
Abstract
Background:
Neutralizing antibodies (NAb) affect efficacy of interferon-beta (IFN-b) treatment in multiple sclerosis (MS) patients. NAbs evolve in up to 44% of treated patients, usually between 6–18 months on therapy.
Objectives:
To investigate whether early binding antibody (BAb) titers or different IFN-b biomarkers predict NAb evolution.
Methods:
We included patients with MS or clinically isolated syndrome (CIS) receiving de novo IFN-b treatment in this prospective European multicenter study. Blood samples were collected at baseline, before and after the first IFN-b administration, and again after 3, 12 and 24 months on that therapy; for determination of NAbs, BAbs, gene expression of MxA and protein concentrations of MMP-9, TIMP-1, sTRAIL, CXCL-10 and CCL-2.
Results:
We found that 22 of 164 (13.4%) patients developed NAbs during a median time of 23.8 months on IFN-b treatment. Of these patients, 78.9% were BAb-positive after 3 months. BAb titers ≥ 1:2400 predicted NAb evolution with a sensitivity of 74.7% and a specificity of 98.5%. Cross-sectionally, MxA levels were significantly diminished in the BAb/NAb-positive samples; similarly, CXCL-10 and sTRAIL concentrations in BAb/NAb-positive and BAb-positive/NAb-negative samples, respectively, were also diminished compared to BAb/NAb-negative samples.
Conclusions:
BAb titers reliably predict NAbs. CXCL-10 is a promising sensitive biomarker for IFN-b response and its abrogation by anti-IFN-b antibodies.
Keywords
Introduction
Neutralizing antibodies (NAb) against interferon-beta (IFN-b) are the best-defined hazard of treatment efficacy in patients with multiple sclerosis (MS). NAbs evolve in up to 44% of treated patients, 1 usually between 6 and 18 months on therapy.2,3 This late development warrants efforts to predict NAbs, in order to prevent ineffective and costly treatment, especially as more MS therapeutics get approved. Since no reliable predictor of NAbs has been detected until now, the aim of this study was to analyze several candidate markers in one prospective cohort of IFN-b treated MS patients. The rationale for each molecule explored will be briefly explained. We included binding antibodies (BAb), which are known to occur in up to 88% of treated patients. Approximately 50% of BAb-positive patients become NAb-positive later on2,4 and there is some evidence that a boost of BAb titers precedes NAb evolution. 5 We included in the IFN-b biomarkers myxovirus protein-A (MxA), 6 which is specifically induced by Type I IFNs 7 and is well-established as a surrogate for in vivo IFN-b bioavailability. 8 Additionally, we investigated several molecules which are involved in the mechanisms of action of IFN-b and can be easily determined in serum: soluble tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL), which is shown to inhibit autoreactive and antigen-specific T cells9–11; the chemokines CXCL-10/IP-10 and CCL-2/MCP-1, which are both involved in the recruitment of leukocytes12,13; as well as matrix metalloproteinase-9 (MMP-9) and its antagonist tissue-inhibitor of matrix metalloproteinase-1 (TIMP-1), which regulate leukocyte trafficking across the blood-brain barrier.14,15
Materials and methods
The prospective European multicenter study NABINMS (Neutralizing antibodies on Interferon beta in Multiple Sclerosis) was conducted by 22 clinical centers in Austria, Denmark, Italy, the Netherlands, Spain and Switzerland. We enrolled 222 patients with clinically isolated syndrome (CIS) or relapsing MS (RMS) fulfilling the revised McDonalds criteria 2005, 16 between June 2006 and August 2009. Inclusion criteria were age of at least 18 years and a medical indication for first-time IFN-b treatment. Exclusion criteria were prior IFN-b treatment or any contraindication to the therapy. Eligible patients received one of the three available IFN-b preparations: intramuscular (IM) IFN-b-1a (Avonex®, Biogen Idec, Cambridge, MA, USA), subcutaneous (SC) IFN-b-1a (Rebif® 22 or 44 µg, Merck Serono, Geneva, Switzerland) or SC IFN-b-1b (Betaferon®, Bayer Schering, Berlin, Germany).
Blood samples were collected by peripheral venous puncture at baseline (BL), immediately before (BL1) and 4–12 hours after (BL2) the first IFN-b injection, and at every follow-up (4–12 hours post-injection) after 3 months ± 2 weeks (Mo3), 12 months ± 4 weeks (Mo12) and 24 months ± 4 weeks (Mo24). Clinical assessments were performed at all visits. We isolated serum from blood by centrifugation, after the blood samples were allowed to clot for ≥ 30 minutes. Whole blood samples were kept at room temperature for ≥ 4 hours after withdrawal. All study samples were stored at −20ºC until analysis, which was centrally performed at the Neuroimmunology Laboratory at Innsbruck Medical University.
Goal
The primary aim of the study was to investigate whether gene expression of MxA and concentrations of MMP-9, TIMP-1, sTRAIL, CXCL-10 and CCL-2 within the first 3 months on IFN-b treatment (i.e. at BL1, BL2 or Mo3), or BAb titers determined after 3 months on therapy (Mo3), predict later NAb development. The secondary aim was to study the relationship of NAb titers to and the impact of BAbs on IFN-b biomarkers.
NAb assay
NAbs were measured in serum by a luciferase assay using human fibrosarcoma cells that express IFN-b receptors on their surface and were stably transfected with a luciferase reporter gene cassette. Upon IFN-b binding, luciferase is produced in a predictable, dose-dependent manner, which becomes diminished in the presence of NAbs17,18 (for assay description, see Supplementary Material). Samples with a titer ≥ 20 neutralizing units (NU) were considered as NAb-positive. For patients receiving IFN-b-1a, NAb titers up to 100 NU were classified as low and > 100 NU, as high; for those on IFN-b-1b, titers up to 400 NU were considered as low and > 400 NU, as high. 8
BAb assay
We measured BAbs by direct enzyme-linked immunosorbent assay (ELISA), as previously described, 19 at the time points designated BL1, Mo3 and Mo12. For details on the sample matrix, see the Supplementary Material.
Measurement of IFN-b biomarkers
We determined MxA at the mRNA level, as previously described. 20 Briefly, RNA was isolated from whole blood samples, converted to cDNA and subjected to real-time polymerase chain reaction (rtPCR) for MxA quantification. We used commercially available ELISA kits to measure serum concentrations of MMP-9, TIMP-1, sTRAIL, CCL-2 and CXCL-10. For details on assay performance, see Supplementary Material.
