
Abstract
Background:
Intestinal microbiota is an important environmental factor in the initiation and progression of autoimmune diseases. However, investigations on the gut microbiome in neuromyelitis optica spectrum disorders (NMOSD) are relatively insufficient, especially for that of the Asia population.
Objectives:
To evaluate whether or not the intestinal microbiota of NMOSD patients had specific microbial signatures.
Methods:
Next-generation sequencing and gas chromatography were employed to compare the fecal microbial composition and short-chain fatty acids (SCFAs) spectrum between patients with NMOSD (n = 84) and healthy controls (n = 54).
Results:
The gut microbial composition of NMOSD distinguished from healthy individuals. Streptococcus, significantly increased in NMOSD, is positively correlated with disease severities (p < 0.05). The use of immunosuppressants results in a decrease of Streptococcus, suggesting that Streptococcus might play a significant role in the pathogenesis of NMOSD. A striking depletion of fecal SCFAs was observed in NMOSD patients (p < 0.0001), with acetate and butyrate showing significantly negative correlation with disease severities (p < 0.05).
Conclusion:
The fecal organismal structures and SCFAs level of patients with NMOSD were distinctive from healthy individuals. These findings not only could be critical events driving the aberrant immune response responsible for the pathogenesis of these disorders but could also provide suggestions for disease therapy.
Keywords
Introduction
The neuromyelitis optica spectrum disorders (NMOSD) are inflammatory autoimmune disorders of the central nervous system (CNS) characterized by severe, immune-mediated demyelination, and axonal damage predominantly affecting optic and spinal cord nerves. 1 Although traditionally considered as a variant of multiple sclerosis (MS), the NMOSD is now recognized as a distinct clinical entity based on unique immunologic findings. In 2004, Lennon et al. described a disease-specific serum autoantibody marker, an autoantibody of aquaporin-4 (AQP4) in NMOSD patients. 2 This discovery has provided a key to a better understanding of the autoimmune features of this diverse spectrum of clinical disorders that comprise the NMOSD. Although studies of the genetic predisposition to NMOSD have implicated genes such as human leukocyte antigen (HLA) and multiple non-HLA disease susceptibility genes,3,4 environmental factors also make up a significant part of the risk in disease initiation and progression, which suggest a critical role of epigenetic mechanisms in the pathogenesis of these disorders. Compared with healthy subjects, antibody responses against Helicobacter pylori and gastrointestinal antigens are more commonly detected in NMOSD patients, suggesting that changes in gastrointestinal environment may be associated with the occurrence and/or progression of the disease.5,6
Over the past few years, a congeries of studies have supported the role of the gastrointestinal microbiome in the pathogenesis of neurologic diseases. 7 However, investigations of the gut microbiome in NMOSD are relatively rare. In 2012, Varrin-Doyer et al. 8 discovered that AQP4 p63–76 contains strong homology to aa 204–217 of an adenosine triphosphate-binding cassette (ABC) transporter permease of a gut microbe—Clostridium perfringens. This provides a possible connection between gastrointestinal microbiota and molecular mimicry in the development of CNS autoimmunity. Cree et al. 9 confirmed that differentially distributed gut microbial composition and a significantly increased proportion of C. perfringens existed in patients with NMOSD. As we have known, NMOSD patients in Asia were associated with HLA-DP1*0501,10,11 but not DRB3*0202 or DRB1*0301, while most patients reported in the earlier work were HLA-DRB3*0202, and all AQP4-specific T cells in Varrin-Doyer et al. 8 were restricted by HLA-DR, not HLA-DP. In addition, as described by Zamvil et al., 12 potential molecular mimicry to one epitope of one organism cannot account for NMOSD in all patients. Therefore, we suggest that an investigative effort of expanded populations should be conducted, so as to have a more comprehensive understanding of the relationship between intestinal microbes and the occurrence of NMOSD.
In addition to gut microbes, dietary fatty acids have now become a primary focus of investigations directed at the role of the pathogenesis of CNS autoimmunity. 13 Of particular interest is the effect of short-chain fatty acids (SCFAs), which are produced by intestinal bacterial metabolism of indigestible carbohydrates, and have been shown to ameliorate disease in allergic asthma, 14 diabetes, 15 MS.13,16 In MS patients, for example, reduced levels of Clostridia clusters XIVa and IV in the gut microbiome have been demonstrated, both known to be bacterial species that can produce SCFAs like butyrate. 17 An ancillary linkage of these SCFAs with important immunomodulatory effects has been suggested by the findings that in an experimental autoimmune encephalomyelitis (EAE) model, SCFAs were correlated not only with an increase of Tregs but also with a suppression of differentiation of Th17 cells. 13 Until now, for NMOSD patients, fecal SCFAs concentration and the correlation between them and the disease severity have not yet been evaluated.
Here, we designed a study of 84 NMOSD patients and 54 healthy individuals in which fecal bacterial populations and SCFAs level were determined by next-generation sequencing (NGS) and gas chromatography (GC) based upon the hypothesis that differential microbial members and different levels of SCFAs exist in NMOSD. These could be critical events driving the aberrant immune response responsible for the pathogenesis of these disorders.
Methods
Research participants and sample collection
A cohort consisting of 84 Chinese NMOSD patients, fulfilling the criteria of Wingerchuk, 16 seropositive for AQP4-IgG, was consecutively recruited from Multiple Sclerosis Center, the Department of Neurology, and 54 healthy controls (HCs) were recruited from the Health Examination Center of the Third Affiliated Hospital, Sun Yat-sen University from November 2016 to May 2017. The flowchart of enrollment is shown in Figure S1. The participants in the patient group had different degrees of disability (Expanded Disability Status Scale (EDSS) score from 0 to 6). Of the 84 NMOSD patients, 66 had been treated by immunosuppressants, including 27 treated by mycophenolate mofetil (mmf), 32 treated by azathioprine, and 7 treated by cyclophosphamide, methotrexate, or rituximab. The control group matched for body mass index (BMI), age, and gender. Subjects consuming probiotics or antibiotics within 1 month before admission were excluded. The study was approved by the Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University and informed consent was obtained from all participants.
