
Abstract
Background:
Myelin oligodendrocyte glycoprotein (MOG) antibodies have been described in children with acute disseminated encephalomyelitis (ADEM), recurrent optic neuritis, neuromyelitis optica spectrum disorders and more recently in children with multiphasic disseminated encephalomyelitis (MDEM).
Objective:
To delineate the clinical, cerebrospinal fluid (CSF) and radiological features of paediatric MDEM with MOG antibodies.
Methods:
Clinical course, serum antibodies, CSF, magnetic resonance imaging (MRI) studies and outcome of paediatric MDEM patients were reviewed.
Results:
A total of 8 children with two or more episodes of ADEM were identified from a cohort of 295 children with acute demyelinating events. All children had persisting MOG antibodies (median titre: 1:1280). All ADEM episodes included encephalopathy, polyfocal neurological signs and a typical MRI. Apart from ADEM episodes, three children had further clinical attacks without encephalopathy. Median age at initial presentation was 3 years (range: 1–7 years) and median follow-up 4 years (range: 1–8 years). New ADEM episodes were associated with new neurological signs and new MRI lesions. Clinical outcome did range from normal (four of the eight) to mild or moderate impairment (four of the eight). A total of four children received monthly immunoglobulin treatment during the disease course.
Conclusion:
Children with MDEM and persisting MOG antibodies constitute a distinct entity of relapsing demyelinating events and extend the spectrum of MOG antibody–associated diseases.
Keywords
Introduction
Acute disseminated encephalomyelitis (ADEM) is characterized by a polyfocal clinical event of the central nervous system (CNS) with encephalopathy ranging from behavioural change to alteration in consciousness. 1 Commonly, ADEM follows a monophasic course with a favourable prognosis.
Recently, it was shown that a subset of children with ADEM had serum myelin oligodendrocyte glycoprotein antibodies (MOG Abs).2,3 Children with ADEM and MOG Abs have a characteristic magnetic resonance imaging (MRI) pattern, monophasic course and a good outcome in combination with undetectable MOG antibody levels in follow-up samples.3,4 Serum MOG Abs have also been detected in paediatric recurrent non-multiple sclerosis (MS) inflammatory-demyelinating diseases such as acute disseminated encephalomyelitis followed by optic neuritis (ADEMON),5,6 aquaporin-4 (AQP4) antibody negative neuromyelitis spectrum disorders (NMOSDs)7–9 and recurrent optic neuritis (ON). 10 More recently, single paediatric patients with multiphasic disseminated encephalomyelitis (MDEM) and MOG Abs were published.4,11–13
In this study, we analysed the clinical, cerebrospinal fluid (CSF) and neuroradiological findings of eight children with two or more ADEM-like events and persisting serum MOG Abs.
Methods
As described recently, we included paediatric patients with an acute inflammatory-demyelinating event in a prospective study investigating the presence of serum antibodies against MOG and AQP4.4,14 This study was approved by the Ethics Committee of the Medical University of Innsbruck (study number AN4059). All included patients or their parents provided written informed consent. In total, 56 different community and university hospitals referred children to our attention. From this cohort, comprising 295 children from Austria, Canada, Germany, Italy, Latvia, Switzerland, and Turkey, more than two-thirds of the patients were referred from 14 large university and community hospitals. The majority of children were presented to us with a first episode, while a subgroup of children already had one or more previous episodes. From the overall cohort, we identified 10 patients who had two or more ADEM episodes and MOG Abs. ADEM was defined as a polyfocal event with encephalopathy ranging from alteration in consciousness (e.g. stupor and lethargy) to behavioural change unexplained by fever, systemic illness or postictal symptoms according to the International Paediatric MS Study Group (IPMSSG) definition provided by Krupp et al. 1 Of these 10 patients, 5 were contributed from the 14 main referring centres. The other five patients originated from centres, which had referred only one or two patients. We excluded two patients with MDEM and MOG antibodies because of insufficient data regarding the initial event and missing follow-up serum samples in one and another because he was previously published. 4 One patient (patient 5) who was previously described was included since new data including follow-up, serum samples and imaging studies were obtained. 5 Finally, eight children with two or more ADEM episodes were selected.
