
Abstract
Background:
Genome-wide association studies (GWAS) have identified over 100 germline variants that influence susceptibility to multiple sclerosis, most of which map within or near to genes with immunological function. However, the role of somatic mutations in multiple sclerosis has not been investigated.
Objective:
The objective of this paper is to explore the role that somatic mutations might play in the development of multiple sclerosis.
Methods:
We exome-sequenced in total 21 individual CD4+ lymphocytes isolated from cerebrospinal fluid of two patients. In addition we sequenced DNA from the patients’ peripheral blood to serve as germline reference.
Results:
In comparison with the respective germline sequence, each cell differed at an average of 1784 positions, but as anticipated subsequent analysis confirms that most, if not all, of these potential mutations are likely to represent artefacts generated during the amplification of a single genome and/or by sequencing. Fifty-six of the potential mutations were predicted to have likely functional effects on genes that have previously been implicated by GWAS, including three in the CD6 gene.
Conclusion:
More robust methods applied to larger numbers of cells will be needed to define the role of somatic mutations.
Introduction
Epidemiological studies have consistently demonstrated that susceptibility to multiple sclerosis (MS), the archetypal chronic inflammatory demyelinating disease of the central nervous system, is influenced by germline genetic variation, 1 and to date genome-wide association studies (GWAS) have identified 110 such variants in addition to the long established influence of the Major Histocompatibility Complex.2,3 Most of the associated variants implicate genes with known immunological functions and collectively suggest that leukocyte dysfunction lies at the heart of aetiology. In this context we hypothesised that somatic mutations in immune cells might contribute to the development of MS and in particular that the expanded lymphocyte clones that so characterise the cerebrospinal fluid (CSF) in the disease might carry such variants. Given the success of recent cancer studies employing exome sequencing in single cells,4–7 we have performed a pilot study using this approach in single lymphocytes isolated from the CSF of patients with relapsing–remitting MS.
Materials and methods
Samples
Samples were obtained from two male subjects undergoing blood testing and lumbar puncture as part of the workup confirming their diagnosis of relapsing–remitting MS. The study was approved by the local ethics committee (REC-11/33/0007). Both patients gave informed consent. A total of 0.5 ml aliquots of fresh CSF were spun for 10 minutes at 500 g after which the supernatant was discarded and cells resuspended in 60 ul of CryoStor® CS5 (Sigma-Aldrich, St. Louis, MO, USA), frozen according to the manufacturer’s instructions