DNA extraction, PCR amplification, and pyrosequencing
The bacterial DNA was extracted from fecal samples with a QIAamp DNA stool Mini Kit (Qiagen, Germany), according to the manufacturer’s instructions. Construction of sequencing libraries and paired-end sequencing (2 × 250 bp) was performed on an Illumina MiSeq platform at Biomarker Technologies Co, Ltd. (Beijing, China) according to standard protocols. Raw sequences were deposited in the Sequence Read Archive database (http://www.ncbinlm.nih.gov/sra), with the accession numbers ranging from SRR6394750 to SRR6394849, and SRR6475107 to SRR6475144.
Determination of SCFAs
Fecal SCFAs concentration was determined by Shenzhen Academy of Metrology & Quality inspection. Briefly, feces (~100 mg) were thawed and suspended in 1 mL of NaOH (0.5 mol/L), and homogenized (Ultra Turrax T 25, Sweden) for about 10 minutes, and then centrifuged at 13,000g for 5 minutes. The supernatant was filtered through a 0.22 μm sterile filter before GC analysis. The injected sample volume for GC analysis was 1 µL. Data handling was carried out with a HP ChemStation Plus software (A.09.01).
Statistical analyses
A value of p < 0.05 was considered statistically significant in the compared groups. Statistical analysis was performed using unpaired two-tailed t test (Mann–Whitney U test) using GraphPad Prism 6.0 software. Concomitant principal coordinate analysis (PCoA) was performed from binary_otu_gain distance with regard to every pairwise combination of all samples. The linear discriminant analysis (LDA) effect size (LEfSe) pipeline 18 and metastats 19 were employed to identify differentially microbes that distinguished patients from HC. Boruta 20 was used to seek the significant genera to distinguish between NMOSD and HC. The correlations between the abundance of microbial and fecal SCFAs levels were calculated by the Pearson correlation coefficient and visualized by heatmap in R using the “corrplot” package. 21
The detailed information for cohort recruitment, sample acquisition and information collection, 16S rRNA pyrosequencing and assembly, SCFAs determination, and statistical analyses are provided in the supplementary materials (Methods).
Results
Intestinal microbiota distinguished NMOSD patients from healthy individuals
To evaluate whether or not the intestinal microbiota of NMOSD patients had specific microbial signatures, fecal samples from each of the collectively 138 subjects were analyzed by sequencing 16S rRNA gene amplicons (Tables 1 and S1). The average sequencing length and effective tags obtained were 418 bp (range 412–424) and 67,327 (range 50,228–71,172), respectively (Table S2). After removing singletons, the sequences were clustered into operational taxonomic units (OTUs) based on 97% sequence similarity. Similar intestinal microbe OTUs types between NMOSD and HCs as well as the remitting state and the relapsing state were observed (Figure S2). The NMOSD patients and the control group exhibited comparable community richness and diversity (α-diversity) (Figure S3, Table S3).
Clinical and demographic features of NMOSD patients and healthy controls.
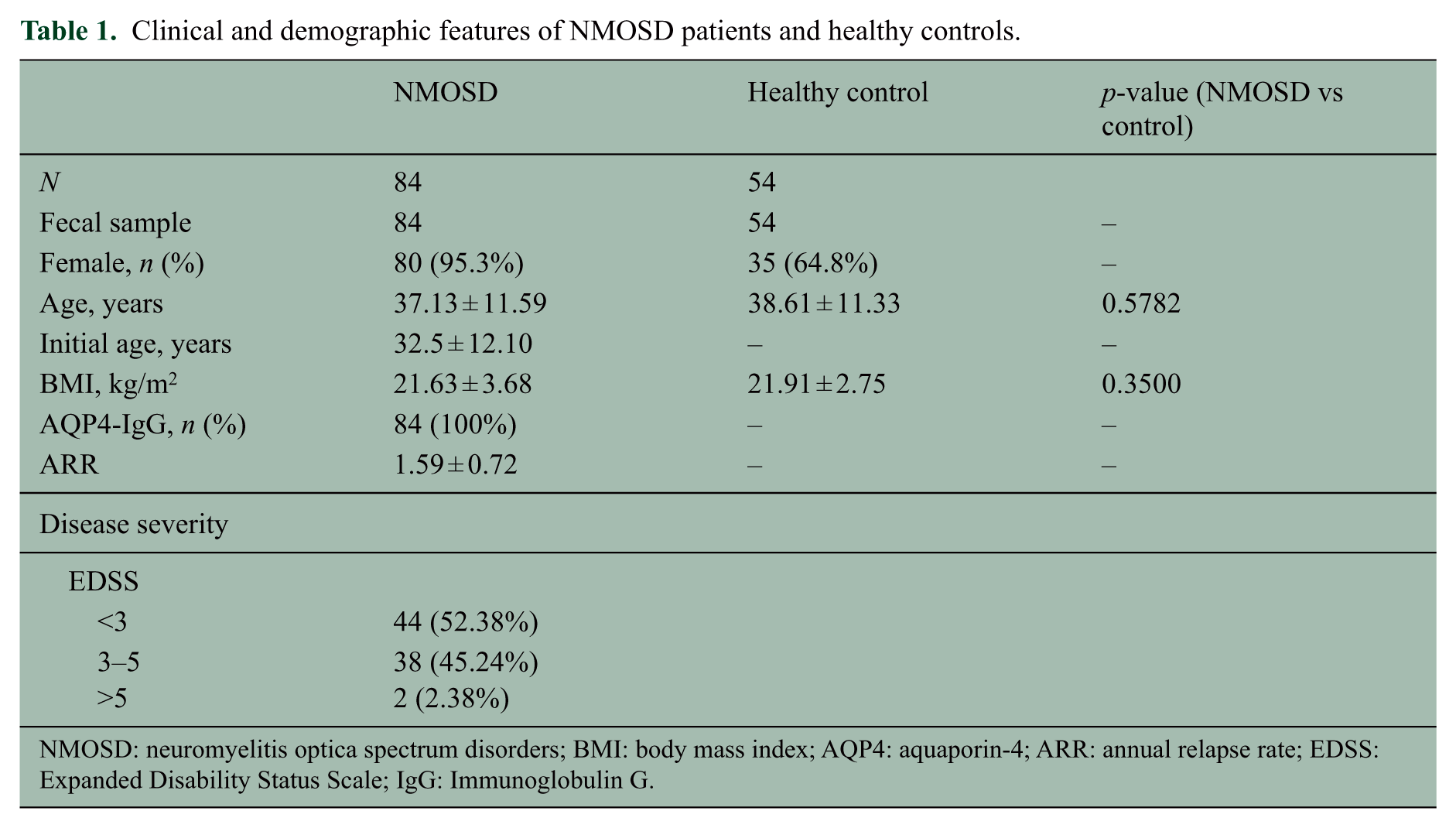
NMOSD: neuromyelitis optica spectrum disorders; BMI: body mass index; AQP4: aquaporin-4; ARR: annual relapse rate; EDSS: Expanded Disability Status Scale; IgG: Immunoglobulin G.
