
Abstract
Background:
Longitudinally extensive transverse myelitis (LETM) is a characteristic manifestation of neuromyelitis optica (NMO). However, not all patients with LETM are positive for aquaporin-4 (AQP4) antibodies. We evaluated the characteristics of idiopathic isolated LETM negative for AQP4 antibodies.
Methods:
From the National Cancer Center registry of inflammatory diseases of the central nervous system, patients with LETM as an initial manifestation and follow-up for at least two years were enrolled. Their medical records and MRIs were reviewed retrospectively. AQP4 antibody was confirmed by three different validated methods at least three times. Cerebrospinal fluid (CSF) glial fibrillary acidic protein (GFAP) levels were measured to investigate astrocyte damage.
Results:
Among 108 patients with first-ever LETM, 55 were positive for AQP4 antibodies (P-LETM) and 53 were consistently negative. Of them, seven were later diagnosed with seronegative NMO, and four were positive for MOG antibodies. The remaining 42 patients (N-LETM) showed several features distinct from P-LETM: male predominance, older age of onset, milder clinical presentation, spinal cord confinement and absence of combined autoimmunity. CSF GFAP levels were not increased in N-LETM but were markedly elevated in P-LETM.
Conclusions:
Idiopathic isolated N-LETM is not that rare among first-ever LETM, and has many features distinct from P-LETM where astrocytic damage is evident.
Keywords
Introduction
Longitudinally extensive transverse myelitis (LETM), defined as a spinal cord inflammatory lesion that extends over three or more vertebral segments, is a syndrome that has various causes.1,2 After excluding vascular, compressive, metabolic, infectious, nutritional, rheumatological, paraneoplastic and radiation-related disorders, most cases are considered to be immune mediated.1,2 LETM is at low risk of developing into multiple sclerosis (MS), 3 while it is a characteristic feature of neuromyeitis optica (NMO), 4 for which aquaporin-4 (AQP4) antibodies are a specific marker. 5 Although NMO is a common underlying aetiology of LETM, 4 not all patients with immune-mediated LETM are positive for AQP4 antibodies.6–9
In clinical practice, AQP4 antibody-negative isolated LETM without a determined cause poses a diagnostic and therapeutic challenge. Some of these patients develop optic neuritis and are diagnosed with seronegative NMO,10,11 and some are positive for myelin-oligodendrocyte glycoprotein (MOG) antibodies.12–14 However, some of the patients are outside the context of the previously defined spectrum of immune-mediated inflammatory diseases of the central nervous system (CNS) and have not been well described.
Previous studies have limitations in their investigations of the features of idiopathic AQP4 antibody-negative LETM because many of these studies were performed before the AQP4 antibody era or relied on the serostatus of the AQP4 antibody that was determined by a single assay at a single time point.15–23 These factors may have caused the low frequency of seropositivity (10–18%) for AQP4 antibodies in previous reports on LETM,16,17,19 and thus patients with false-negative results may compromise the identification of the characteristics of true AQP4 antibody negative LETM. Recent studies have used more than one validated assay. However, they have focused mainly on seropositive LETM or included various causes of seronegative LETM, thus without focusing on ‘idiopathic’ AQP4 antibody negative LETM.8,9,24
In this study, we sought to establish a well-defined cohort who presented with isolated LETM without a determined cause and who were consistently negative for AQP4 antibodies after serial testing at different time points using three different validated assays, and to compare their characteristics to those of AQP4 antibody positive LETM. We also compared cerebrospinal fluid (CSF) levels of glial fibrillary acidic protein (GFAP) as a marker of astrocytic damage 25 in order to investigate discriminative pathophysiological features.
Methods
Patients and assays
Patients with LETM as an initial manifestation and follow-up for at least two years from the National Cancer Center (NCC) registry for inflammatory diseases of the CNS from September 2005 to April 2013 were enrolled. To ensure the idiopathic nature of the patients, we excluded vascular and compressive lesions identified by spinal MRI, and performed infectious, metabolic and autoimmune screening. Patients with a history of cancer and/or radiation were excluded. Patients with insidious clinical manifestation were also excluded.