Statistics
We performed our statistical analysis using SPSS 18.0 (SPSS Inc, Chicago, IL) and GraphPad Prism 5.03 (GraphPad Inc, La Jolla, CA). Mann-Whitney-U or Kruskal-Wallis test was used for group comparisons of non-parametric data such as MxA, MMP-9, TIMP-1, sTRAIL, CCL-2, CXCL-10 concentrations or BAb titers, and t-test was used for parametric data such as patients’ age. Qualitative variables, e.g. NAb or BAb frequencies among the treatment groups, were compared with χ2-test. We analyzed repeated measurements by either Wilcoxon or Friedman test. Logarithmic transformation of data was performed to achieve normal distribution for univariate variance analysis with repeated measurement. We used Spearman coefficient for correlation analysis. We assessed sensitivities and specificities by receiver operating characteristics (ROC) analysis. Two-tailed P values < 0.05 were considered as statistically significant and were adjusted according to Bonferroni’s correction, in the case of multiple comparisons.
Standard protocol approvals and patient consents
The study protocol was approved by the local ethic committees (approval number AM2538 239/4.8) of all participating centers. Prior to any study-related investigations, written informed consent was obtained from all patients.
Results
Study population
We enrolled 222 patients into the study. 190 (85.6%) patients completed follow-up at Mo24 and of those, 150 were still on IFN-b treatment (Figure 1). Demographic and baseline clinical data are shown in Table 1. Overall, 920 blood samples were collected throughout the study, 667 samples while a patient was on treatment after a median time of 4.3 hours following the previous IFN-b injection (Table 2, Supplementary Material).
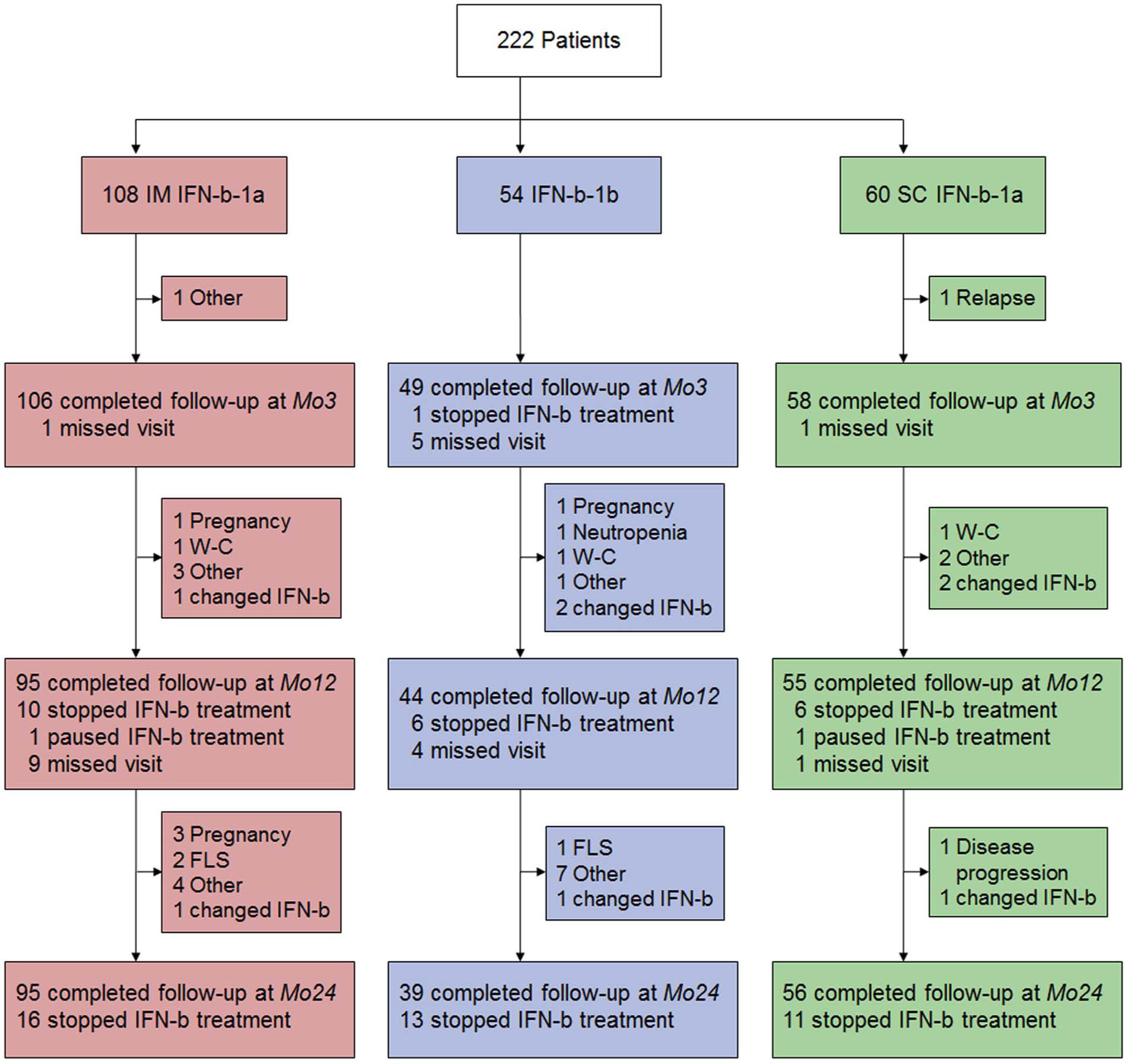
Patient flow chart.
Demographic and baseline clinical data.