From these children, clinical data, MRI, serum antibody and CSF studies were evaluated. CSF studies included white blood cell count, total protein and oligoclonal bands (OCBs). MRI was analysed by M.B. and K.R. Magnetic resonance (MR) sequences included T2-axial, fluid-attenuated inversion recovery (FLAIR)-axial, T2-sagittal, T1-axial with contrast medium, myelin T2-sagittal and T1-sagittal with contrast medium. In five of the eight children, MRI was performed on scanners with field strength of 1.5 Tesla and in three of the eight children with 1.5- and 3.0-Tesla scanners.
Serum samples were analysed for the presence of IgG antibodies to MOG and AQP4 by recombinant live cell–based immunofluorescence assays with human embryonic kidney (HEK) 293A cells as previously described.2,15 Screening was performed at dilutions of 1:20 and 1:40 by at least two independent clinically blinded investigators (M.R. and K.S.), and antibody titres of positive serum samples were determined by serial dilutions. MOG-IgG antibody titre levels of ⩾1:160 were classified as seropositive as previously described. 2
Results
Clinical data
From our cohort of 295 children, which included 59 children with ADEM, we identified 10 children (5 girls and 5 boys) with two or more episodes of ADEM, fulfilling the criteria of the IPMSSG. 1 After exclusion of two patients (see above) finally eight children with two or more ADEM episodes were selected (five girls and three boys). A total of six children were of Caucasian origin, and two children had a West Asian, respectively, and a East Asian ethnic background. Median age at onset was 3 years (range: 1–7 years). A total of four patients had a preceding febrile illness within the last 4 weeks before onset. Median follow-up was 4 years (range: 1–8 years). Mean number of ADEM episodes was 3 (range: 2–4), and the median inter-attack interval was 4 months (range: 1 month up to 4 years). According to the definition of MDEM, all patients had at least two ADEM episodes separated by 3 or more months. All ADEM episodes were associated with new symptoms and new lesions on cerebral MRI. In addition, three patients (patients 3, 4 and 5) had further distinct attacks with new neurologic symptoms but without encephalopathy. Two children (patients 3 and 5) had an episode of isolated ON without signs of encephalopathy.
All children except one patient who received intravenous dexamethasone during the first two episodes were treated with intravenous methylprednisolone during the acute attacks (Figure 2 and Supplementary Table). A total of six children received in addition intravenous immunoglobulins (IVIGs) during the acute attack. A total of four children (patients 3, 5, 7 and 8) received IVIG as monthly immunomodulatory treatment at some point during the disease course. One child (patient 3) was treated with plasmapheresis during an acute attack of ADEM.
CSF studies
All children had a lumbar puncture performed at disease onset and at additional time points later in the disease course. A total of seven of the eight children showed a CSF pleocytosis (>5 cells/µL) with a range of 9–91 cells/µL. Only in one child, OCBs were detected in the initial CSF sample (patient 4; Table 1). In two patients (patients 7 and 8), OCBs became positive in subsequent CSF studies performed in both children after the third event.
Demographic, clinical and paraclinical features of eight patients with multiphasic disseminated encephalomyelitis and MOG antibodies.