AQP4 antibody serostatus was confirmed rigorously using three different methods: a commercial cell-based assay (CBA; Euroimmun, Luebeck, Germany), 26 an in-house CBA at Tohoku University, 27 and an in-house enzyme-linked immunosorbent assay (ELISA) at the NCC. 28 In particular, we performed repeated tests during follow-up using at least three different time points, particularly at the time of any acute exacerbation in patients who were seronegative in the initial test. AQP4 antibodies were also assessed in CSF to exclude the possibility of AQP4 antibodies undetectable in serum but present in CSF. 29 To exclude MOG antibody-mediated disease, which was recently reported in AQP4 antibody-negative NMO/NMOSD, serum MOG antibodies were tested using a CBA with live transfected cells with full-length MOG at Tohoku University. 14 We analysed demographic, clinical, laboratory and MRI findings retrospectively and compared them between AQP4 antibody negative and positive patients. GFAP levels in CSF, obtained during acute attacks of LETM before administration of high-dose methylprednisolone, were measured using a human GFAP ELISA kit according to the manufacturer’s protocol (BioVendor, Minneapolis, MN). Patients with MS were also tested as a control.
The Institutional Review Board of the NCC approved the study protocol and waived the need for informed consent due to de-identified data used.
Statistical analysis
Statistical analysis was performed using GraphPad Prism v4. We performed two-sample t-tests or Mann–Whitney U-tests for continuous data and chi-square or Fisher’s exact tests for categorical data for comparisons of patients with and without AQP4 antibodies. Two-sided p-values of <.05 were considered statistically significant.
Results
Among the 108 enrolled patients with first-ever LETM, 55 were positive for AQP4 antibodies (P-LETM) and 53 were consistently negative. Of the 53 AQP4 antibody-negative patients, seven (13.2%) patients developed optic neuritis within the follow-up period and were diagnosed with seronegative NMO. Four (7.5%) of the 53 AQP4 antibody-negative patients were positive for MOG antibodies, while none of the P-LETM patients was positive. Thus, 42 (38.9%) patients were finally identified as having idiopathic isolated AQP4 antibody-negative LETM (N-LETM). Figure 1 provides a classification of patients with first-ever LETM.
