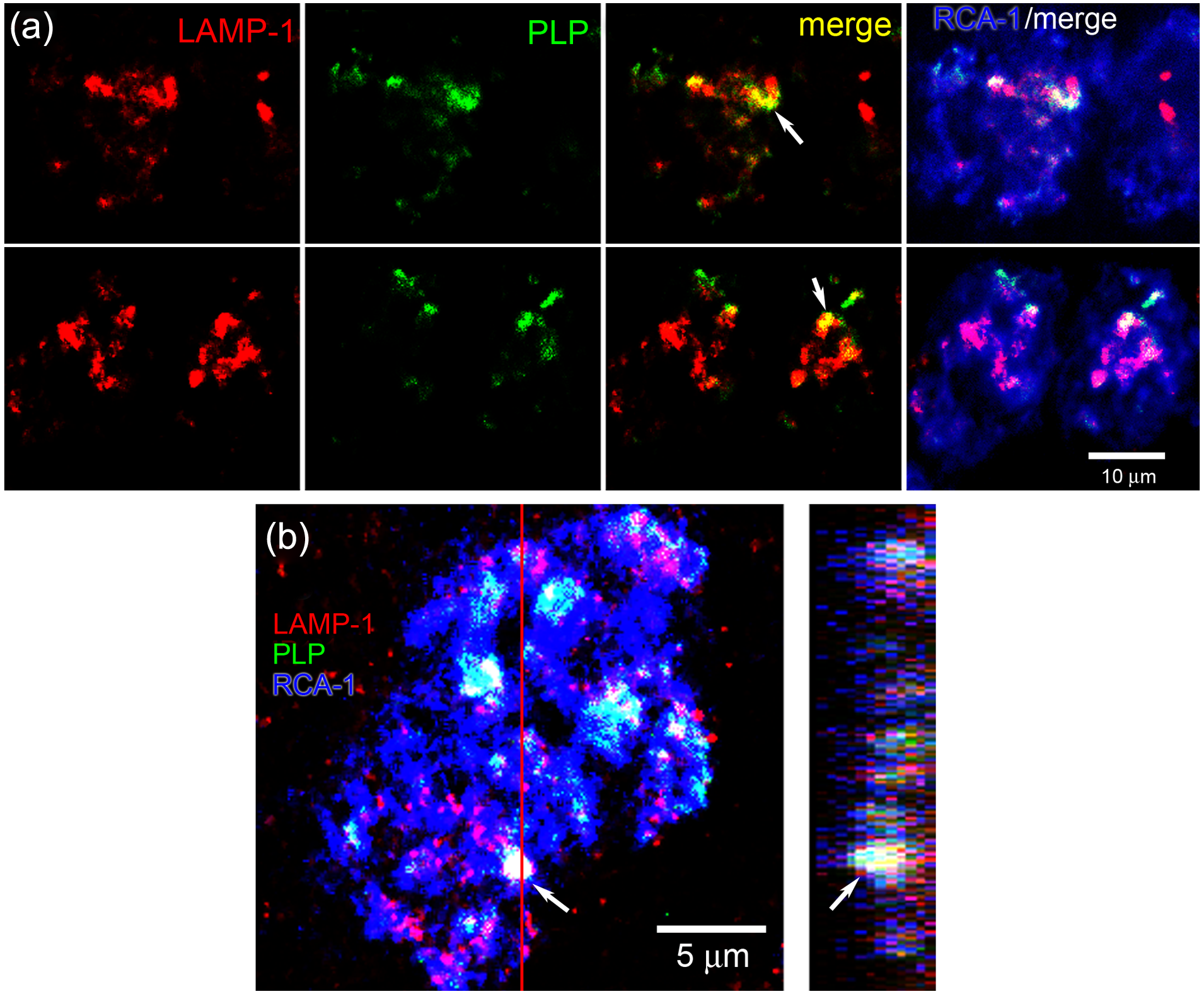
Co-localization of LAMP-1 and myelin fragments in macrophages within MS lesion. (a) Two examples of individual RCA+ macrophages are shown. Fragments of myelin (labeled for PLP (green)) are engulfed within RCA-1+ macrophages (blue). LAMP-1+ (red) endosomes are present in the macrophages; note co-localization (yellow) of LAMP-1 and PLP. (b) Confocal Z-stack reconstruction (left panel) and orthogonal view (right panel) of macrophage labeled with LAMP-1 (red), PLP (green), and RCA-1 (blue). Arrow indicates one region of co-localization of PLP and LAMP-1 in RCA-1+ macrophage. Red line indicates plane of section for orthogonal view.