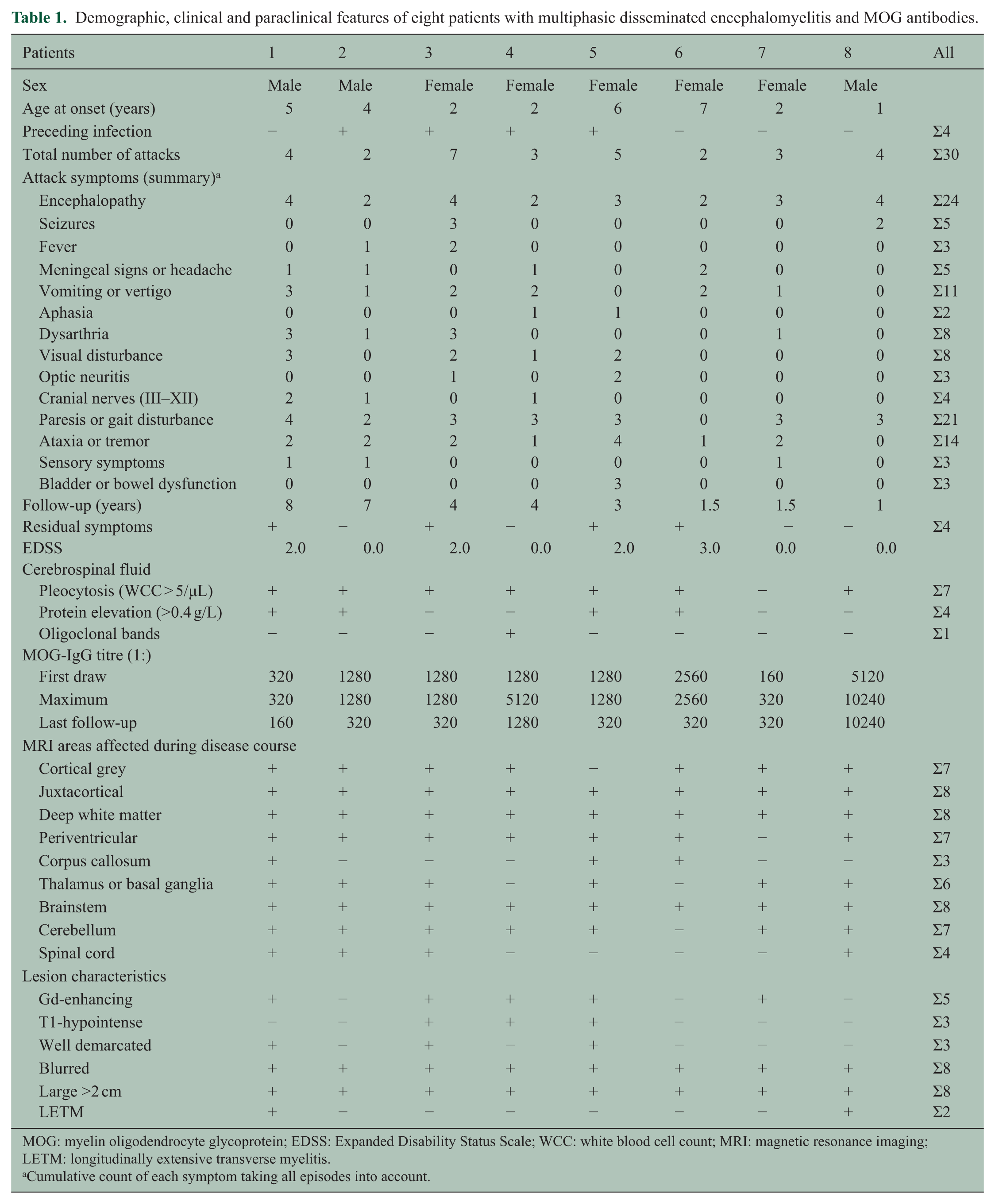
MOG: myelin oligodendrocyte glycoprotein; EDSS: Expanded Disability Status Scale; WCC: white blood cell count; MRI: magnetic resonance imaging; LETM: longitudinally extensive transverse myelitis.
Cumulative count of each symptom taking all episodes into account.