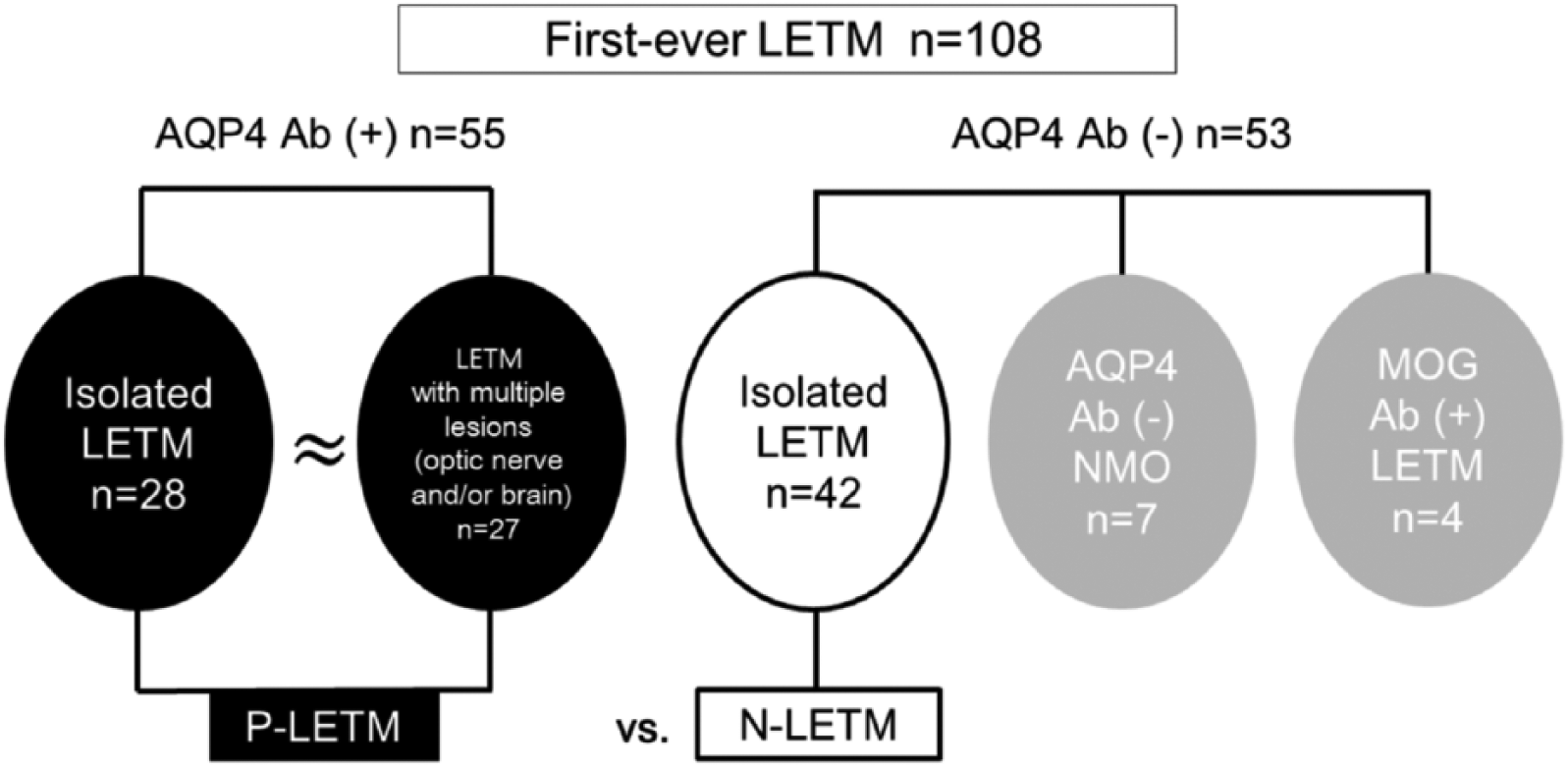
Classification of longitudinally extensive transverse myelitis (LETM). Ab, antibody; P-LETM, AQP4 antibody-positive LETM; NMO, neuromyelitis optica; N-LETM, idiopathic isolated AQP4 antibody and MOG antibody-negative LETM.
As shown in Table 1, N-LETM showed demographic and clinical characteristics different from those of P-LETM. The male:female ratio (33:9 vs. 11:44; p < .001) and mean age at onset (43.1 ± 9.8 (range 14–62) vs. 37.4 ± 10.1 (range 17–59); p = .006) were higher in N-LETM than P-LETM. The clinical presentation of N-LETM during the acute stage tended to be milder than that of P-LETM. N-LETM showed a lower median EDSS score at the nadir (3.0 (range 3.0–8.5)) than did P-LETM (8.0 (range 2.5–9.5); p < .001). Partial myelitis (76.2%) and pure sensory symptoms (57.1%) were the most common presenting features in N-LETM, whereas almost two-thirds of P-LETM (63.6%) cases presented with complete myelitis. Severe initial symptoms, such as inability to walk, need for catheterization and respiratory insufficiency, were more common in P-LETM than in N-LETM (65.5% vs. 26.2%; p < .001). The clinical course of N-LETM also tended to be more benign than that of P-LETM. There was no difference in the mean follow-up periods between groups (5.4 ± 2.6 vs. 7.0 ± 4.4 years; p = .121). During follow-up, 71% of N-LETM and 87% of P-LETM cases had relapses. N-LETM showed a longer median time until the second attack (11.5 (range 2–72) vs. 4.5 (range 1-41) months; p = .013) and a lower median annualised relapse rate (ARR) before treatment (0.5 (range 0–4) vs. 1.8 (range 0–5); p < .001) than did P-LETM. At the final follow-up, 57% of patients with N-LETM (83% of monophasic N-LETM patients) were untreated, while all P-LETM patients were treated with an immunosuppressive agent. The median ARR after treatment and the median EDSS score at the last visit for N-LETM patients were similar to those of P-LETM patients (0 (range 0–2) vs. 0 (range 0–1); mean: 0.2 ± 0.5 vs. 0.2 ± 0.3, 2.5 (range 1.0–6.0) vs. 3.0 (range 0–7.5), respectively).
Comparison of clinical manifestations of AQP4 antibody-negative and -positive LETM.