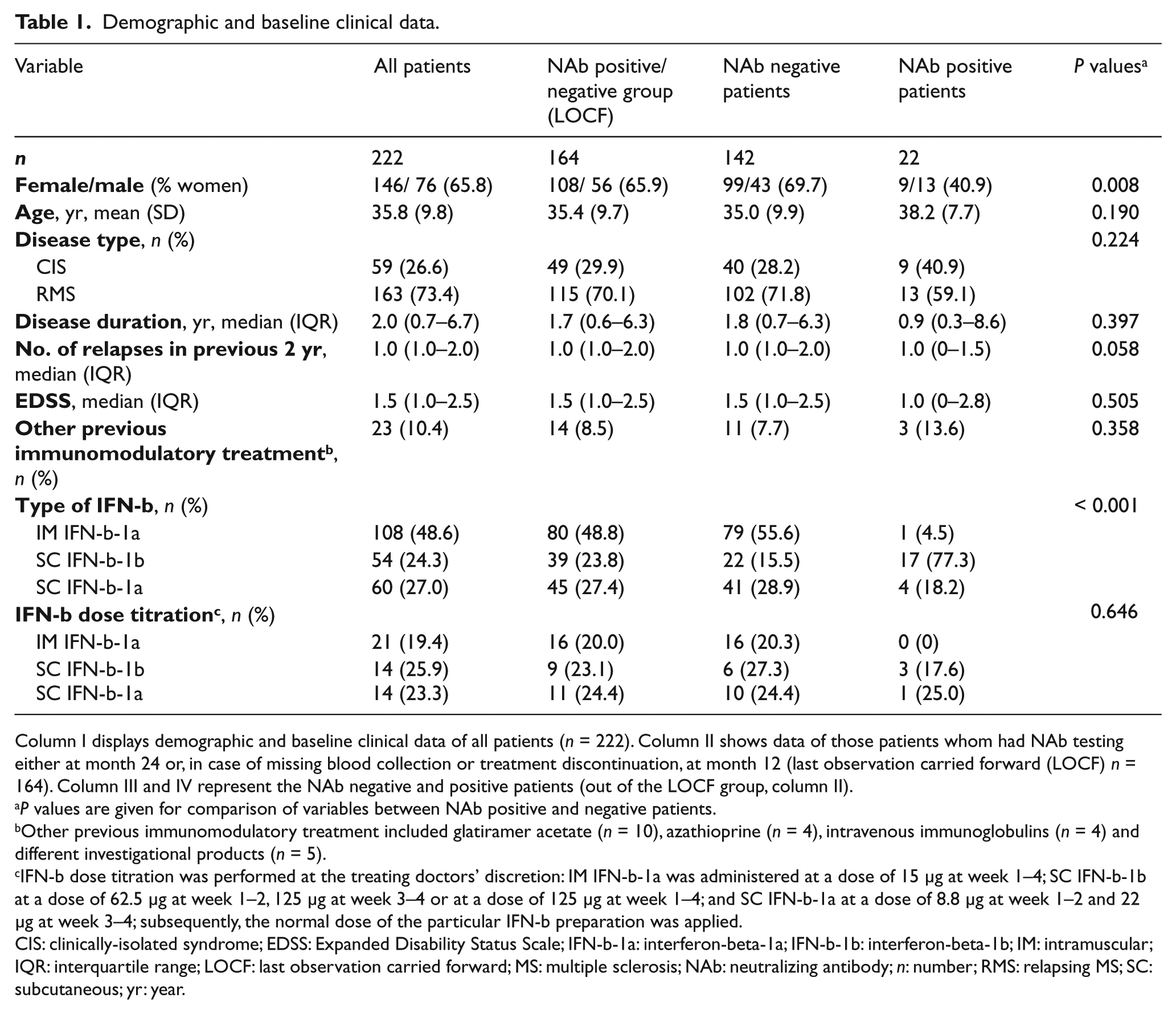
Column I displays demographic and baseline clinical data of all patients (n = 222). Column II shows data of those patients whom had NAb testing either at month 24 or, in case of missing blood collection or treatment discontinuation, at month 12 (last observation carried forward (LOCF) n = 164). Column III and IV represent the NAb negative and positive patients (out of the LOCF group, column II).
P values are given for comparison of variables between NAb positive and negative patients.
Other previous immunomodulatory treatment included glatiramer acetate (n = 10), azathioprine (n = 4), intravenous immunoglobulins (n = 4) and different investigational products (n = 5).
IFN-b dose titration was performed at the treating doctors’ discretion: IM IFN-b-1a was administered at a dose of 15 μg at week 1–4; SC IFN-b-1b at a dose of 62.5 μg at week 1–2, 125 μg at week 3–4 or at a dose of 125 μg at week 1–4; and SC IFN-b-1a at a dose of 8.8 μg at week 1–2 and 22 μg at week 3–4; subsequently, the normal dose of the particular IFN-b preparation was applied.
CIS: clinically-isolated syndrome; EDSS: Expanded Disability Status Scale; IFN-b-1a: interferon-beta-1a; IFN-b-1b: interferon-beta-1b; IM: intramuscular; IQR: interquartile range; LOCF: last observation carried forward; MS: multiple sclerosis; NAb: neutralizing antibody; n: number; RMS: relapsing MS; SC: subcutaneous; yr: year.
Neutralizing antibodies
All patients were NAb-negative at baseline (BL1 and BL2). We stratified patients who were on IFN-b treatment throughout the whole study into NAb-positive and negative groups, according to their NAb status at Mo24 (total n = 120). In the case of a missing blood collection (n = 31) or treatment discontinuation (n = 13), we used NAb status at Mo12 (last observation carried forward (LOCF)), resulting in 164 patients available for NAb status stratification. Demographic and baseline clinical data are shown in Table 1. Of the 164 patients, 22 (13.4%) were NAb-positive after a median time of 23.8 months, with a median titer of 184.5 NU. We found 9 of the 22 (40.9%) NAb-positive patients had high NAb titers. NAb frequency was significantly higher in patients treated with SC-IFN-b-1b (43.6%) than SC-IFN-b-1a (8.9%) or IM-IFN-b-1a (1.3%). A total of 149 of 164 patients had NAb testing at Mo3, of which three (2.0%) were NAb-positive, showing a median titer of 48 NU. We found that the 13 NAb-positive patients at Mo12 remained NAb-positive at Mo24, i.e. there were no transient NAbs. In comparing NAb titers at Mo12 and Mo24, eight patients showed increasing median titers (from 496 NU towards 2235.5 NU) and five patients had declining titers (from 166 NU towards 52 NU). Of the 106 NAb-negative patients at Mo12, only two patients had developed NAbs by Mo24, with a median titer of 74.5 NU.