Serum antibodies to MOG and AQP4
Serum samples were available from all children. Only from one child (patient 8), a serum sample from the first ADEM episode was available. In the other children, the first serum sample was obtained 5 weeks after the first episode (patient 6), 9 months after the second episode (patient 2), from the third episode (patients 1, 4 and 5), 3 months after the third episode (patient 7) or between the fifth and sixth episodes (patient 3). All children were MOG-IgG antibody positive with a median MOG-IgG antibody level of 1:1280 (range: 1:160–1:10,240; Table 1). Follow-up MOG-IgG Abs from one to five different time points were available from all eight children. MOG-IgG antibody titres remained positive in all samples. Nevertheless, a decrease in MOG-IgG antibody levels over time could be seen (Figure 2; patients 2, 3 and 6). Only three children showed an increase in MOG-IgG antibody titres during acute attacks (Figure 2; patients 4, 5 and 8). All children were tested negative for AQP4-IgG antibodies.
In addition to the above-described MDEM patients, 66 children from the initial cohort of 295 were found to be MOG antibody positive. In 19 of the 66 patients, no follow-up samples were collected, 14 of the 66 patients became MOG antibody negative on follow-up testing and 33 of the 66 patients were still MOG antibody positive on subsequent serum samples. Of the 33 patients who were still MOG antibody positive on follow-up, 19 patients had negative neuromyelitis (NMO) or limited forms of the disease such as recurrent ON or longitudinally extensive transverse myelitis (LETM) 14 and 11 monophasic ADEM or ADEMON.
Neuroimaging
In all patients, serial cerebral and spinal cord imaging was performed. The median number of MR imaging studies was 7 (range: 4–11), performed over a time period of 1–8 years. Representative MR images of four patients are shown in Figure 1. MR imaging of the first and subsequent ADEM episodes showed in general multiple hazy and large (>2 cm) lesions (Figure 1(c), (e) and (o)), which resolved or diminished in size before the next episode of ADEM occurred (Figure 1(p)). Only in one child, new asymptomatic lesions were detected on follow-up (Figure 1(n)). In three of the eight patients, lesions were well demarcated, and in three of the eight patients, T1-hypointense lesions appeared (Figure 1(g)). Gadolinium-enhancing lesions were found in five of the eight children, mostly with diffuse enhancement, beside rim-enhancing cerebellar lesions in one child in the sixth episode (Figure 1(h)). During the course of the disease, different anatomical areas were affected in the event of new attacks. In half of the children, all brain regions (cortical grey matter, supratentorial white matter, thalamus or basal ganglia, brainstem and cerebellum) and spinal cord were affected (Table 1). Of the four of the eight children with spinal cord lesions, two had an LETM spanning over more than three vertebral segments (Figure 1(d)). Cortical grey matter was affected in seven of the eight children over the course of the disease (Figure 1(c) and (e)). All children had lesions in the juxtacortical and deep white matter and seven of the eight children in the periventricular white matter. The lesions involved to a greater extend the juxtacortical or deep white matter and to a lesser extend the periventricular zone (Figure 1(l) and (m)). Of the eight children, six had lesions in thalamus or basal ganglia (Figure 1(e), (f), (i) and (j)). Infratentorial brainstem lesions (Figure 1(a) and (k)) occurred in all children and cerebellar lesions (Figure 1(b) and (g)) in seven of the eight children. Also, in the majority of non-encephalopathic events, new and large lesions compatible with ADEM appeared (Figure 1(g), (h) and (k)). Only in an episode with pure ON (patient 5), the enhancing optic nerve lesion was the only new lesion detectable on MRI. From seven of the eight children, follow-up MRIs 3–13 months (median: 7 months) after the last event were available. On the last follow-up MRI available, there was a decrease in number and size of lesions noted, but none of the MRIs showed complete resolution of lesions. Follow-up MRI in four children showed minor residuals such as few remaining T2 signal changes but much improved and in three children improvement but still moderate residual lesions.