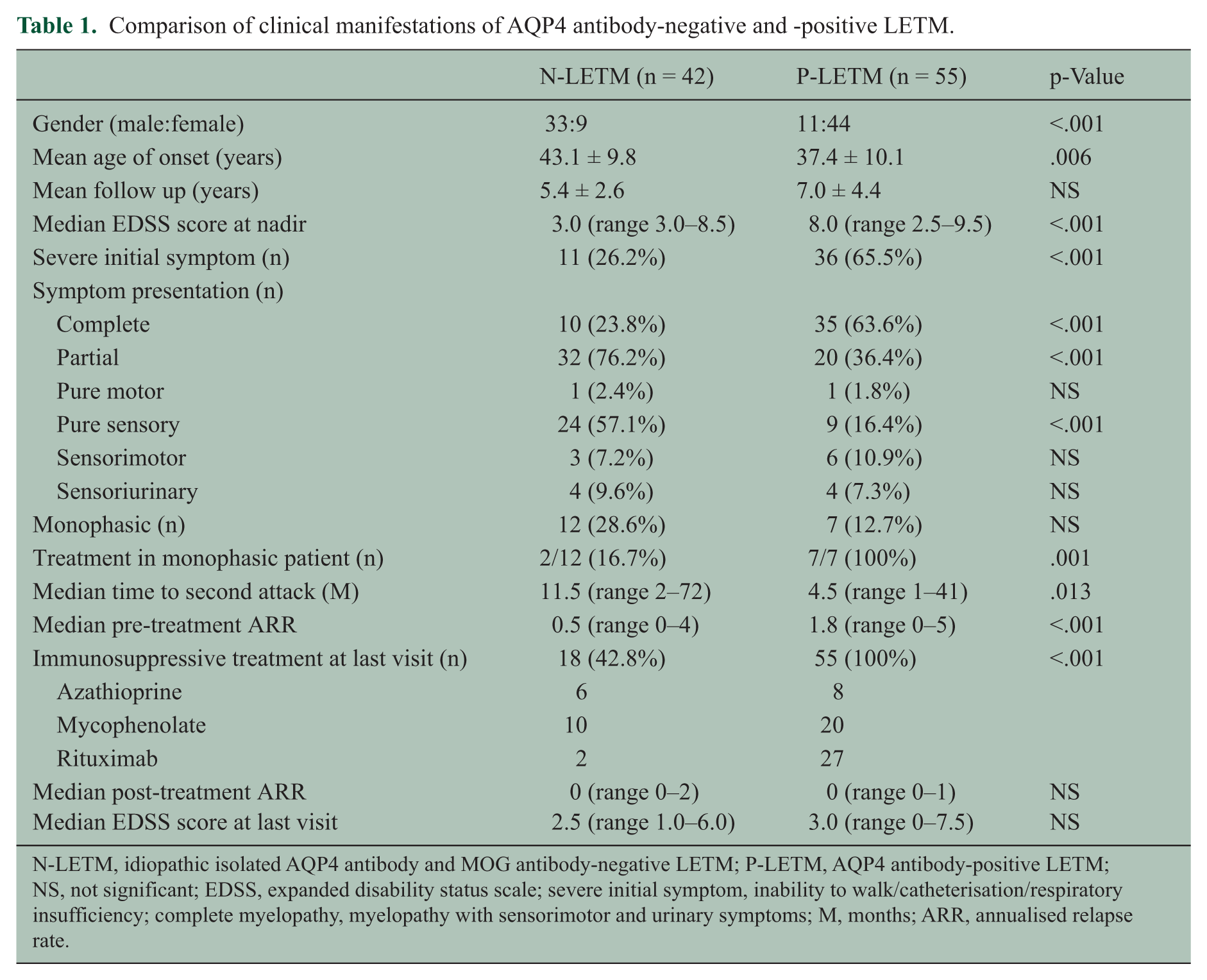
N-LETM, idiopathic isolated AQP4 antibody and MOG antibody-negative LETM; P-LETM, AQP4 antibody-positive LETM; NS, not significant; EDSS, expanded disability status scale; severe initial symptom, inability to walk/catheterisation/respiratory insufficiency; complete myelopathy, myelopathy with sensorimotor and urinary symptoms; M, months; ARR, annualised relapse rate.
In spinal MRI, performed at the acute phase, there were some differences between the two groups. In sagittal views, N-LETM showed a shorter mean length of the lesion (4.0 ± 1.6 vs. 7.4 ± 4.5; p < .001) and a lower prevalence of medullary expansion of cervical lesions than did P-LETM (1/20 (5.0%) vs. 19/36 (52.8%); p < .001). On axial scans, centrally located T2 hyper-intense lesions occupying more than two-thirds of the cross-sectional area of the cord were seen very frequently in both N-LETM and P-LETM (32/33 (97.0%) vs. 41/44 (93.2%)), whereas T1-hypointense lesions were more common in P-LETM than N-LETM (40/44 (90.9%) vs. 20/32 (62.5%); p = .004). In brain MRI during follow-up, N-LETM showed markedly fewer white matter lesions (3/37 (8.1%) vs. 31/55 (56.4%); p < .001) than did P-LETM, and no N-LETM cases showed brain lesions characteristic of NMO or MS (Table 2).
Comparison of paraclinical findings in AQP4 antibody-negative and -positive LETM.