Taxonomic classification showed that most of the intestinal bacteria detected fell into three phyla: Bacteroidetes, Firmicutes, and Proteobacteria. At the genus level, the top 25 genera (mainly Bacteroides, Prevotella_9, Shigella, Faecalibacterium) compose more than 70% of the total microbiota (Figure 1). A concomitant PCoA pointed to disease-dependent clustering of samples (Figure 2(a)), and immunosuppressive therapy did not generate extra clusters (Figure 2(b)). Significant separation was only observed on the PC1 between NMOSD and HC (p < 0.0001) (Figure 2(c) and (d)), indicating that the use of immunosuppressants did not significantly influence the gut microbial composition. In addition, the impact of hormone treatment on gut microbial community structure was also evaluated, and no significant difference was found (Figure S4).
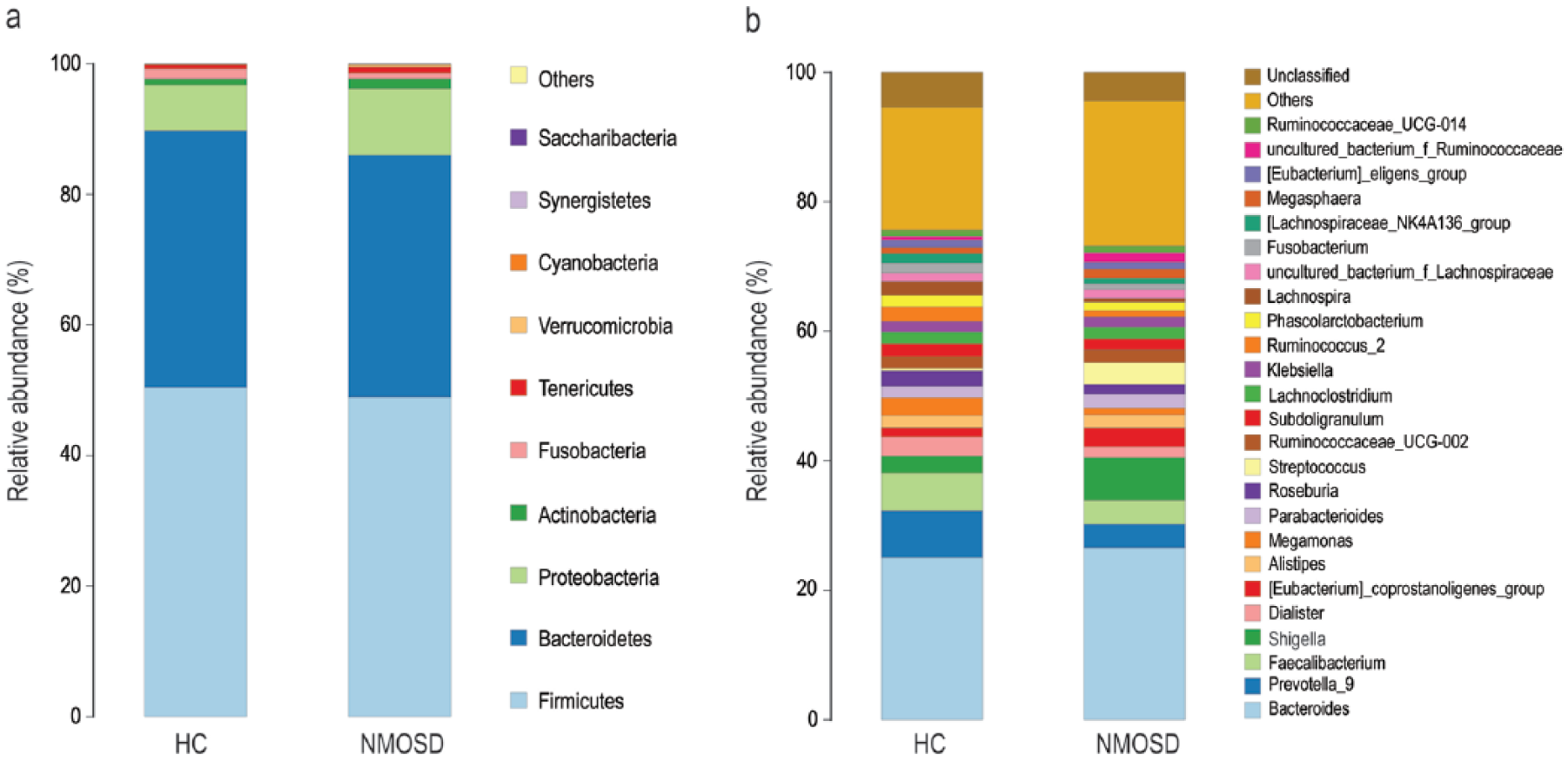
Taxonomic summary of the gut microbiota of NMOSD patients and healthy control (HC) at (a) phylum level and (b) genus level.