Low CXCL-10 and sTRAIL expression after 3 months on IFN-b treatment predict NAb development
After beginning IFN-b treatment, overall MxA, sTRAIL, CXCL-10 and CCL-2 concentrations were significantly elevated at every following visit, as compared to BL1. At Mo24, the levels of TIMP-1 were significantly increased and MMP-9 levels decreased, as compared to BL1. Whereas MMP-9, TIMP-1 and CCL-2 values did not differ between the NAb-positive and NAb-negative treated patient groups at any time point; MxA, sTRAIL and CXCL-10 concentrations were significantly lower in the eventually NAb-positive group, at Mo12 and Mo24. At Mo3, sTRAIL and CXCL-10 levels were even diminished, and exclusion of the three patients who were already NAb-positive at Mo3 did not affect these results (Figure 2A-F).
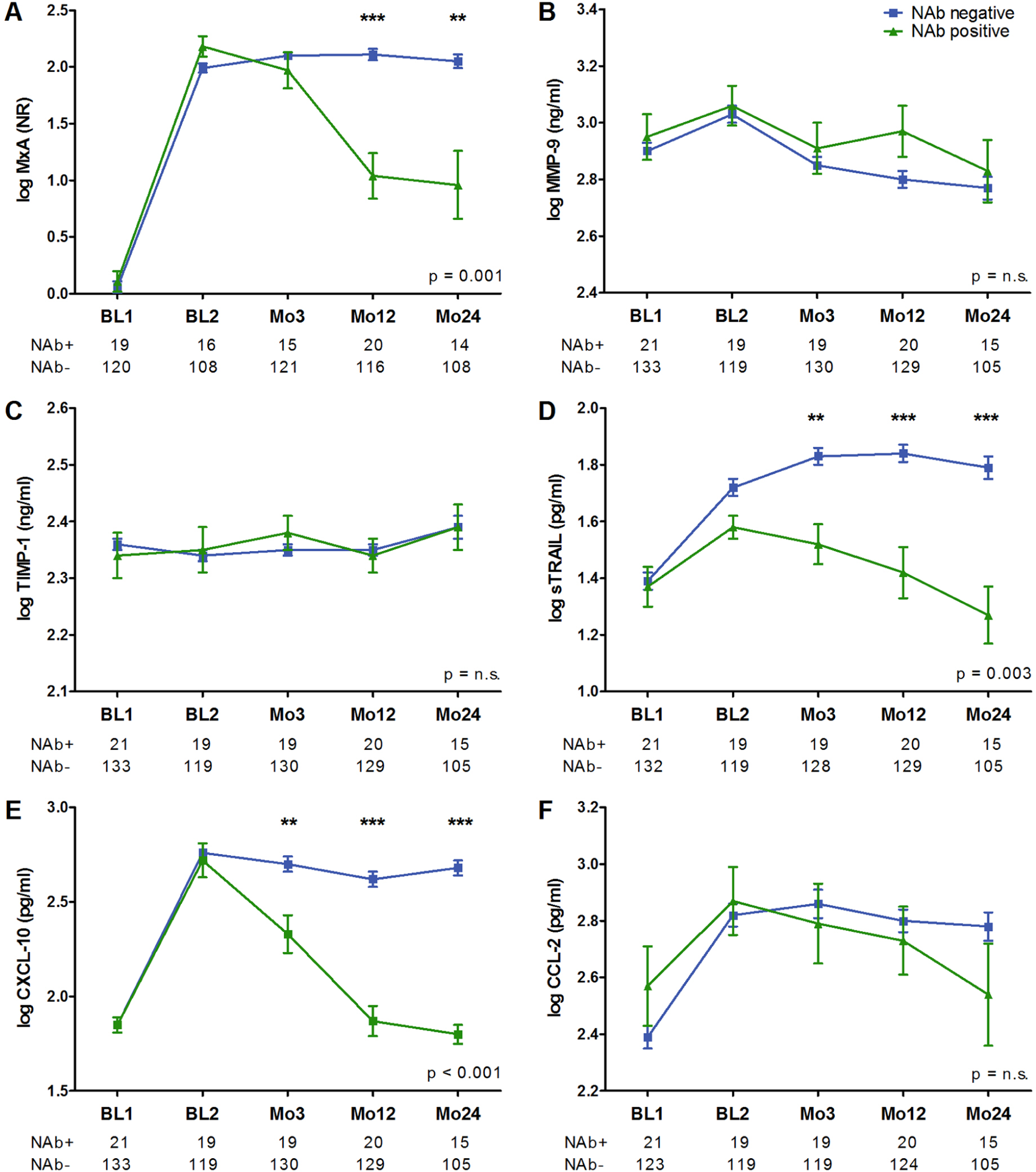
IFN-b biomarker concentrations in NAb positive and NAb negative MS patients.
ROC analysis of sTRAIL and CXCL-10 levels at Mo3 were performed, to calculate their predictive value for NAb evolution. The sTRAIL levels ≤ 42.2 pg/ml showed a sensitivity of 63.2% and a specificity of 72.2% (area under the curve (AUC) = 0.731; p = 0.001), and the CXCL-10 levels ≤ 173.5 pg/ml showed a sensitivity of 57.9% and a specificity of 80.5% (AUC = 0.727; p = 0.001).
BAb titers after 3 months on IFN-b treatment predict NAb development
At BL1, we found that 208/210 (99.0%) patients were BAb-negative. The remaining two patients had a median titer of 1:50 and were BAb- and NAb-negative in the subsequent tests at Mo3 and Mo12. Out of the 164 patients tested for NAbs (LOCF), 149 patients were also tested for BAbs at Mo3. Of those, 35 (23.5%) were BAb-positive. BAb frequencies and titers among the different treatment groups are shown in Table 3 of the Supplementary Material. At Mo3, we found that 42.9% of BAb-positive patients and only 3.5% of BAb-negative patients turned NAb-positive at follow-up, i.e. 78.9% of eventually NAb-positive patients and 15.4% (20 of 130) of NAb-negative patients were BAb-positive by Mo3. ROC analysis revealed a BAb cut-off titer ≥ 1:2400 with a sensitivity of 73.7% and a specificity of 98.5%, able to predict the later presence of NAbs (AUC = 0.874, Figure 3). Because most BAbs at Mo3 (91.4%) occurred within the SC-IFN-b-1b treatment group, and 46.9% of those developed NAbs later on, we performed ROC analyses also for this treatment group only. Similarly, a BAb cut-off titer ≥ 1:2400 showed a sensitivity of 87.5% and a specificity of 90% to predict NAbs. Furthermore, BAb titers at Mo3 significantly correlated with eventual NAb titers (LOCF: R = 0.719; p < 0.001); and different BAb cut-off titers were calculated for the prediction of NAbs or high-titer NAbs (Table 4, Supplementary Material). BAbs at Mo3 did not differ between patients showing increasing or declining NAb titers between Mo12 and Mo24.