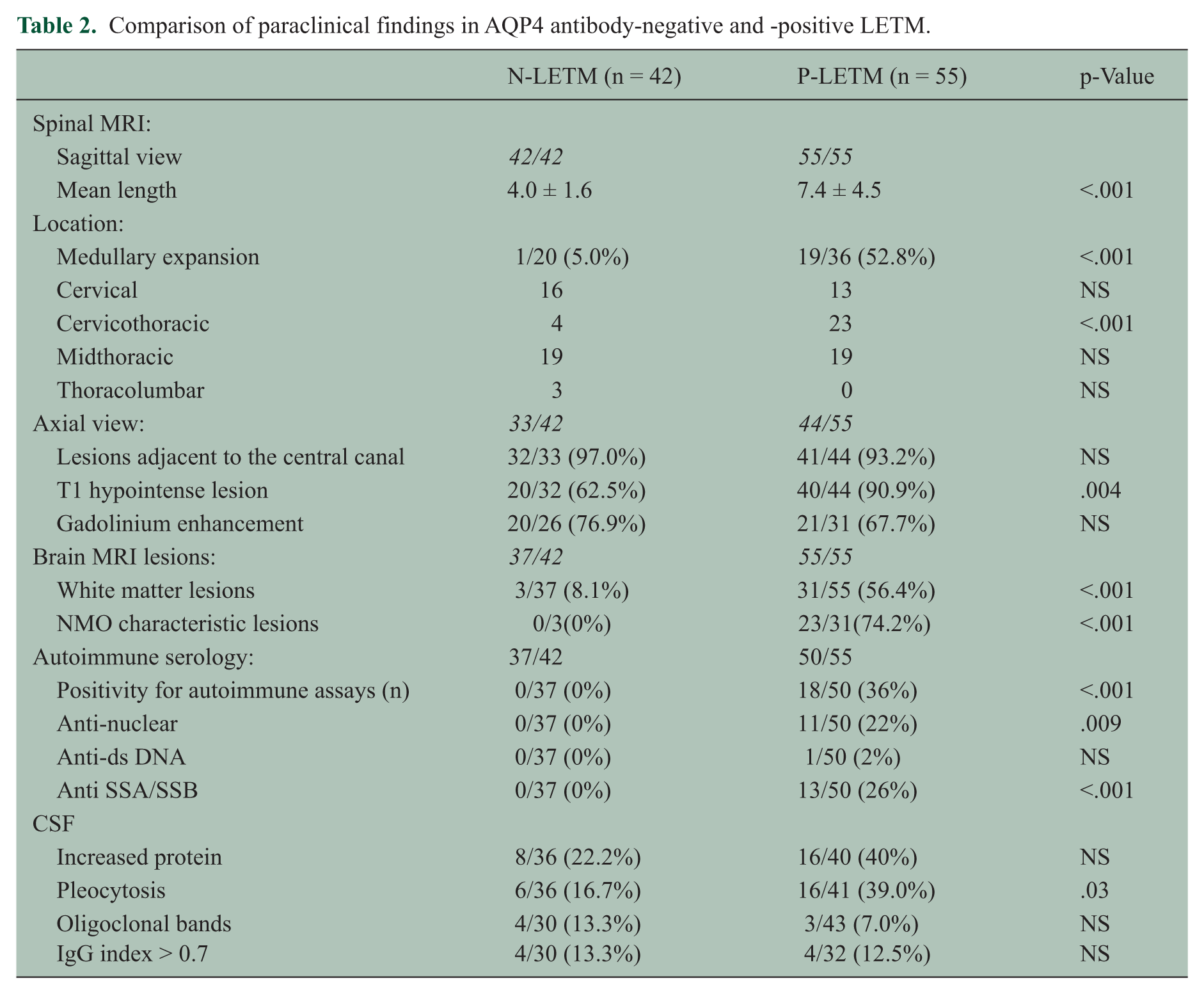
In laboratory tests during follow-up, no N-LETM cases were positive for non-organ specific autoantibodies, whereas 36% of P-LETM cases showed positivity for anti-nuclear, anti-ds DNA or anti-SSA/SSB antibodies. General CSF findings were not significantly different between the two groups, except that less pleocytosis was seen in N-LETM than P-LETM (6/36 (16.7%) vs. 16/41 (39.0%); p = .03; Table 2). CSF GFAP levels showed significant differences between N-LETM and P-LETM. CSF GFAP levels were significantly higher in P-LETM (n = 17, 2078.0 ± 3805.1 ng/mL) than in N-LETM (n = 13, 1.4 ± 1.7 ng/mL; p < .001) or MS (n = 13, 0.5 ± 0.6 ng/mL, p < .001; Figure 2).