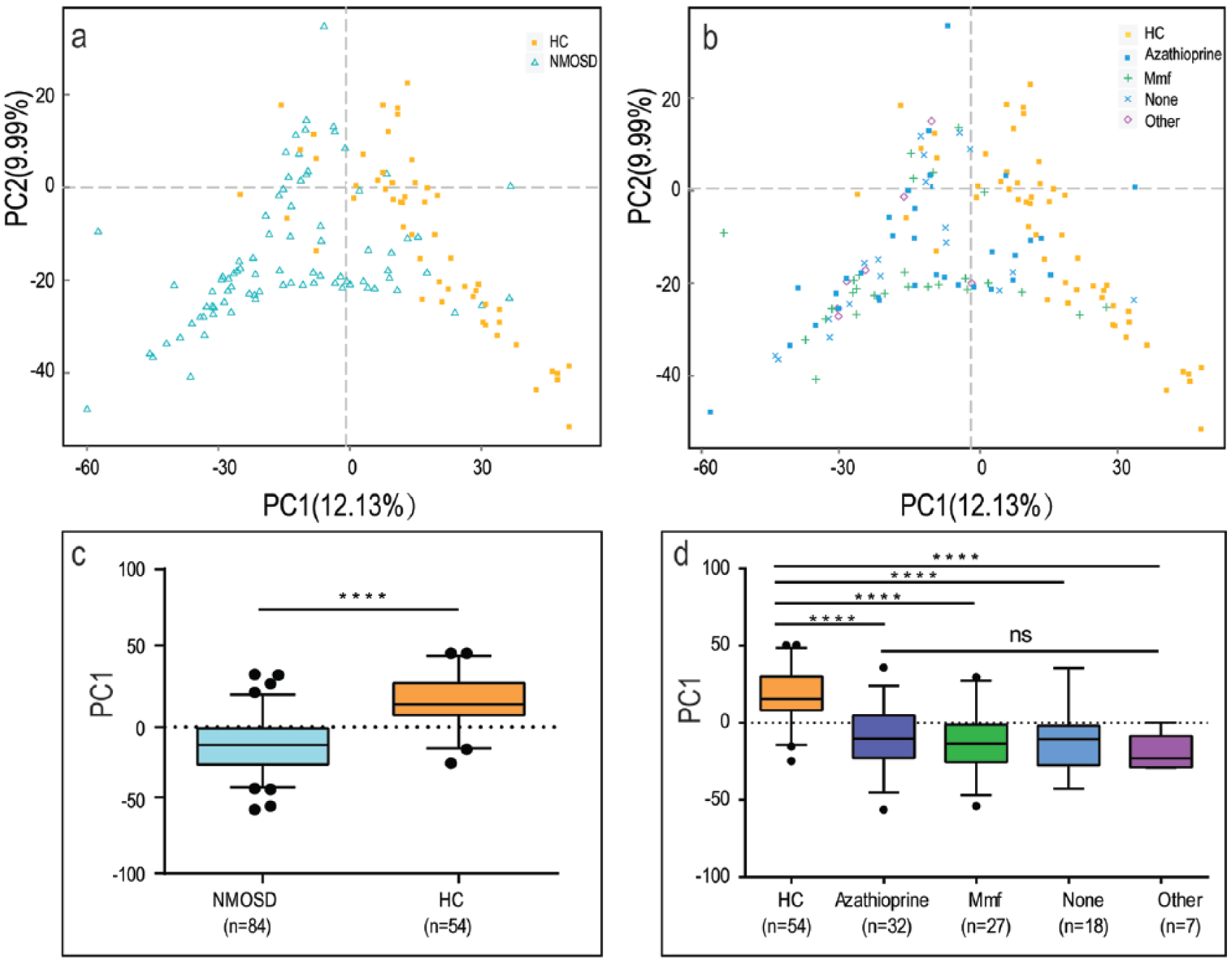
Principal coordinate analysis illustrating the grouping patterns for NMOSD and HC (a, c), and for HC, mmf-/azathioprine-/other immunosuppressant-treated and non-treated patients (b, d). Each point represents the composition of the intestinal microbiota of one participant (a, b). The boxplots display the 95% confidence intervals, and the dots lying outside the whiskers are referred to as outliers (c, d) (****, p < 0.0001; ns, p > 0.05, by the Mann–Whitney U test).
Differential distribution of intestinal microbiota in NMOSD patients
‘LEfSe’ and ‘Metastats’ algorithms were employed to identify the differential distribution of microbiota between NMOSD and HC. Three genera, Streptococcus, Shigella, and Faecalibacterium, were found to be differentially distributed between NMOSD and HC by LEfSe analysis (LDA Score (log 10) = 4) (Figure 3). Sixty differential distributed genera were identified by Metastats analysis (p < 0.05). However, most of them were characterized by low abundance, with only 17 of them reaching a level of 10−3 (Table S4; Figure S5). On the whole, a lack of SCFAs-producing bacteria (e.g. Feacalibacterium, Roseburia, Lachnospira) 22 and an overload of pathogens or opportunistic pathogens define dysbiosis in patients with NMOSD. Boruta algorithm was used to seek the significant genera to distinguish between NMOSD and HC. Of the 17 differential distributed genera, Streptococcus showed the highest importance Z-score (Figure 4). Numerical covariation analysis between differential microbes and clinical indices revealed that the abundance of Streptococcus is significantly positively correlated with disease severities (EDSS) (p < 0.05) (Figure S6). A further characterization of the Streptococcus strains by metagenome sequencing revealed that S. oralis, S. salivarius, S. parasanguinis, S. pneumonia, and S. mitis were the five dominant strains, with S. oralis, S. pneumonia, and S. mitis being significantly different between NMOSD and HC (Table S5, Figure S7).