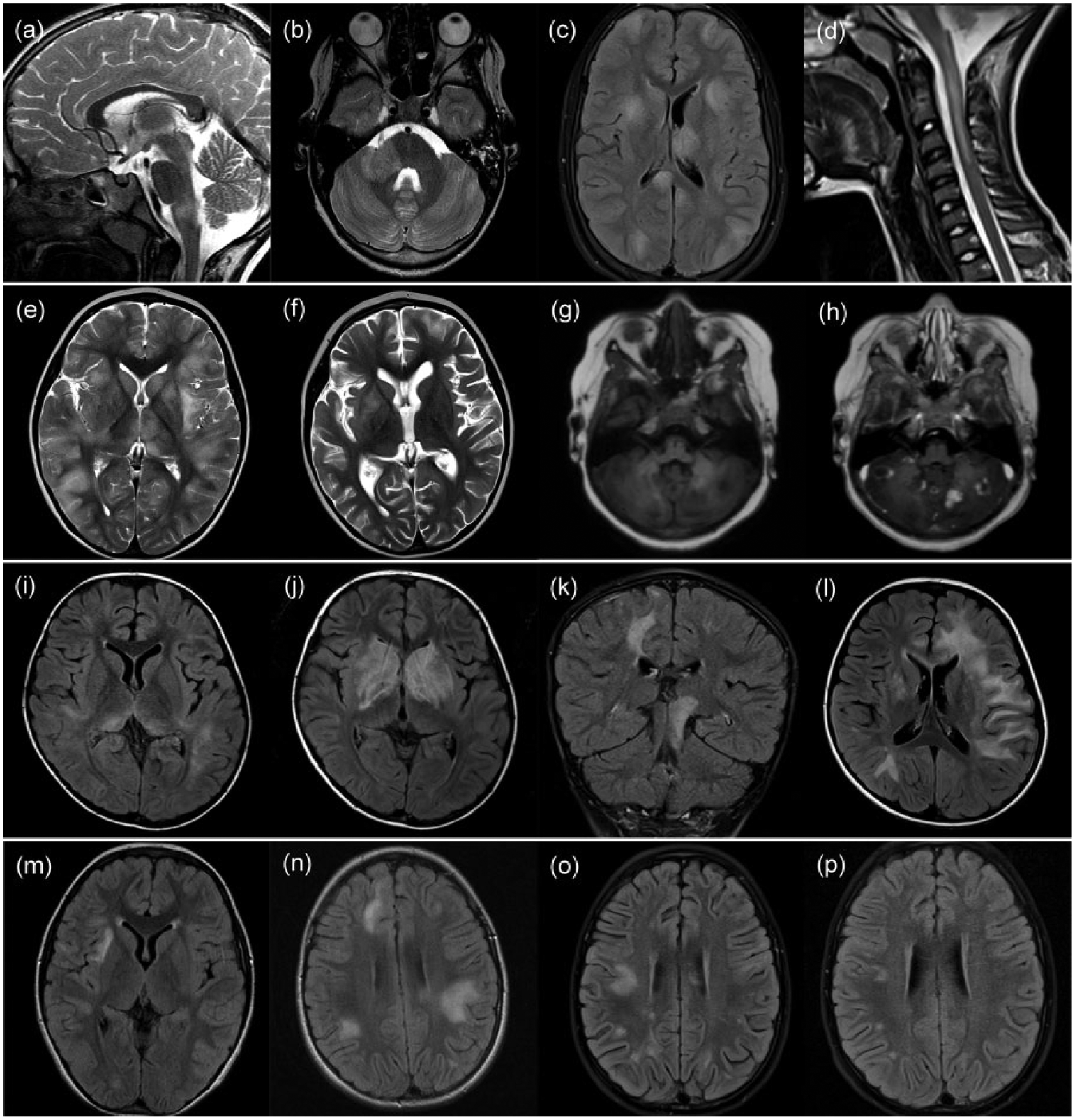
Magnetic resonance imaging of four of the eight paediatric patients with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein. (a–d) Cerebral and spinal MRI of a boy (patient 1) who had the first ADEM episode at the age of 5 years and in total four ADEM episodes (3 months, 3 years and 7 years after the first episode). The MRI of the second episode showed lesions in the brainstem ((a), sagittal-T2) and spinal cord. In the third episode, beside supratentorial lesions, lesions in the cerebellum and cerebellar peduncle ((b), axial-T2) appeared. At the fourth episode, MRI showed multiple blurred supratentorial lesions involving cortex, white matter, basal ganglia, thalamus ((c), axial-FLAIR), brainstem and spinal cord ((d), sagittal-T2). (e–h) MRI of a girl (patient 3) who had the first ADEM episode at the age of 2 years and in total four ADEM episodes (after 2, 6 and 8 months), as well as three additional clinical attacks without encephalopathy (9, 22 and 33 months after the first episode). The MRI of the first episode showed multiple lesions of the cortex, supratentorial white matter, basal ganglia, thalamus ((e), axial-T2), brainstem and cerebellum. In the third ADEM episode, new lesions in the cortex, supratentorial white matter, basal ganglia ((f), axial-T2), brainstem and cerebellum appeared. In the second non-encephalopathic episode, she presented with dysarthria and gait disturbance. The MRI showed beside lesions in the cortex, supratentorial white matter and brainstem multiple T1-hypotintense ((g), axial-T1) and gadolinium-enhancing lesions in the cerebellum ((h), axial-gadolinium-enhanced-T1). (i–l) MRI of a girl (patient 5) who had the first ADEM episode at the age of 6 years and in total three episodes of ADEM (2 and 18 months after the first episode) and two further non-encephalopathic events (9 and 20 months after the first episode). The MRI of the first episode showed lesions in the supratentorial white matter and thalamus ((i), axial-FLAIR) beside brainstem and cerebellar lesions. At the second ADEM episode, the MRI showed large basal ganglia lesions ((j), axial-FLAIR), supratentorial white matter and cerebellar lesions. In the first non-encephalopathic episode, she presented with an optic neuritis and gait disturbance. The MRI showed lesions in the supratentorial white matter, basal ganglia, thalamus, brainstem ((k), coronal-FLAIR) and cerebellum. At the third ADEM episode, MRI showed extensive white matter lesions ((l), axial-FLAIR). (m–p) Cerebral MRI of a girl (patient 6) who had the first ADEM episode at the age of 7 years and a second episode 4 months later. The MRI of the first episode showed lesions in the cortex, supratentorial white matter ((m), axial-FLAIR) and brainstem. An MRI done 2 months after the first event, while she was asymptomatic, showed new large white matter lesions ((n), axial-FLAIR). The MRI of the second ADEM episode showed new blurred white matter lesions ((o), axial-FLAIR), which regressed on follow-up MRI 1 month later ((p), axial-FLAIR).