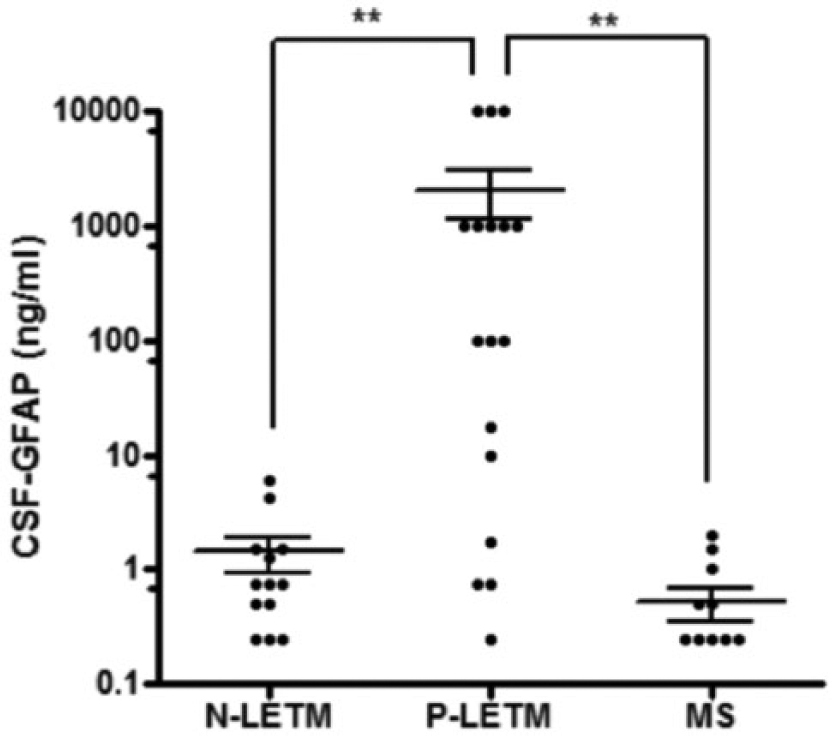
Cerebrospinal fluid (CSF) glial fibrillary acidic protein (GFAP) levels. MS, multiple sclerosis; **p < 0.001).
Seven patients with seronegative NMO showed no female predominance, and two of these showed simultaneous development of myelitis and optic neuritis. Four patients with MOG antibodies also showed no female predominance and good clinical recovery. Half of them presented a monophasic clinical course and sensoriurinary symptom at onset (Table 3).
Characteristics of seven seronegative NMO and four MOG antibody-positive patients.
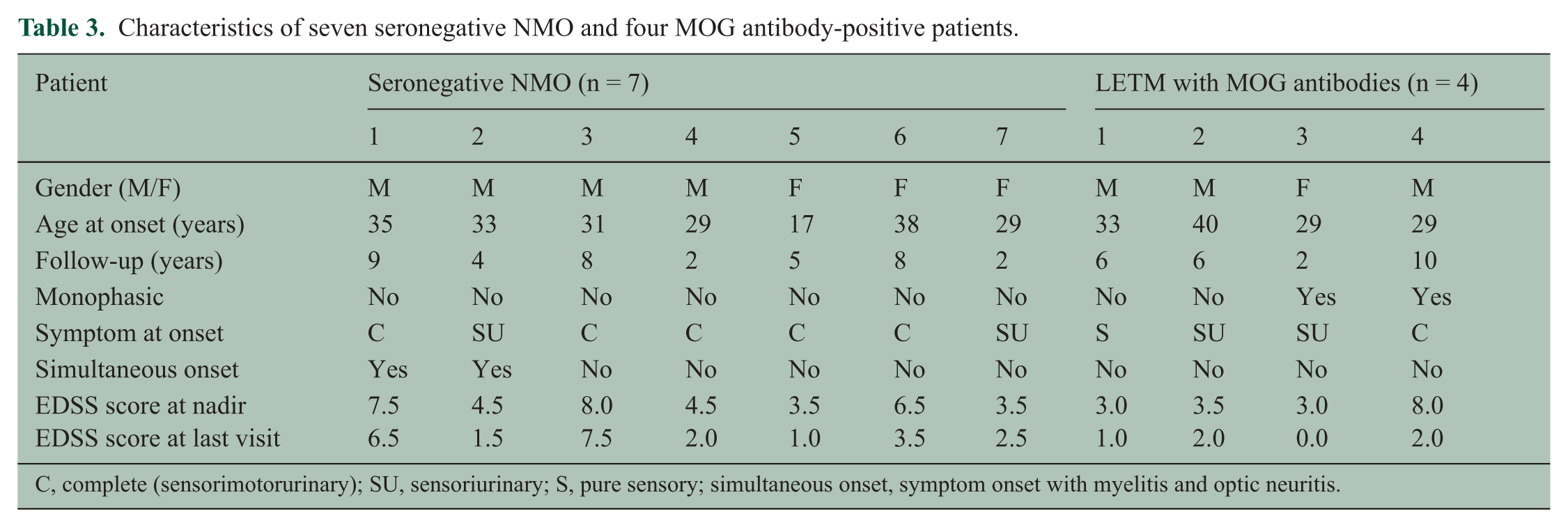
C, complete (sensorimotorurinary); SU, sensoriurinary; S, pure sensory; simultaneous onset, symptom onset with myelitis and optic neuritis.