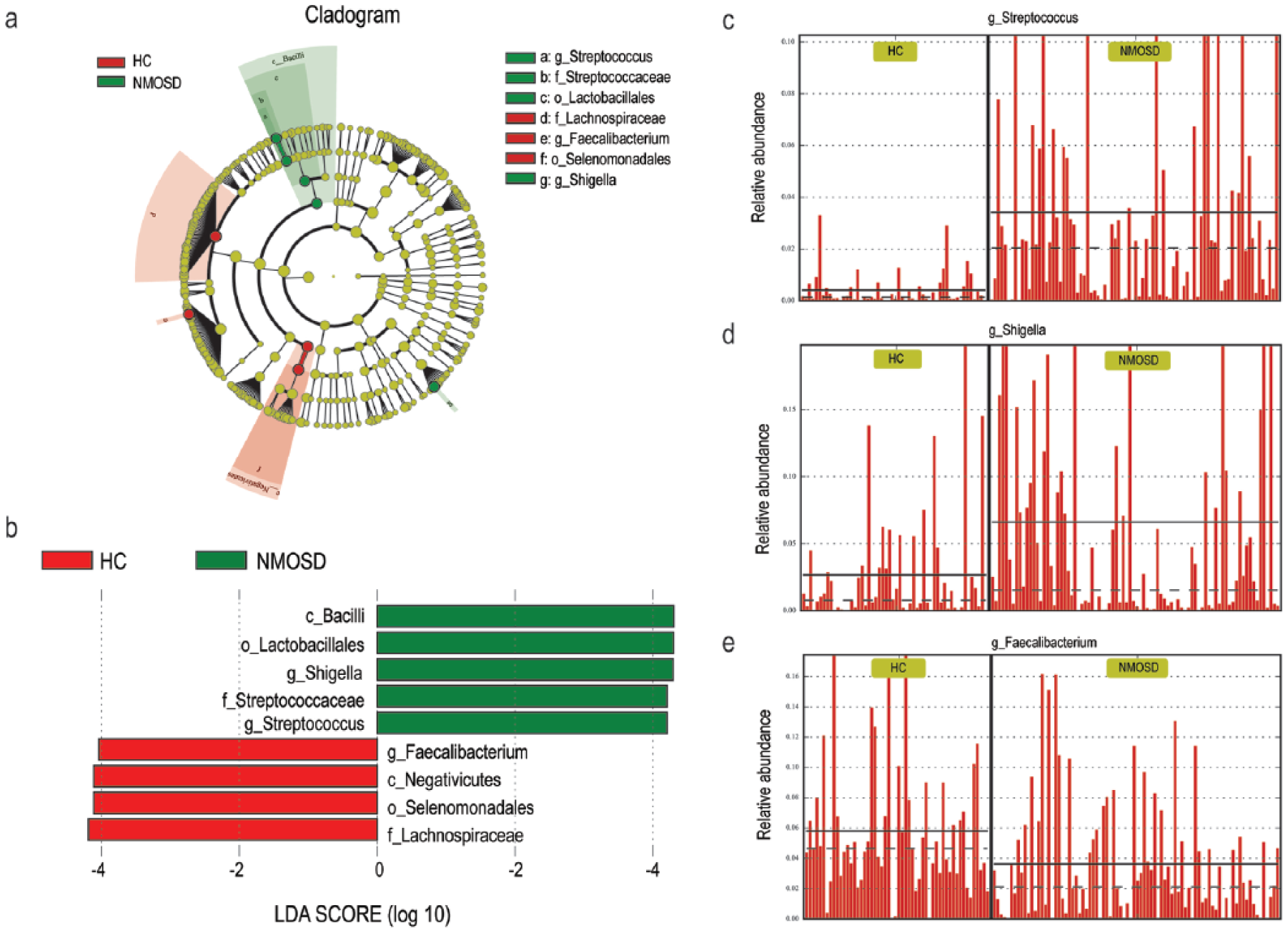
Identification of differential microbes based on the linear discriminant analysis (LDA) and effect size (LEfSe) pipeline. (a) Cladogram using LEfSe method indicating the phylogenetic distribution of intestinal microbiota associated with patients with NMOSD (green) and healthy controls (red). (b) LDA scores showed the significant bacterial difference between NMOSD and HC. (c)–(e) The relative abundance of Streptococcus, Shigella, and Faecalibacterium in each participant from the two groups.
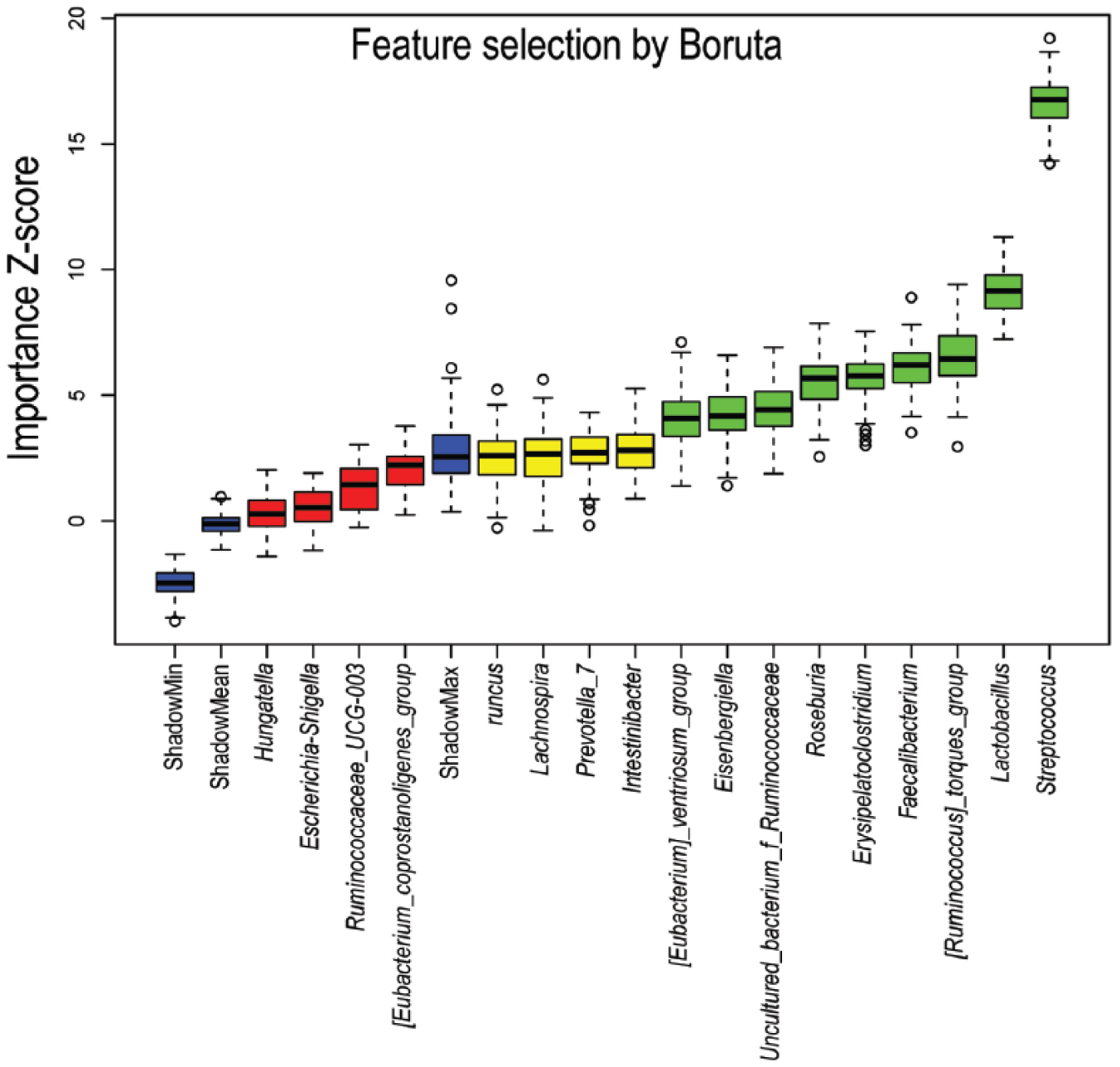
Predictive power of individual genera assessed by Boruta feature selection algorithm. Blue boxplots correspond to minimal, average, and maximum Z-score of shadow genera, which are shuffled version of real genera introduced to RF classifier and act as benchmarks to detect truly predictive genera. Red, yellow, and green colors represent rejected, suggestive, and confirmed genera by Boruta Selection.