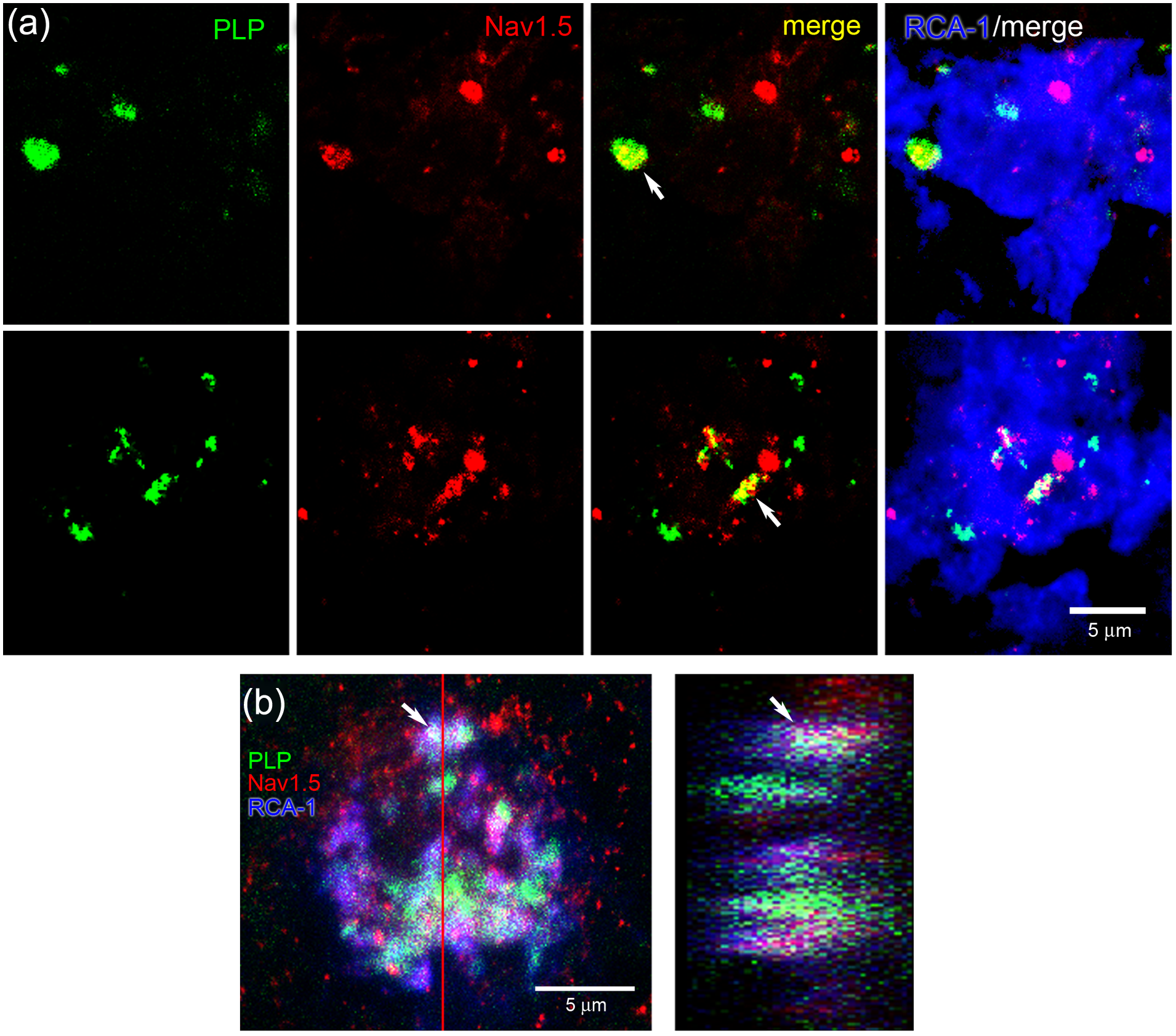
Co-localization of engulfed myelin fragments and Nav1.5 in macrophages. Two examples of individual RCA+ macrophages are shown. PLP (green) and Nav1.5 (red) are co-localized (yellow) in RCA-1+ macrophages (blue) within MS lesions. (b) Confocal Z-stack reconstruction (left panel) and orthogonal view (right panel) of macrophage labeled with PLP (green), Nav1.5 (red) and RCA-1 (blue). Arrow indicates one region of co-localization of PLP and Nav1.5 in RCA-1+ macrophage. Red line indicates plane of section for orthogonal view.