Discussion
To determine the characteristics of N-LETM, it is critical to confirm true AQP4 antibody-negativity. First, we used three different validated assays, which proved high sensitivity and specificity.26–28,30–32 Second, we performed serial assays, particularly at times of exacerbations to increase the detection rate, because some patients who are negative initially may become positive during subsequent attacks.27,30–33 Third, we also performed AQP4 antibody assays on CSF because AQP4 antibodies may be undetectable in serum but present in CSF in 4–5% of patients. 29 Finally, we excluded other known causative factors, including MOG antibody-associated disease.12–14 Through such rigorous confirmations, we established the largest well-defined cohort of idiopathic isolated LETM negative for AQP4 antibodies.
This unique cohort of N-LETM showed several distinct features, compared to P-LETM: (a) a male predominance, (b) older age at onset, (c) milder clinical presentation, (d) less frequent relapse, (e) phenotypic confinement to the spinal cord, (f) absence of combined autoimmunity, and (g) less frequent T1 hypointense lesions and shorter lengths of spinal cord lesions. Furthermore, the CSF GFAP level, a marker for astrocytopathy, 25 was low in N-LETM but was very high in P-LETM, indicating severe astrocytic damage only in P-LETM. These findings suggest that N-LETM have clinical and laboratory features differ from P-LETM.
Notably, idiopathic isolated N-LETM among first-ever LETM cases was actually not so rare (39%) in our cohort, despite applying multiple and serial assays for AQP4 antibodies. This finding is consistent with a recent Italian study, which reported 41% idiopathic N-LETM among 37 first-ever LETM. 8 However, other recent studies have reported relatively low frequencies of idiopathic N-LETM (10.4% of recurrent LETM from the USA and 6.6% of all LETM from the UK).9,24 The cause of these differences remains unclear, but it is likely related to the differences in enrolled populations (first-ever LETM vs. all or recurrent LETM during the course of disease) and/or referral patterns and/or ethnicity of patients. Previous studies in Asia have also suggested that idiopathic recurrent TM or LETM without extra spinal lesions was common, although these studies did not apply multiple and serial assays for AQP4 antibodies.16,20,34 By contrast, seronegative NMO was relatively rare in our cohort (6.5% of all first - ever LETM and 13.2% of AQP4 antibody-negative first-ever LETM), consistent with recent studies (8.1% of all first-ever LETM from Italy, 6.6% of all LETM from the UK, respectively).8,9
Another finding of note was that a relapsing course was quite common (71%) in N-LETM, although the risk of relapse was lower in N-LETM than in P-LETM. This suggests that while AQP4 antibody positivity predicts a high risk of relapses after LETM, AQP4 antibody negativity does not necessarily mean no risk of relapse. Monophasic LETM was more common in N-LETM, which showed a longer median time until the second attack and a lower median ARR before treatment, even though fewer patients were treated with immunosuppressants than were those with P-LETM. The higher frequency of recurrent N-LETM in the current study compared to previous studies might be associated with the longer follow-up duration in our cohort (64.8 vs. 25–38 months).8,9
Many features of our N-LETM patients, including absence of female predominance, less frequent relapse, benign clinical course and the absence of combined autoimmunity, are consistent with previous studies.8,9,16–20,22,24 Nevertheless, some features, such as older age of onset, milder clinical presentation, shorter spinal cord lesion lengths and similar frequency of central grey matter involvement on MRI compared to P-LETM, were discrepant from previous studies.9,15–19,24 These may result from the uncertainty regarding AQP4 antibody serostatus in earlier studies performed before the ‘AQP4 antibody era’ or which only used a single assay at a single time point.15–23 They may also result from the heterogeneous N-LETM populations that included idiopathic LETM and other aetiologies evaluated in the previous studies.8,9,24 Further studies are required to define these features in larger cohorts.