Comparative analysis among immunosuppressant-treated groups was conducted (Figure 5). Overall, Streptococcus and Shigella were found enriched in NMOSD, while Faecalibacterium was more abundantly detected in HC. Compared with non-treated group, a reduced proportion of Streptococcus was observed in mmf- and azathioprine-treated groups (Figures 5(c) and S8(a)), suggesting that the enrichment of Streptococcus in NMOSD was primarily affected by disease but not immunosuppressants treatment. Shigella was more abundant in mmf-/other immunosuppressant-treated group than that in azathioprine-/non-treated groups, indicating that immunosuppressant treatment may disproportionately influenced the distribution of Shigella (Figures 5(d) and S8(b)). Compared with HC, the abundance of Faecalibacterium tended to be lower in non-treated group, but the difference was not significant (Figure S8(c)). However, a significant reduction of Faecalibacterium was observed in mmf- or azathioprine-treated groups (Figure 5(e)), indicating that immunosuppressant treatment may further reduce the abundance of Faecalibacterium. On the whole, immunosuppressive therapy did not cause a significant difference in bacterial populations, except that azathioprine significantly reduced the abundance of Streptococcus in patients with NMOSD (Figure S8(a)). Ruminococcus_torques_group and Veillonella were detected enriched in patients treated by other immunosuppressants (Figure S9). But due to too few cases, these members will not be discussed any further.
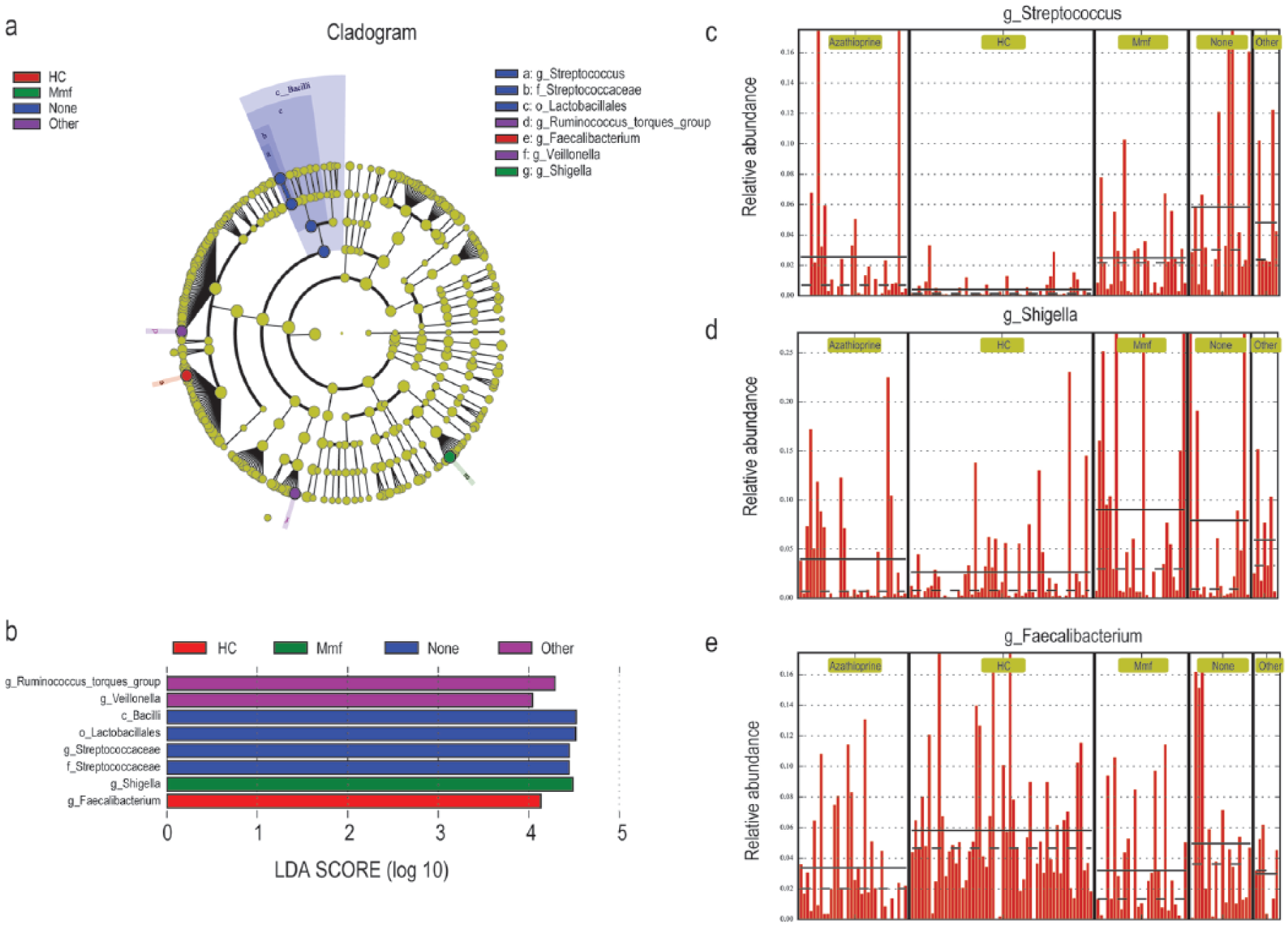
Identification of differential microbes based on the linear discriminant analysis (LDA) and effect size (LEfSe) pipeline. (a) Cladogram using LEfSe method indicating the phylogenetic distribution of intestinal microbiota associated with HC (red), mmf-treated (green), non-treated (none, blue) and other immunosuppressant-treated (other, violet) groups. (b) LDA scores showed the significant bacterial difference among HC, mmf-treated, non-treated, and other immunosuppressant-treated groups. (c)–(e) The relative abundance of Streptococcus, Shigella, and Faecalibacterium in HC, mmf-treated, non-treated, and other immunosuppressant-treated groups.