and stored at −80°C until staining. Thawed samples were stained with CD4+ fluorescein isothiocyanate (FITC) antibody (BD Biosciences, San Jose, CA, USA) and one or 10 CD4+ cells were sorted by flow cytometry analysis (FACS) onto 96-well plate wells with 5 ul of PicoPLEX™ WGA kit Cell Extraction Buffer per well. Amplification was performed according to the manufacturer’s instructions (Rubicon Genomics, Ann Arbor, MI, USA) and amplified samples were purified with GenElute™ polymerase chain reaction (PCR) Clean-Up kit (Sigma-Aldrich). Exome sequencing was performed on samples from 13 single cell amplifications (12 from Patient A and one from Patient B) and eight 10-cell amplifications (six from Patient A and two from Patient B) (see Table S1
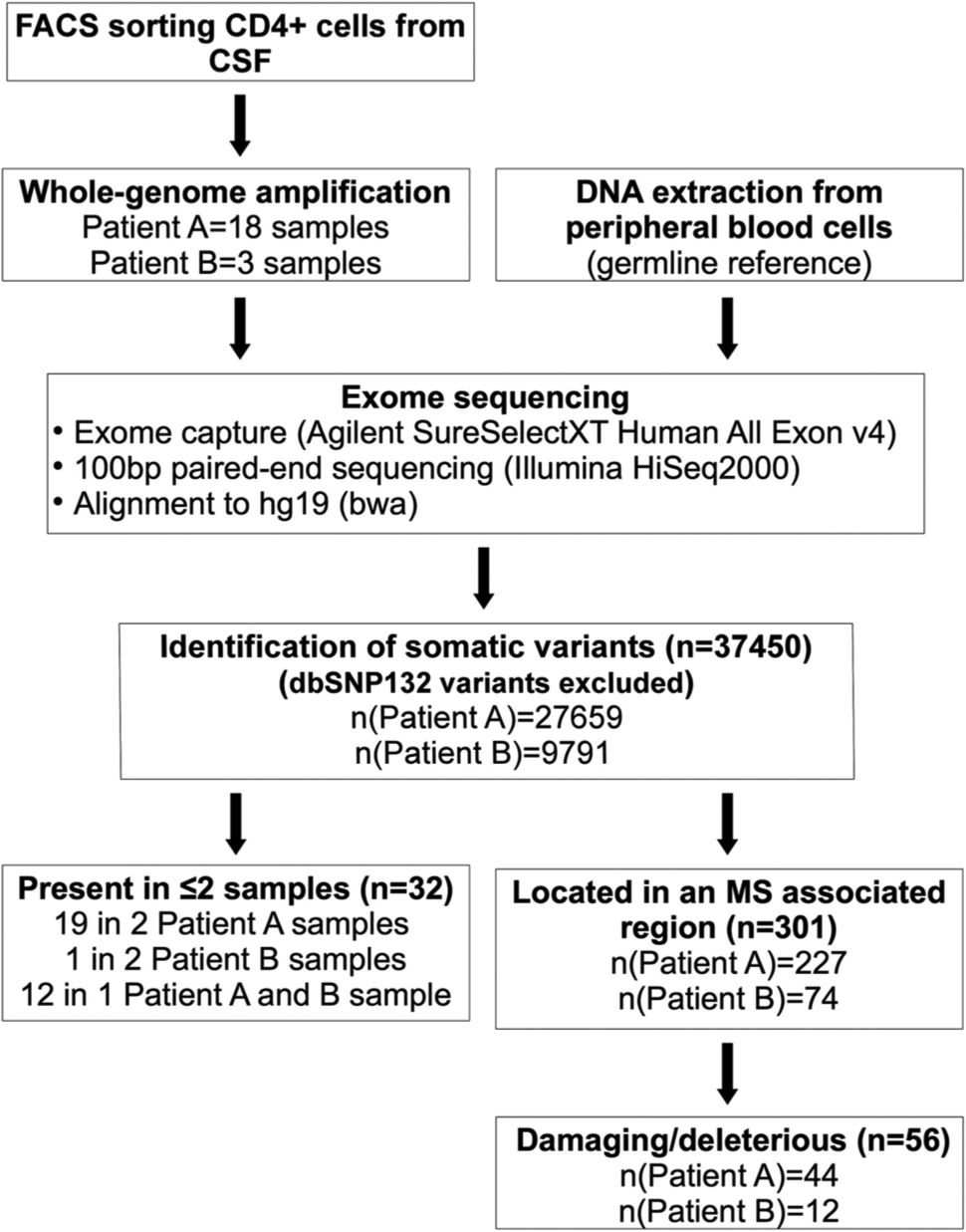
Overview of the study design.
Exome sequencing
Exome capture was performed using Agilent SureSelectXT Human All Exon v4 kit following the manufacturer’s procedures (Agilent, Santa Clara, CA, USA) and sequenced with Illumina paired-end sequencing (protocol v1.2). Briefly, DNA was fragmented by shearing (Covaris, Woburn, MA, USA) and purified using Agencourt AMPure XP beads (Beckman Coulter, Fullerton, CA, USA). Fragment ends were repaired and adaptors were ligated to the fragments. The library was purified using Agencourt AMPure XP beads, amplified by PCR and hybridised with biotinylated RNA baits. Bound genomic DNA was purified with streptavidin-coated magnetic Dynabeads (Invitrogen, Carlsbad, CA, USA) and re-amplified to include barcoding tags before pooling for sequencing 100 bp paired-end reads on an Illumina HiSeq 2000. Read files (Fastq) were generated from the sequencing platform via the manufacturer’s proprietary software. Reads were mapped to the human genome build version hg19/b37 using the Burrows-Wheeler Aligner (BWA) package, version 0.6.1. Local realignment of the mapped reads around potential insertion/deletion (indel) sites was carried out with Genome Analysis Tool Kit (GATK) version 1.6. Duplicate reads were marked using Picard version 1.62 and were not considered further in the analysis. Base quality (Phred scale) scores were recalibrated using GATK’s covariance recalibration. Single-nucleotide polymorphism (SNP) and indel variants were called using the GATK Unified Genotyper for each sample. Potential somatic mutations in CSF samples were called against PBMC (Patient A) or whole blood (Patient B) samples using SomaticSniper. 8 Because our hypothesis was that pathogenic somatic mutations would have a major impact on gene and cell function, they would be unlikely to exist in germline and we therefore excluded all variants found in dbSNP (Release 132). However, we did not exclude variants previously reported in Exome Variant Server or more recent versions of dbSNP. All exome sequencing, alignment and variant calling were performed by the Oxford Gene Technology (Begbroke, Oxfordshire, UK).