In the current study, overall clinical features of N-LETM tended to be milder and more benign compared to those of P-LETM. However, not all patients with N-LETM showed such features. Some patients with N-LETM exhibited severe attacks (up to EDSS 8.5), complete myelitis (24%), frequent relapses before treatment (up to ARR 4.0) and T1-hypointense lesions on MRI (37%). Thus, N-LETM cannot be distinguished from P-LETM solely on the presenting features and severity. These variable features hold the possibility of remaining heterogeneity in N-LETM and emphasise the need to find undiscovered marker(s) that may be associated with other potential CNS-specific epitopes other than AQP4 and MOG, or even beyond humoral immunity.
Although treatment was not the main consideration in this study, the median post-treatment ARR was significantly lower than pre-treatment ARR in both N-LETM and P-LETM. Patients with AQP4 antibodies received more frequently immunosuppressive drugs, but these treatments produced a remarkable reduction of relapses in both groups.
The proportion of MOG antibody positive patients in first-ever LETM was quite low (3.7% of all first-ever LETM and 7.5% of AQP4 antibody-negative first-ever LETM). However, not all sera tested for MOG antibodies were collected during the first attack, and these antibodies may disappear after some months. Consistent with a previous report, none of the P-LETM cases was positive for MOG antibodies, whereas four N-LETM patients were positive. 14 Those patients with MOG antibodies had good recovery after attacks. However, we were unable to evaluate whether there were any differences between patients with MOG antibodies and N-LETM due to the small number of cases.
The retrospective nature of this study involved methodological shortcomings and some limitations due to its evaluation of only a single referral centre. Some of the patients were not checked for the AQP4 and MOG antibodies at initial presentation, and thus the serostatus of the first attack was unknown. In addition, all our patients were Asian, and the majority were adults. Therefore, racial and age differences were not broadly reflected in this study. Additionally, as our study was not designed to evaluate treatment responses, we were unable to consider therapeutic aspects when analysing clinical outcomes. Lastly, CSF GFAP could not be tested in all acute attacks because we were unable to perform the CSF study in every attack. Considering the potential heterogeneity in N-LETM, the question of whether an increased level of CSF GFAP is an exclusive finding in NMOSD remains to be determined.
In conclusion, we demonstrated that patients with idiopathic isolated LETM, who were consistently negative for AQP4 antibodies despite repeated testing, were not rare and had many distinct features compared to AQP4 antibody-positive LETM patients. Additionally, the recurrent form of N-LETM may be more common than recognised previously. Since the identification of AQP4 antibody-mediated pathogenesis in NMOSD began with the question of whether NMOSD could be a disease distinct from MS, understanding the clinical features of N-LETM that differ from those of seropositive NMOSD could herald clarifying whether N-LETM has a distinct disease mechanism.
Footnotes
Acknowledgements
We thank Drs Toshiyuki Takahashi and Ichiro Nakashima, Department of Neurology, Tohoku University, for assistance with the serological assays.
Conflict of interest
Drs Hyun, S-H Kim, Huh, W Kim, Ms Yun, RN Joung report no disclosures.
Dr Sato receives scholarship from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and has received research support from Ichiro Kanehara Foundation.
Dr Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Merck Serono, Alexion Pharmaceuticals, Medimmune and Medical Review; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai, Inc., Mitsubishi Tanabe Pharma Corp., Novartis Pharma, Astellas Pharma, Inc., Takeda Pharmaceutical Co. Ltd, Asahi Kasei Medical Co., Daiichi Sankyo and Nihon Pharmaceutical; serves as an editorial board member of Clinical and Experimental Neuroimmunology (2009–present) and a advisory board member of Sri Lanka Journal of Neurology; has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan; is funded as the secondary investigator (#22229008, 2010–2015) by the Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology of Japan and as the secondary investigator by the Grants-in-Aid for Scientific Research from the Ministry of Health, Welfare and Labor of Japan (2010–present).
Dr HJ Kim has received honoraria for speaking or consulting from Bayer Schering Pharma, Biogen Idec, Genzyme, Merck Serono, Novartis, MedImmune and Teva-Handok, and has received research grants from Genzyme, Merck Serono and Kael-GemVax.
Funding
There was no funding source to conduct this research.