Determination of fecal SCFAs level in NMOSD patients and HC
Due to the fact that a lack of SCFAs-producing bacteria was observed in the microbiome of NMOSD, fecal acetate, propionate, and butyrate of NMOSD and HC were determined. A striking deficiency of fecal SCFAs was observed in NMOSD patients (p < 0.0001) (Figure 6). The average concentration of acetate, propionate, and butyrate in NMOSD was 2862, 1332, and 487 mmol/g, respectively, while the average concentration of acetate, propionate, and butyrate in HC was 16339, 4414, and 2623 mmol/g, respectively (Table S6). Spearman correlations showed that the concentration of SCFAs was negatively correlated with EDSS, with acetate and butyrate being significantly correlated with EDSS (p < 0.05). Significant positive correlations were observed between the abundance of specific SCFAs-producing bacteria and fecal SCFAs concentration. Besides, a few significant differences on fecal SCFAs concentration were observed between immunosuppressive treatments, including fecal acetate level between mmf-/azathioprine-treated and other immunosuppressants-treated groups as well as fecal propionate level between mmf-treated and azathioprine-treated groups (Figure S10)
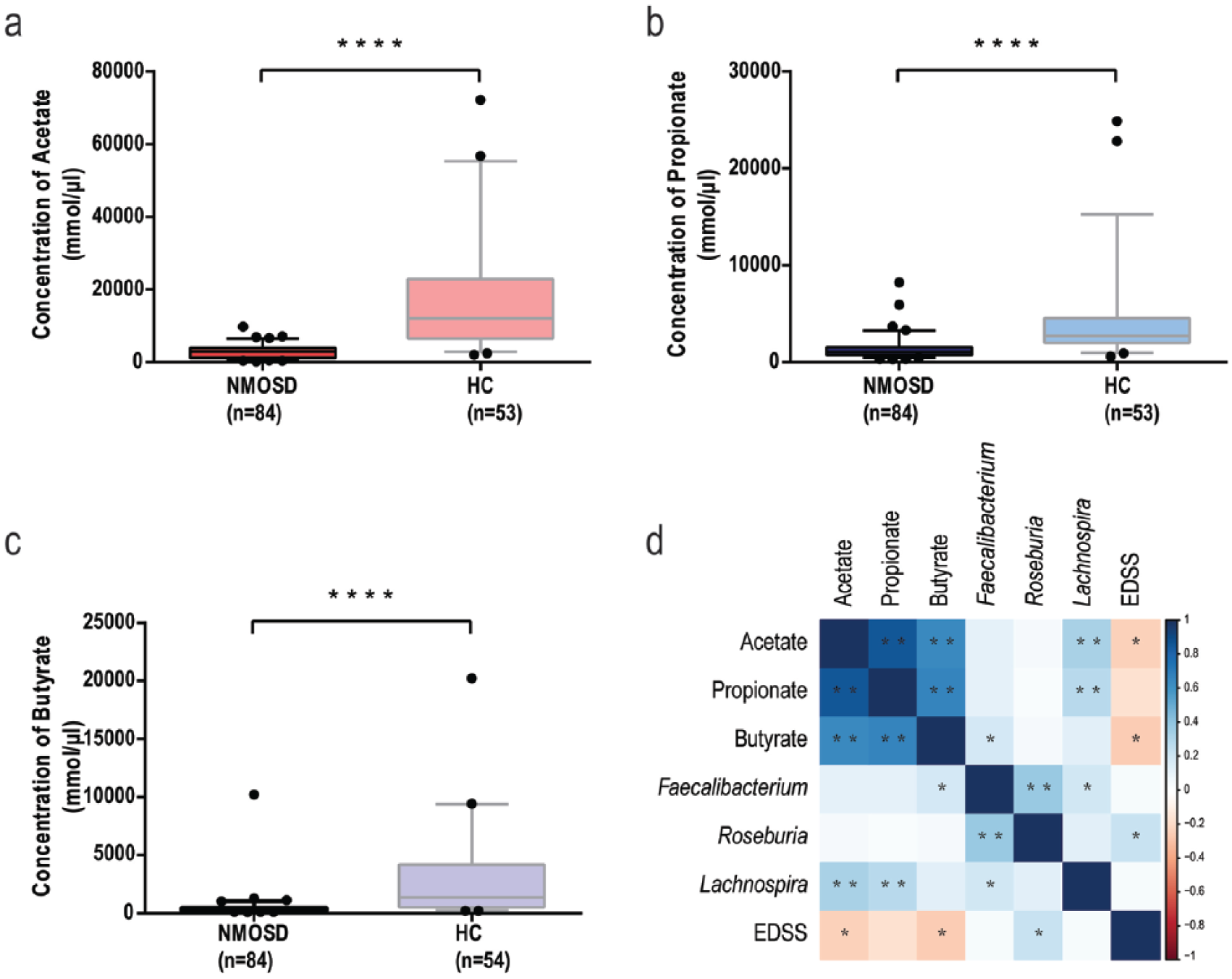
Concentration of fecal short-chain fatty acids in NMOSD and HC (a, b, c), and their correlations with Feacalibacterium, Roseburia, Lachnospira, and EDSS (d). (a) Fecal acetate concentration in NMOSD and HC, (b) fecal propionate concentration in NMOSD and HC, (c) fecal butyrate concentration in NMOSD and HC, and (d) Spearman’s correlation coefficient was calculated between the concentration of SCFAs and the abundance of Faecalibacterium, Lachnospira, and Roseburia and EDSS. The boxplots display the 95% confidence intervals, and the dots lying outside the whiskers are referred to as outliers (a–c) (****, p < 0.0001; *, p < 0.05; **, p < 0.01, by Mann–Whitney U test).