As illustrated in Figure 7, PLP+ myelin fragments within macrophages exhibited co-localization with Nav1.5. To more definitively determine whether Nav1.5 and myelin fragments were co-localized in identified endosomal compartments, we performed triple-label studies with antibodies to Nav1.5, PLP, and Rab7. An example is shown in Figure 8 where a macrophage is located immediately adjacent to a segment of a PLP+ myelinated fiber, in which it appears that myelin has been stripped from a portion of the fiber. Nav1.5 is co-localized in many of the Rab7+ endosomes within the macrophage. In addition, some Rab7+ endosomes exhibit co-localization with Nav1.5 and PLP.
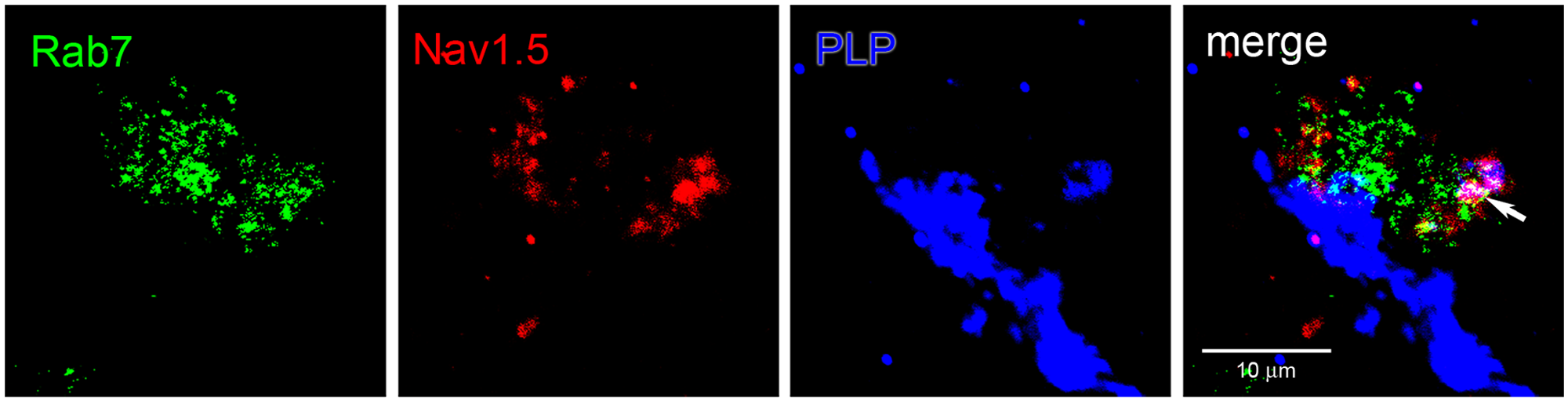
Immunolabeling for Rab7, Nav1.5, and PLP in macrophages within MS lesion. Section of an active MS lesion was triple-immunolabeled for the late endosomal marker Rab7, Nav1.5, and myelin fragments (PLP). Vesicular labeling for Rab7 (green) and Nav1.5 (red) within the macrophage that is immediately adjacent to a disrupted PLP+ (blue) myelinated fiber. Fragments of engulfed PLP+ myelin are co-localized (white) within Rab7+/Nav1.5+ vesicles (arrow).