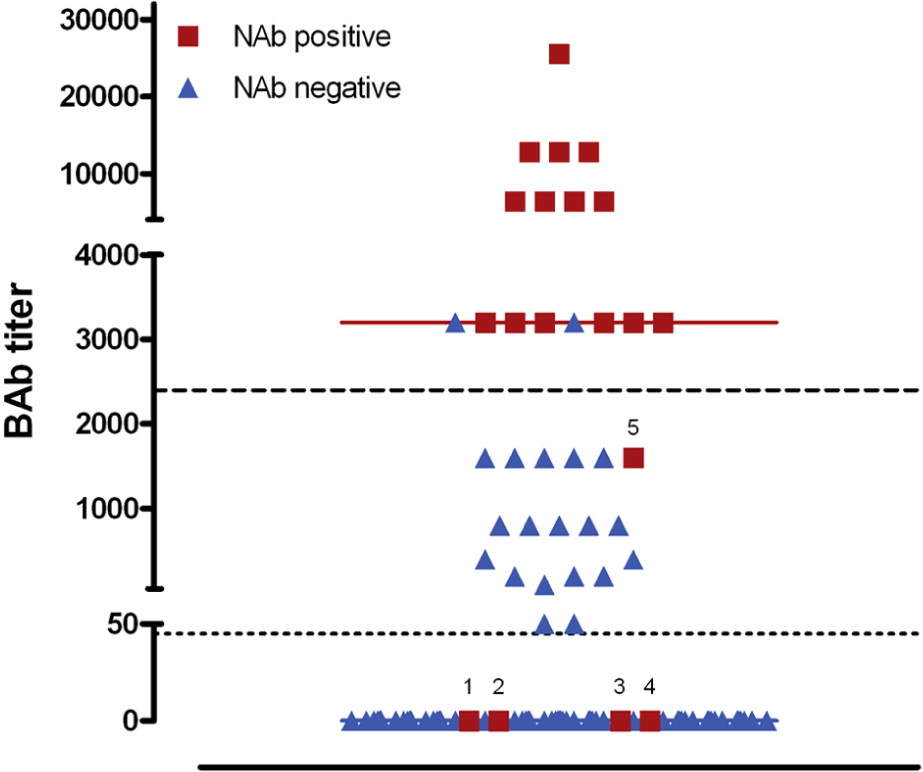
BAb titers predict NAb evolution.
CXCL-10 and sTRAIL concentrations are diminished in BAb-positive but NAb-negative patients
The following sections refer to all available samples during IFN-b therapy at designated time points, i.e. at Mo3, Mo12 or Mo24 (Table 2 of the Supplementary Material). At Mo3, a total of 197 samples from patients on IFN-b treatment were measured for BAbs. Concentrations of sTRAIL (p < 0.001) and CXCL-10 (p = 0.001) were significantly lower in the BAb-positive than BAb-negative group; whereas the MxA, MMP-9, TIMP-1 and CCL-2 levels were similar. To exclude the influence of NAbs on biomarker levels, we then analyzed only NAb-negative samples (n = 193) and essentially observed the same results (Figure 4A).
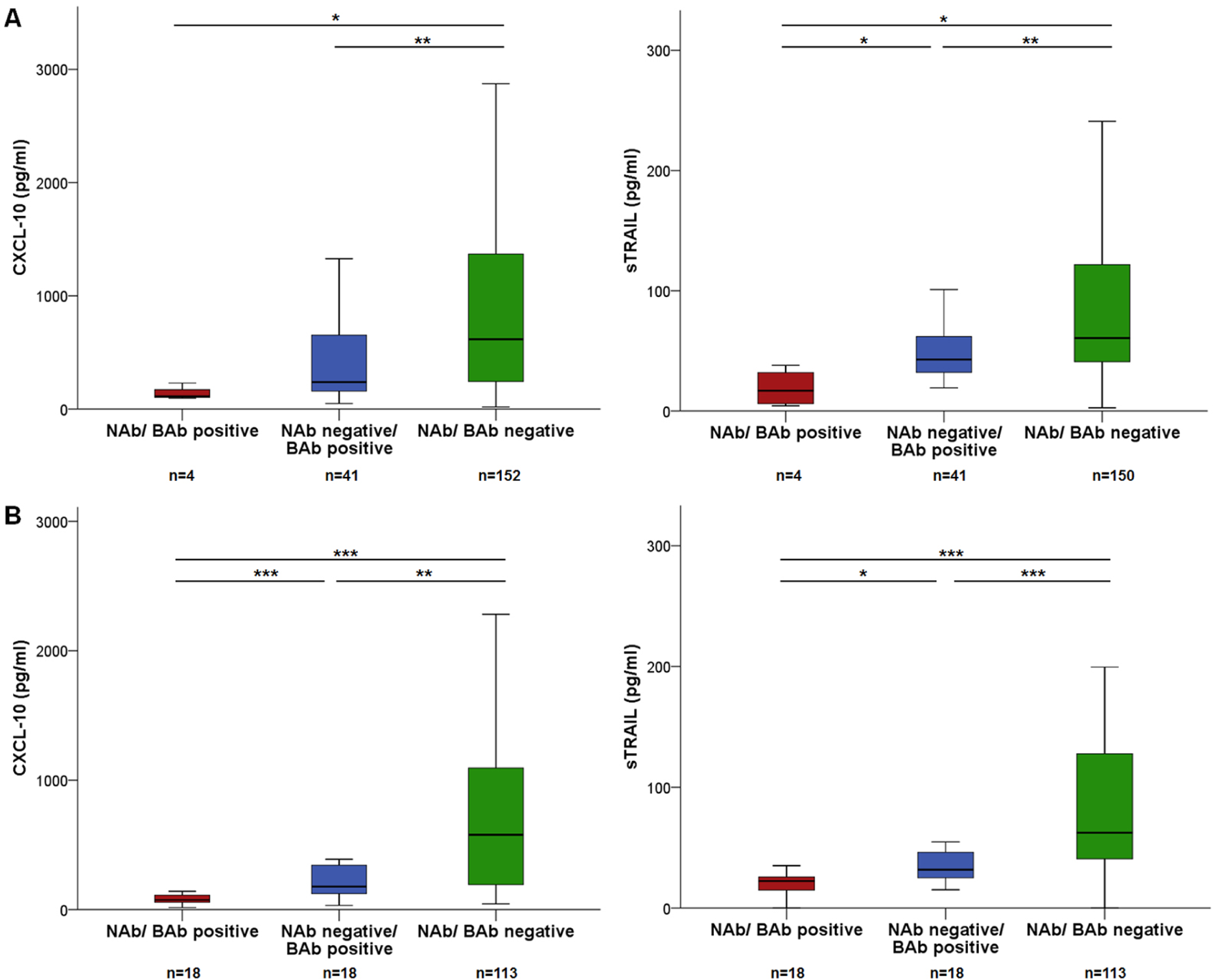
CXCL-10 and sTRAIL concentrations diminished in patients with BAbs and NAbs.