Outcome
On the last follow-up, 8 months–4 years after the last episode, outcome did range from normal (four of the eight) to mild or moderate impairment (Expanded Disability Status Scale (EDSS): 2.0–3.0) in four of the eight patients with cognitive deficits (attention deficit, memory deficit and learning disorder), motor deficits (coordination disorder and pyramidal signs) or seizures.
Discussion
Here, we describe the clinical and neuroradiological features of eight paediatric patients with two or more ADEM-like episodes qualifying for the diagnosis of MDEM with persisting serum MOG Abs. This is the largest group of MOG antibody–associated MDEM with detailed clinical, serological, CSF and MRI follow-up over a period of 1–8 years. Previous studies have described single patients with MDEM and MOG Abs with a comparable age of onset (3–6 years) and also attacks with ON without signs of encephalopathy.4,11–13
Serum MOG Abs have been described in adults and children with monophasic and relapsing forms of acute inflammatory-demyelinating diseases such as ADEM, ADEMON, AQP4 antibody negative NMOSD or recurrent ON. Although serum MOG antibodies have been initially thought to play a role in the disease process of MS, two recent large paediatric studies showed that the presence of serum MOG Abs strongly predicts a non-MS course and a favourable outcome and is only rarely detectable in children with MS.6,16
Rising evidence suggests that MOG Abs may contribute to the pathogenic events leading to different clinical presentations. MOG protein resides at the cell surface of oligodendrocytes, although despite their proposed importance in CNS demyelination, the function of MOG Abs is not well understood. MOG Abs are predominantly from the IgG1-subtype and induce complement-mediated cytotoxicity in vitro. 15 They further recognize different conformational epitopes and have recently been shown to lead to temporal loss of organization of the microtubule cytoskeleton of oligodendrocytes.17–19 The neuropathology of three cases with MOG antibody–associated demyelination demonstrated MS pattern II pathology with antibody and complement deposition.20–22
In addition to the presence of MOG Abs in this unique group of children, several other clinical findings are of note. Children with MDEM at disease onset were younger than children with recurrent ON or NMOSD as previously reported,6–8,14,23 and interestingly, three of the eight children had additional acute demyelinating events with a wide range of neurological symptoms but without signs of encephalopathy. The MRI obtained during these attacks without encephalopathy revealed a similar MRI pattern with widespread lesions in different anatomical areas.