Discussion
Macrophages are active participants in the destruction of white matter within active/chronic active plaques in MS.2,3,5 Phagocytosis and degradation of myelin by macrophages is a complex and highly-regulated process that involves multiple protein–protein interactions and significant ionic fluxes across limiting membranes of maturing endosomes, resulting from the activity of ion channels, pumps, transporters, and exchangers on these membranes.6,7 Acidification of endosomes is critical for functioning of the endosomal pathway, being requisite for endosomal sorting, activity of degradative enzymes, and the formation of intraluminal vesicles. 7 Molecular mechanisms that regulate proton accumulation within the lumena of endosomes are incompletely understood. However, it is known that the macrophage plasma membrane depolarizes during endocytosis and activation, 24 suggesting involvement of voltage-gated sodium channels, particularly in light of observations that macrophages/microglia can express sodium currents. 25
The sodium channel family includes nine isoforms (Nav1.1–Nav1.9), each of which exhibits distinct voltage-dependence, kinetic, and pharmacological. In neurons and muscle cells, sodium channels are responsible for the initiation and propagation of action potentials. However, sodium channel isoforms are also expressed in a variety of cells generally considered to be non-excitable, including astrocytes, chondrocytes, osteoblasts, lymphocytes, endothelial cells, keratinocytes, and some metastatic cancer cells.26,27 Contribution(s) of sodium channels to cellular functions in these non-excitable cells are generally not well-understood, although the activity of the cardiac sodium channel Nav1.5 in breast cancer cells 28 and T-lymphocytes 16 and Nav1.7 in prostate cancer cells 29 and a subset of dendritic cells 18 has been linked to the invasiveness of these cells. Interestingly, it was recently demonstrated that Nav1.5 is upregulated in human reactive astrocytes in MS plaques and in the glial scar surrounding new and old cerebrovascular accidents and brain tumors, 30 though the functional consequences of this upregulation are not clear. In the present study, Nav1.5+ astrocytes were easily distinguishable from microglia and macrophages by morphology and cell-specific markers, in addition to the more robust and widespread cytoplasmic expression of Nav1.5 in hypertrophic astrocytes. 30 In addition, while RCA-1 has been reported to label reactive astrocytes in pathological brain tissue, 31 we did not observe RCA-1 labeling above background levels within GFAP+ astrocytes in our samples (data not shown).
Carrithers et al.8,9 have provided evidence for a novel role for sodium channels in the regulation of phagocytosis by macrophages in vitro. Utilizing cultured THP-1 cells, a monocyte-derived cell line, and primary monocyte-derived macrophages studied in vitro, Carrithers et al. 8 have shown that the activity of Nav1.5 contributes to the regulation of endosomal acidification by providing a route for the movement of positive charges from the endosomal lumen to the cytoplasm, thus offsetting the entry of protons into the lumen. Significantly, Nav1.5 has been shown to be preferentially co-localized with markers of late endosomes/phagolysosomes (e.g. Rab7, LAMP-1) rather than early endosomal markers 8 (EEA-1), an expression pattern that parallels the increasing acidification of late endosomes. This study also showed that non-selective sodium channel blockade with TTX, and selective shRNA knockdown of Nav1.5, reduce phagocytosis by cultured macrophages. More recently, it was reported by Carrithers et al. 9 that the knockdown of Nav1.5 attenuates the initial calcium response to bacterial challenge in macrophages, and prevents extended and localized calcium oscillations in maturing endosomes. These observations suggest the possibility that Nav1.5 channels may contribute to macrophage-mediated phagocytosis events in vivo.