At Mo12, MxA, sTRAIL, CXCL-10 (all p < 0.001) and CCL-2 (p = 0.013) concentrations were significantly lower in BAb-positive than BAb-negative patients (n = 149), after exclusion of NAb-positive samples (n = 131), still sTRAIL (p < 0.001), CXCL-10 (p = 0.001) and CCL-2 (p = 0.003) levels were significantly decreased in BAb-positive patients (Figure 4B). Correlations of IFN-b biomarkers with NAb and BAb titers (at Mo12) are shown in Table 5 of the Supplementary Material.
CXCL-10 is more sensitive than MxA in detecting NAbs
We found that 18 of 149 (12.1%) patients at Mo12 and 15 of 122 (12.3%) patients at Mo24 were NAb-positive. We performed ROC analyses, to find the best biomarker to discriminate between NAb-positive and NAb-negative status. At both visits, CXCL-10 showed a higher sensitivity and specificity than MxA and sTRAIL (Figure 5). CXCL-10 and MxA levels in the NAb-negative, low NAb and high NAb titer patients at Mo12 and Mo24 are displayed in Figure 6 of the Supplementary Material.
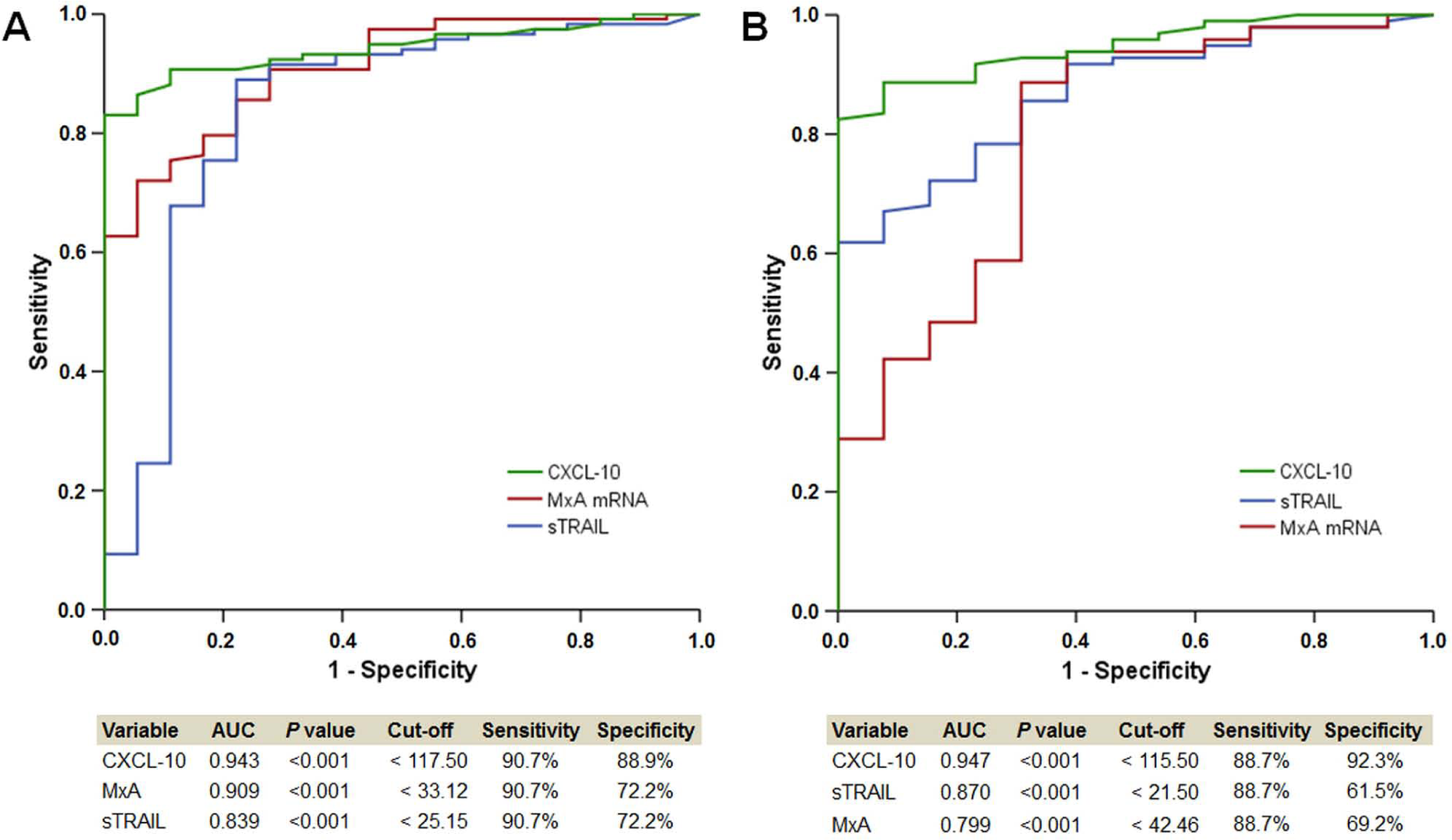
IFN-b biomarkers discriminate between NAb positive and negative samples.