Results
CD4+ lymphocytes were FACS sorted from the CSF of two patients (A and B) and dispensed into 96 well plates at one or 10 cells per well. Whole-genome amplification was successful in 13 wells containing a single cell (12 from Patient A and one from Patient B) and eight wells containing 10 cells (six from Patient A and two from Patient B). As anticipated, exome sequencing in these 21 amplified samples had lower sequencing coverage than reference blood samples (Table 1, Table S1). Depths at target regions were highly correlated across the 21 amplified samples (average pair-wise correlation coefficient r = 0.797), suggesting that amplification efficiency of the PicoPLEX™ varies systematically rather than stochastically across the genome. We did not see correlation between target region depth and GC content (r = 0.09).
Sequencing coverage and depth.
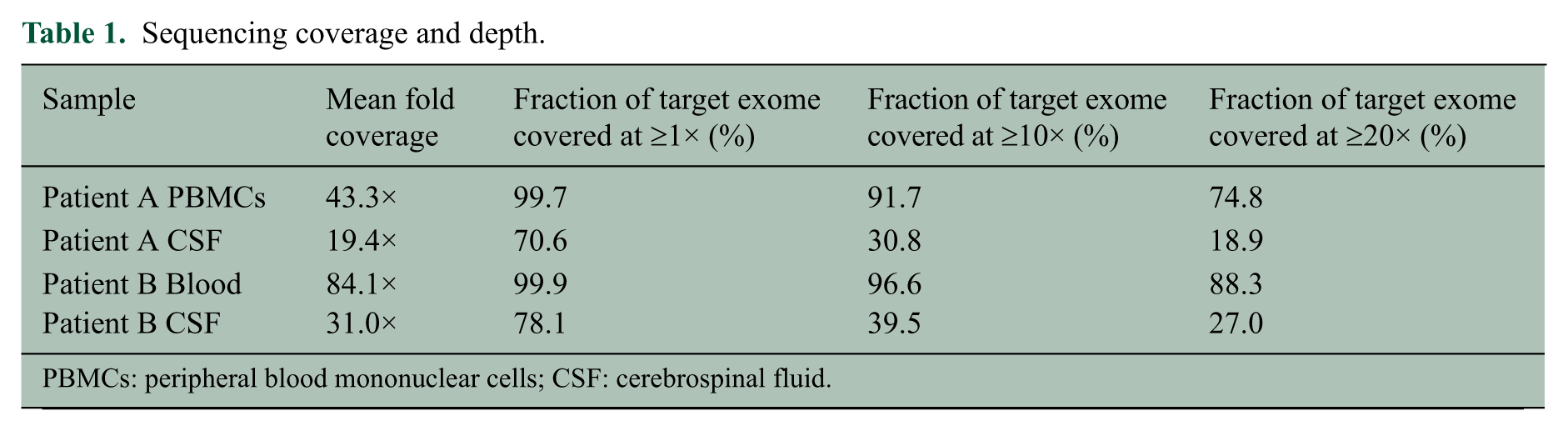
PBMCs: peripheral blood mononuclear cells; CSF: cerebrospinal fluid.