The presence of an encephalopathy as a criterion for ADEM was 2007 added in the IPMSSG definitions. 24 In clinical practice, the presence of encephalopathy at clinical presentation reduced the number of children who were assigned the diagnosis of ADEM but were subsequently diagnosed as MS. Beside encephalopathy, seizures, bilateral ON, brainstem symptoms and headache with or without meningeal signs are reported to be more likely associated with a diagnosis of ADEM than MS. 25 According to the latest definition proposed by the IPMSSG children with a second episode of ADEM with new symptoms and MRI lesions should be termed MDEM. 1 Disease courses with more than two attacks indicate a chronic disorder such as MS or NMO. In our group, the patients with more than two attacks did not show an MS course or fulfilled the criteria for NMO.
Another important observation was that all children with multiple episodes of ADEM in our cohort of 295 children had persisting MOG Abs. The occurrence of further acute demyelinating attacks reminiscent of ADEM in the absence of MOG Abs according to our experience is associated with an alternative diagnosis. Further of note is that four of the eight children had or are still on immunomodulatory therapy with monthly IVIG. All four children had no further attacks after treatment with IVIG was initiated (Figure 2).
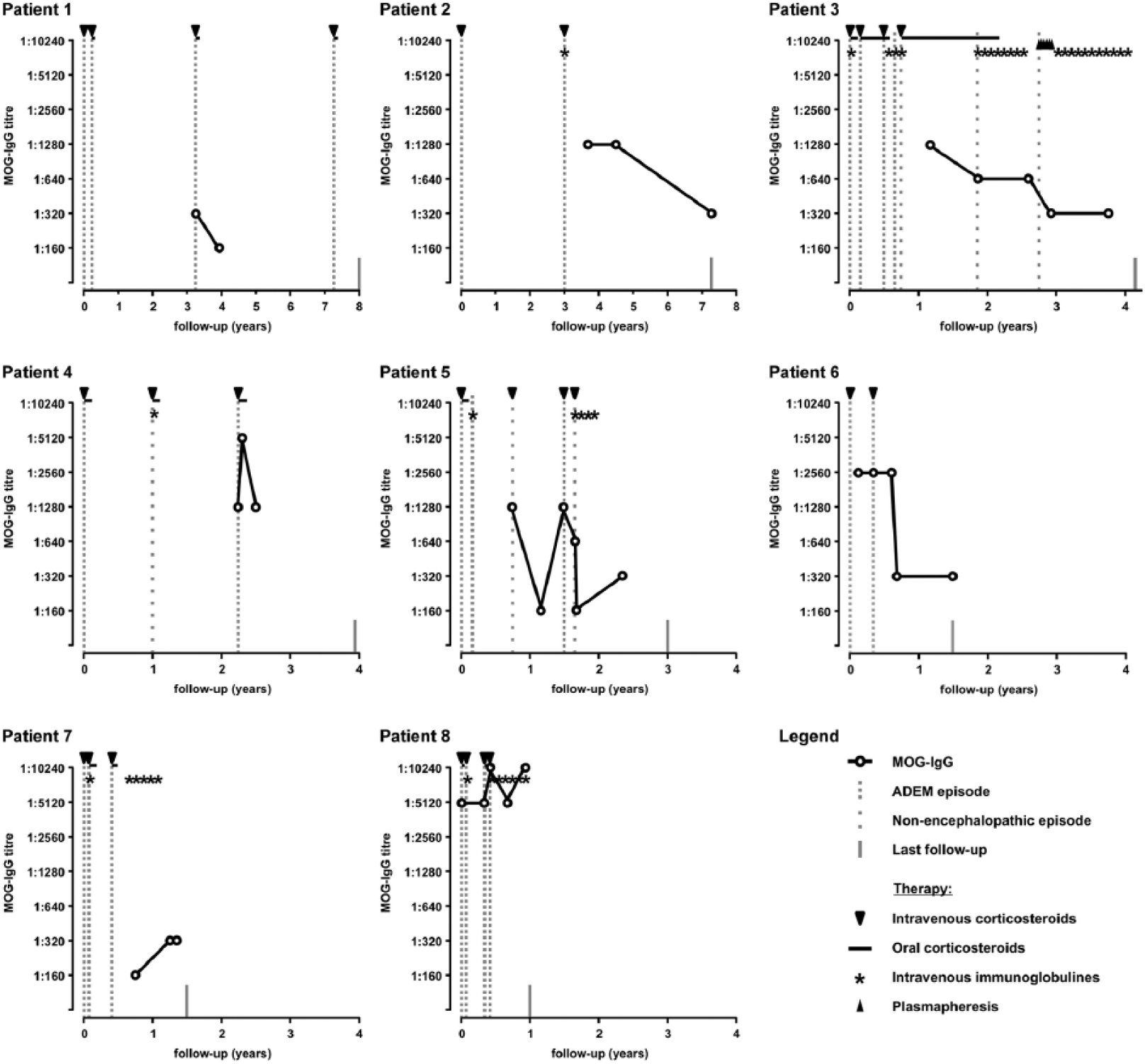
Myelin oligodendrocyte glycoprotein-IgG serum antibody titres, relapse rates and therapy of eight children with multiphasic acute disseminated encephalomyelitis.
Several limitations need to be addressed such as the small number of patients and the variable follow-up period between the different patients. Third, serum samples from the initial ADEM event were not available from the majority of patients. However, serial measurements subsequently performed from each child showed persistently elevated MOG Abs levels over time. Further of note is also that in our overall cohort, monophasic ADEM patients