The present results demonstrate that Nav1.5 is expressed by macrophages within parenchyma of active MS lesions, where it is preferentially localized within Rab7+ and LAMP-1+ late endosomal/phagolysosomal compartments and is only sparsely present within EEA-1+ early endosomes. We did not examine the expression of Nav1.5 in lipid-laden macrophages in perivascular cuffs, 32 which often displayed high levels of background fluorescence that precluded definitive immunocytochemical demonstration of Nav1.5. Moreover, while we recognize that heterogeneity exists in macrophage phenotype between perivascular and parenchymal sites in MS lesions, 33 and during stages of plaque development,2,3 quadruple-stain immunocytochemistry was not feasible, so we could not distinguish subpopulations of macrophages. Therefore, only macrophages within lesion parenchyma were examined, without further classification of their phenotypes. Nevertheless, our results provide for the first time data on the expression of Nav1.5 within human macrophages in situ.
Nav1.5 labeling was not observed in microglia ~500 µm from the active lesion border or within control tissue, suggesting that detectable expression of Nav1.5 in late endosomes and phagolysosomes occurs in parallel with active phagocytosis by macrophages. It is likely that microglia, which are the main population of phagocytosing cells during early demyelination in MS lesions and are also termed macrophages at this time, 34 also exhibit Nav1.5 expression within late endosomal/phagolysosomal compartments when undergoing active phagocytosis. However, our studies could not distinguish whether the macrophages exhibiting Nav1.5 expression within endosomal compartments in the active MS lesions were peripherally- or resident-derived.
The observation of co-localization of Nav1.5, Rab7, and PLP, a myelin marker, in macrophages within active MS lesions adds to the evidence for a role of Nav1.5 in the regulation of phagocytic engulfment and degradation of myelin in active plaques. In this context, it might be expected that blockade of Nav1.5 in phagocytosing macrophages could contribute to the neuroprotection observed in rodent models of MS (EAE) following administration of sodium channel blockers. Treatment with phenytoin,11,13 lamotrigine, 12 and carbamazepine, 35 which are blockers of TTX-sensitive and -resistant sodium currents, 10 result in axonal preservation and improved clinical status in rats and mice with EAE. These sodium channel blockers may protect axons via multiple converging pathways. One pathway involves a direct action on axonal sodium channels that prevents an influx of Na+ into axoplasm that would otherwise lead to reverse operation of the sodium-calcium exchanger and importation of damaging levels of Ca2+ and initiation of a cascade of Ca2+-induced degradative pathways.14,15 A second pathway involves an effect on the mobility of immune cells. Systemic treatment with phenytoin significantly attenuates immune cell infiltrates in the spinal cords of mice with EAE compared to untreated mice with EAE.13,35 Consistent with an action of sodium channel blockade on the mobility of immune cells, TTX and phenytoin have been shown to significantly reduce migration of cultured microglia. 17 In the context of the observations of Carrithers et al. 8 , the present results suggest that sodium channel blockade may provide axonal protection by blocking Nav1.5-regulated acidification of endosomal compartments, thus limiting effective functioning of the endosomal degradative pathway.
Together with earlier observations, which showed that knockdown of Nav1.5 attenuates phagocytosis by macrophages in vitro, 8 the present results suggest that, in future studies of sodium channel blockers in MS, macrophage activity within the CNS should be monitored. Our observations also raise the possibility that, if methods for cell type-specific blockade of Nav1.5 can be developed, it might be possible to attenuate macrophage-mediated CNS injury via macrophage-specific targeting of Nav1.5.
Footnotes
Funding
This work was supported in part by grants from the National Multiple Sclerosis Society (grant number RG1912 to SGW), and the Medical Research Service and Rehabilitation Service, Department of Veterans Affairs (grant number B7521R to SGW). The Center for Neuroscience and Regeneration Research is a collaboration of the Paralyzed Veterans of America with Yale University.
Conflict of interest statement
The authors declare that there are no conflicts of interest.