We detected an average of 1784 apparent single nucleotide differences between each CSF sample and its germline reference (Table S2). In order to estimate a lower limit for the false discovery rate (FDR), we investigated correlation between these potential somatic mutations and nearby heterozygous germline variants in single-cell samples; for a true somatic mutation we would expect to see a perfect correlation with the nearby germline variant while artefacts arising during genomic amplification or next-generation sequencing would usually show imperfect correlation. We considered potential mutations located within 100 bp of a heterozygous germline variant and lying in regions where there were at least 10 sequence reads carrying both the germline variant and the candidate somatic mutation. We further required that at both the germline variant and the potential somatic mutation, both alleles were represented by at least five reads. We found that only in 9% of cases the alleles at the candidate mutation correlated perfectly with alleles at the nearby germline variant. This observation would suggest that at least ~90% of the observed differences between single cells and germline are artefacts rather than true somatic mutations. To increase the likelihood of detecting genuine somatic mutations, previous studies have only considered differences present in several cells as candidates.4–7 Thirty-two of all observed potential mutations were present in more than one sample: 19 in two samples from Patient A, one in two samples from Patient B and 12 in one sample from each patient (Table S3). These numbers are close to what we would expect by chance taking into account the number of pairwise comparisons and frequencies provided in Table S2 (14, one and nine expected by chance, respectively). Six of the 32 potential mutations were predicted to be damaging by at least one of the three prediction programs used (Sift, Polyphen, Condel) (Table S4). No variants were detected in more than two CSF samples.
Given the higher probability of relevance associated with changes in genes already implicated by GWAS, we next considered potential mutations in such previously reported genes 2 and found in total 56 potential mutations that were considered to be possibly or probably damaging or deleterious according to Sift, Polyphen and Condel (Table S5). This number is, however, similar to what we would expect based on the number of observed potential mutations filling the same criteria in the entire exome. Each of the 56 mutations was found in only one sample. Three potential mutations were seen in each of CD6, CLEC16A, DAB2 and SDK1. Because of the established role of CD6 in T lymphocyte activation and proliferation, 9 we attempted to validate the three candidate mutations detected in this gene and successfully confirmed them by Sanger sequencing them in the original whole-genome amplified single-cell DNA sample. All three were successfully validated and we can therefore exclude the possibility that they were sequencing errors. However, we cannot exclude the possibility that they are errors introduced during whole-genome amplification. Interestingly, all three alter the amino acid sequence in the cytoplasmic tail of the protein product and thereby might alter signal transduction via this surface receptor. 10
Discussion
Here, we report the first exome sequencing study in single CSF lymphocytes from patients with MS intending to explore the role of somatic mutations in the development of the disease. By focusing on CD4+ lymphocytes isolated from CSF at early stages of MS, we hoped to increase the likelihood of detecting pathogenically relevant mutations. Unfortunately, the number of errors introduced in whole-genome amplification and sequencing are overwhelming, making it impossible to conclusively distinguish genuine somatic mutations. We therefore attempted to increase the likelihood of detecting true mutations by focusing on regions previously implicated by MS GWAS and by looking for variants present in more than one sample. However, we found no enrichment of potential somatic mutations with predicted damaging effects in previously reported MS susceptibility genes compared to the rest of the exome. The slight increase in number of variants observed in more than one sample compared to the expected amount could implicate a slightly higher probability of these being true mutations but could equally be explained by a non-uniform probability distribution of amplification or sequencing errors. Therefore the two filtering approaches we employed are unlikely to have significantly increased our chances of observing true mutations. Nevertheless, it could be of interest that we found several potential somatic mutations in certain genes previously implicated in the aetiology of MS by GWAS; in particular three variants in the cytoplasmic tail of CD6, which were confirmed not to be sequencing errors. Sequencing of many additional subjects would be required to establish whether somatic mutations in CD6 are commonly found in MS.
There could be several reasons for why we failed to confidently identify somatic mutations. First, if a somatic mutation is rare, for example present only in a single clone, a very large number of cells would need to be screened to detect it. Second, although there is plenty of evidence supporting the role of CD4+ cells in MS, it could be that somatic mutations play a role in other cell populations or that they are not relevant in MS at all. Finally, unlike for germline variants, validation for variants identified in whole-genome amplified material from single cells is virtually impossible because the original single-cell DNA is not available. Looking for variants present in at least three or four samples would likely be the best approach for identifying true mutations. Future studies will therefore need to employ substantially more robust amplification and sequencing methods and very much larger sample sizes in order to overcome the extremely high FDR inherent in the approach we have employed and to ensure detection of rare somatic mutations.
Footnotes
Acknowledgements
We gratefully acknowledge the help and efforts of the staff at Oxford Gene Technology, especially Amy Gravestock and Daniel Swan.
Conflict of interest
None declared.
Funding
This work was supported by the Cambridge NIHR Biomedical Research Centre. AK was funded by the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) Postdoctoral Research Fellowship Programme and BF is supported by a Raymond and Beverley Sackler studentship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.