Discussion
In this study, we investigated whether early BAb titers or IFN-b biomarkers predicted later NAb development. We demonstrated that BAb titers ≥ 1:2400 in patient serum after 3 months on IFN-b treatment reliably predicted the eventual presence of NAbs, after roughly 2 years. We showed that BAbs, independently from NAbs, are associated with diminished IFN-b bioavailability, as measured by reduced protein concentrations of CXCL-10 and sTRAIL. Furthermore, CXCL-10 showed a considerably higher sensitivity and specificity to discriminate between NAb-positive and NAb-negative samples than the well-established MxA.
Although several factors contributing to the immunogenicity of recombinant IFN-b are already known, 21 there are still unresolved issues concerning immune responses against the drug, e.g. it has remained unclear why BAbs evolve into neutralizing activity after a certain time on therapy, in a subset of patients. In the present study, we found up to 89% of patients being BAb-positive after 3 months (SC-IFN-b-1b treatment group) and overall, 43% of the BAb-positive patients went on to develop NAbs. These figures are almost identical with previous reports;2, 4, 22, 23 however, the detection of neutralization by the measurement of IFN-b-inducible gene products either in vitro or in vivo depends upon the sensitivity of the assay used, thereby determining frequency of NAbs within BAb-positive samples. For the assessment of in vivo IFN-b bioavailability, current guidelines recommend measurement of MxA levels. 8 We confirm that MxA is induced by IFN-b treatment, is clearly abrogated in patients with high NAb titers, and might range from absent to fully expressed in patients with low titers.24,25 We defined low/ high NAb titers as recently recommended (cut-off of 400 NU for IFN-b-1b and 100 NU for IFN-b-1a), 8 after a previous study report of higher neutralizing activity of anti-IFN-b antibodies against IFN-b-1a than IFN-b-1b. 26 We further confirmed that CXCL-10 and sTRAIL levels are strongly upregulated by IFN-b and diminished by NAbs.12,13,25,27,28 Although the relative up-regulation of CXCL-10 is less than that of MxA, CXCL-10 achieved the best discrimination between NAb-positive and NAb-negative samples, possibly because of its clear abrogation in patients even with low NAb titers, in contrast to MxA.13,29
We showed that IFN-b bioavailability, as indicated by CXCL-10 and sTRAIL concentrations, is significantly lower in BAb-positive/NAb-negative patients, as compared to antibody-negative patients. In contrast, MxA mRNA levels were not affected by BAbs, which is in line with a previous study determining MxA at the protein level. 6 Only one study reports diminished MxA expression in four BAb-positive/NAb-negative patients. 30 Taken together, these findings suggested that CXCL-10 is a more sensitive marker for reduced in vivo IFN-b bioavailability than is MxA. Further evidence comes from a recent study, which reports fully abrogated CXCL-10 gene expression in patients with low NAb titers and remaining MxA expression. 13 One could conclude that some IFN-b stimulated genes are less affected by anti-IFN-b antibodies than others, a factor that might contribute to the “grey zone” of NAb assays; however, in the present study comparison is limited, as MxA was determined at the RNA and CXCL-10 at the protein level.
Our observation that BAb titers predict later NAb evolution is in line with a previous preliminary study, in which a boost of BAb titers is observed after 6 months on IFN-b treatment, only in those patients who developed NAbs later on. 5 The affinity of anti-IFN-b antibodies is significantly higher in NAb-positive than in NAb-negative patients, correlates with NAb titers and increases with treatment duration. 31 If IFN-b-1b is used as an antigen in the affinity assay, there is also a significant correlation with BAb titers. 31 These findings support the hypothesis that anti-IFN-b antibodies undergo affinity maturation in a titer-dependent manner 32 in those patients who develop NAbs. Furthermore, one might speculate that the increase of neutralizing activity of anti-IFN-b antibodies over time is accompanied by a gradual decrease of IFN-b bioavailability, and that different sensitivities of IFN-b biomarkers account for their decrease at different time points during antibody development. In clinical practice, prediction of persistent NAbs might be even more interesting and it is known that NAb persistency strongly depends on NAb titer. 33 We observed that BAb titers at Mo3 correlate with NAb titers after roughly 2 years, and that calculation with different BAb cut-off titers predicted NAb positivity or high titer NAbs (within the IFN-b-1b treatment group). Taken together, one could conclude that BAb titers also qualify for prediction of persistent NAbs.
In this study, approximately 50% of patients received treatment with IM IFN-b-1a, whereas only 25% were treated with SC IFN-b-1a and SC IFN-b-1b, respectively. This might account for the relatively low NAb frequency in this study of 13%, as compared to most other studies reporting 21% on average 1 ; however, the lower NAb frequency certainly does not influence the impact of NAbs on IFN-b bioavailability. We want to stress that the present results mainly apply to patients receiving SC-IFN-b-1b, as the vast majority was on this type of IFN-b. Antibodies against IFN-b-1a develop later than those against IFN-b-1b, 2 probably due to the lower protein load of IFN-b-1a or higher structural similarity to endogenous IFN-b; therefore, a different predictive time point of BAb titers could apply in patients treated with IFN-b-1a. The BAb cut-off titer might also vary, depending upon the assay used. Although previous studies found fair inter- and intra-assay variability in terms of BAb frequency,19,34 there are some hints that BAb titers differ between the different immunoassays. 19 The chosen time point for blood collection of 4–12 hours after the last IFN-b injection seemed appropriate, as previous studies show that different IFN-b-gene products are already induced after 3 hours and peak around 12 hours.12,20,28 We want to stress that this investigation was not intended to study the impact of anti-IFN-b antibodies on clinical outcome parameters. The LOCF procedure did most likely not influence our study findings, as the separate analysis of BAb titers (at Mo3) for prediction of NAbs either at Mo12 or Mo24 essentially returned the same results (data not shown). A further limitation of this study is that the low number of antibody-positive patients did not allow an adjustment for different co-variables, such as gender or type of IFN-b.
We feel that BAb titers may be a predictive tool for NAb evolution. In practical terms, one could recommend switching to a non-IFN-b disease-modifying treatment, for those patients who develop high BAb titers, ideally before NAbs develop and IFN-b loses its treatment effects. The findings on CXCL-10 further supported the hypothesis of a titer-dependent continuum of neutralizing activity between BAbs and NAbs and that all anti-IFN-b antibodies are neutralizing, at least to some extent. 35 Because in some patients with low NAb titers, measurement of IFN-b bioactivity is recommended, 8 one could use CXCL-10, as we showed that this biomarker has a higher sensitivity than MxA and can be determined by ELISA in serum samples, which is logistically and technically less demanding as well as less cost-intensive than the measurement of MxA mRNA by rtPCR 20 or MxA protein out of lysed blood from a citrate tube. 6
To confirm these results, we and others have launched another prospective European multicenter study, ABIRISK, which is currently ongoing.