Discussion
Previously, it has been reported that infectious agents, including gastrointestinal pathogens (e.g. H. pylori) have an impact on the pathogenesis of NMOSD.6,23 Moreover, antibodies against gastrointestinal antigens were more commonly found in NMOSD patients. 5 Thus, we suggest that changes in gastrointestinal environment are associated with the occurrence and/or progression of NMOSD.
In the present study, we confirmed that a striking imbalance of intestinal flora accompanied by a lack of SCFAs existed in this cohort of Chinese patients with NMOSD. A lack of fecal SCFAs and an overgrowth of opportunistic pathogens define dysbiosis in patients with NMOSD. Streptococcus, Shigella, and Faecalibacterium were the three differential distributed genera detected between NMOSD and HC. Of them, Streptococcus was primarily affected by disease and the use of immunosuppressive therapy inhibited the expansion of this bacteria. Meanwhile, Streptococcus showed the highest importance Z-score to distinguish between NMOSD and HC, and the proportion of Streptococcus was significantly positively correlated with EDSS. Thus, we suggested that Streptococcus might play a significant role in the pathogenesis of NMOSD. This result is inconsistent with findings from previous studies, which proposed that C. perfringens could be a specific pathogen in the promotion of NMOSD through a molecular mimicry hypothesis.11,12 However, since the NMOSD patients involved in the previous studies were mainly from western countries, we suggested that the difference in genetic and environmental background might contribute to the difference in results.
S. oralis, S. pneumonia, and S. mitis were the three dominant strains distributed differentially between NMOSD and HC. Among them, S. oralis and S. mitis are normal residents of oral cavity, which have been found selectively increased in the commensal microbiota of small intestine of MS patients. 24 S. pneumoniae has been identified as part of the normal upper respiratory tract flora, which was able to cause meningitis 25 and brain abscess 26 other than pneumonia. Until now, the mechanism of Streptococcus in triggering immunity remains unclear; studies showed that they were capable of inducing Th17 cell differentiation in human. 27 Therefore, we suggest that the progressive of NMOSD might be relevant with an aberrant immune response to these members. Shigella has also been reported to be increased in autoimmune diseases, including reactive arthritis 28 and Sjögren syndrome. 29 Besides, Escherichia coli, which is phylogenetically close to Shigella, has been reported to be associated with the pathogenesis of NMOSD via influencing of the differentiation of CD4+ T cells. 30 However, in this study, a significant increase of Shigella was observed in mmf-treated group (p < 0.01), but not in non-treated group. Similarly, Faecalibacterium, which has been reported to be butyrate-producing bacteria, was found significantly decreased in mmf-/azathioprine-treated groups. Therefore, the use of immunosuppressants led to a decrease of Streptococcus (or reducing the EDSS) and dysbiosis, and this may be associated with the clinically reported severe gastrointestinal side effects. 31
In addition, the significant reduction of fecal SCFAs level is another important feature of NMOSD patients. Correlation analysis showed that acetate and butyrate were significantly negatively correlated with EDSS, indicating that they may play important roles in regulating the occurrence of NMOSD. SCFAs have anti-inflammatory effects that go beyond the gut, which help increase Tregs and suppress of differentiation of Th17 cells. 17 Besides, Treg cells play an important role in the prevention and treatment of autoimmune diseases.32–35 Therefore, the lack of protection from anti-inflammatory metabolites of beneficial bacteria also plays a key role in the pathogenesis of NMOSD.
In summary, this study used a large cohort to offer a first investigation of the intestinal microbiota and fecal SCFAs spectrum in Chinese NMOSD patients and HC. An increase of immunogenic microbial and a decrease of anti-inflammatory metabolites were suggested to be a driving force for the development and progression of NMOSD. This is different from the viewpoint of molecular mimicry. Given that the control used in this study is HCs but not unaffected controls, the differentially distributed microbes may offer limited assistance for early diagnosis, but they provide suggestions for disease treatment from the perspective of gut microbiota. We further suggest that an integrated analysis of immunological, microbial as well as metabolic features should be conducted, which could be necessary in revealing the microbial functions involved in the generation or promotion of NMOSD.
Supplemental Material
MSJ790396_materials_and_methods – Supplemental material for Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study
Supplemental material, MSJ790396_materials_and_methods for Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study by Junli Gong, Wei Qiu, Qin Zeng, Xiyuan Liu, Xiaobo Sun, Huijuan Li, Yu Yang, Aimin Wu, Jian Bao, Yuge Wang, Yaqing Shu, Xueqiang Hu, Joseph A Bellanti, Song Guo Zheng, Yongjun Lu and Zhengqi Lu in Multiple Sclerosis Journal
Supplemental Material
MSJ790396_supplementary_figures – Supplemental material for Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study
Supplemental material, MSJ790396_supplementary_figures for Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study by Junli Gong, Wei Qiu, Qin Zeng, Xiyuan Liu, Xiaobo Sun, Huijuan Li, Yu Yang, Aimin Wu, Jian Bao, Yuge Wang, Yaqing Shu, Xueqiang Hu, Joseph A Bellanti, Song Guo Zheng, Yongjun Lu and Zhengqi Lu in Multiple Sclerosis Journal
Supplemental Material
MSJ790396_supplementary_tables – Supplemental material for Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study
Supplemental material, MSJ790396_supplementary_tables for Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study by Junli Gong, Wei Qiu, Qin Zeng, Xiyuan Liu, Xiaobo Sun, Huijuan Li, Yu Yang, Aimin Wu, Jian Bao, Yuge Wang, Yaqing Shu, Xueqiang Hu, Joseph A Bellanti, Song Guo Zheng, Yongjun Lu and Zhengqi Lu in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors thank Dr Ji and Eng. Deng at Shenzhen Academy of Metrology & Quality Inspection, Guangdong, China, for technical support in SCFAs determination in this work. J.G. and W.Q. were co-first authors
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was partly funded by the National Natural Science Foundation of China (NSFC) (no. J1310025) and Program from Guangdong Introducing Innovative and Entrepreneurial Teams (2016ZT06S252).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.