who were MOG antibody negative did not become MOG antibody positive on follow-up testing (data available from 12 patients). Finally, we found a high percentage of children with MDEM in our overall cohort, which is most likely due to a referral bias. Although the majority of children in our initial cohort was included after the first episode, we observed that in particular children with recurrent demyelinating episodes highly suggestive of MDEM were referred after the initial event or even later most likely overestimating the true incidence of this disorder.
The findings of our study and previous reports suggest that MDEM associated with MOG Abs represents a distinct disorder, which should be included under the umbrella of MOG antibody–associated diseases. Furthermore, according to our experience, the presence of MOG Abs in children with MDEM argues against an alternative diagnosis such as MS. Third, children without MOG Abs and recurrent demyelinating events are assigned often a different diagnosis (e.g. CNS vasculitis). 4
Several questions remain and should be addressed in future studies. Why do children with MOG Abs present with different clinical entities? What is the precise role of MOG Abs? Is there a need for, and if yes, which is the best long-term treatment? Due to the rarity of these diseases, only large collaborative project will help to address these questions in the future.
In summary, children with MDEM and persisting MOG antibodies constitute a distinct entity of relapsing demyelinating events and extend the spectrum of MOG antibody–associated diseases. Overall, prognosis appears to be less severe than previously assumed.
Footnotes
Acknowledgements
Matthias Baumann and Kevin Rostásy had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This work was supported by grant from the Jubilaeumsfonds of the Austrian National Bank (K.R.; grant number 14158) and research grant ‘BIG WIG MS’ from the Austrian Federal Ministry of Science, Research and Economy (M.R.).