Footnotes
Acknowledgements
In addition to the authors, the following contributors participated in patient recruitment in the NABINMS study: LF Van der Voort (VU Medical Center, Amsterdam, The Netherlands), A Skrobal (Landesklinikum Waldviertel Horn, Austria), C Adelwoehrer (Landesnervenklinik Wagner Jauregg, Linz, Austria), D Debelic (Landeskrankenhaus Weinviertel Mistelbach, Austria), D Deuretzbacher (Landeskrankenhaus Bruck an der Mur, Austria), Sabine Graeser-Lang (Hanusch-Krankenhaus, Wien, Austria), G Morgenstern (Bezirkskrankenhaus Lienz, Austria), I Soeser-Brence (Krankenhaus der Barmherzigen Brüder Graz-Eggenberg, Austria), M Mayr (Bezirkskrankenhaus Kufstein, Austria), M Jeschow (Krankenhaus der Barmherzigen Schwestern Ried, Austria), N Albrecht (Konventhospital Barmherzige Brüder Linz, Austria), R Resch (Konventhospital Barmherzige Brüder Linz, Austria) and C Tilz (Konventhospital Barmherzige Brüder Linz, Austria).
Conflict of interest
A Millonig, M Guger, M Hoelzl, M Mehling, D Rudzki, F Schautzer report no disclosures.
H Hegen received honoraria for lectures from pharmaceutical companies marketing treatment for multiple sclerosis (Bayer Schering, Biogen Idec and Merck Serono).
A Bertolotto was on steering committees in clinical trials sponsored by Biogen Idec and Roche; received speaker honoraria from BiogenIdec, Merck Serono, TEVA, Bayer Schering, Sanofi-Aventis and Novartis; was a member of scientific boards supported by Allergan and Almirall; received research support from Biogen Idec, Biogen-Dompè, Bayer Schering, Merck Serono, Sanofi-Aventis, the Italian Multiple Sclerosis Society and Associazione Ricerca Biomedica Onlus.
M Comabella received compensation for consulting services and speaking honoraria from Bayer Schering Pharma, Merk Serono, Biogen-Idec, Teva Pharmaceuticals, Sanofi-Aventis and Novartis.
G Giovannoni received consulting fees for participating on advisory boards in relation to clinical trial design, trial steering committees and data and safety monitoring committees from: Bayer-Schering Healthcare, Biogen-Idec, Five Prime, Genzyme, Ironwood, Merck-Serono, Novartis, Teva, Sanofi-Aventis and Vertex Pharmaceuticals; lecture fees from Bayer-Schering Healthcare, Merck-Serono and Vertex Pharmaceuticals. He received compensation from Elsevier for being co-chief editor of the journal MS and Related Disorders. He received research grant support from Biogen-Idec, Ironwood, Merck-Serono, Merz and Novartis.
M Khalil received research support from the Austrian Science Fund (FWF) [J2992-B09].
J Killestein received consultation fees from Novartis, Merck-Serono and Biogen Idec. His institution received research support from Biogen Idec, Bayer Schering, Teva, Merck-Serono, Novartis, Glaxo SK and UCB.
R Lindberg received research support from Swiss MS Society, Swiss National Science Foundation, Roche Postdoc Fellowship Program, and unrestricted research grants from Novartis and Biogen.
S Malucchi was reimbursed by Merck-Serono, Sanofi-Aventis, Biogen Dompe, Biogen Idec, Novartis and Bayer Schering for attending conferences; Malucchi received fees for lectures by Merck-Serono and Biogen Idec.
X Montalban received speaking honoraria and travel expenses for scientific meetings, has been a steering committee member of clinical trials or participated in advisory boards of clinical trials in the past for Bayer Schering Pharma, Biogen Idec, EMD, Merck Serono, Genentech, Genzyme, Novartis, Sanofi-Aventis, Teva Pharmaceuticals and Almirall.
CH Polman received honoraria/consultation fees/research support from Actelion, Biogen Idec, Bayer Schering, Glaxo Smith Kline, Merck Serono, Novartis, TEVA, UCB and Roche.
F Sellebjerg served on scientific advisory boards for and received funding for travel from Biogen Idec, Merck-Serono, Novartis, Sanofi-Aventis and Teva; served as a consultant for Biogen Idec and Novo Nordisk; received speaker honoraria from Bayer-Schering, Biogen Idec, Merck-Serono, Novartis, Sanofi-Aventis and Schering-Plough; and received research support from Biogen Idec, Merck-Serono, Novartis and Sanofi-Aventis.
PS Sorensen was on scientific advisory boards for Biogen Idec, Merck Serono, Novartis, Genmab, TEVA, Elan and GSK; was on steering committees or independent data monitoring boards in clinical trials sponsored by Merck Serono, Genmab, TEVA, GSK, Bayer Schering, and received travel funding for these activities; served as Editor-in-Chief of the European Journal of Neurology and is currently an editorial board member for Multiple Sclerosis Journal, European Journal of Neurology, Therapeutic Advances in Neurological Disorders; and received speaker honoraria from Biogen Idec, Merck Serono, TEVA, Bayer Schering, Sanofi-Aventis and Novartis. His department received research support from Biogen Idec, Bayer Schering, Merck Serono, TEVA, Baxter, Sanofi-Aventis, BioMS, Novartis, Bayer, RoFAR, Roche, Genzyme, the Danish Multiple Sclerosis Society, the Danish Medical Research Council, and the European Union Sixth Framework Programme in the areas of Life sciences, Genomics and Biotechnology for Health.
F Deisenhammer participated in meetings sponsored by, or received honoraria for acting as an advisor for: Biogen Idec, Merck Serono, Bayer and Sanofi. His institution received financial support for participation in randomized controlled trials in MS from Bayer Schering Pharma, Biogen Idec, Merck Serono and Teva Pharmaceuticals.
Funding
This work was supported by the European Union (grant number LSHB-CT-2005-018926).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.