
Abstract
Traditionally, multiple sclerosis (MS) specialists have been the go-to providers for managing certain treatable non-demyelinating inflammatory or autoimmune central nervous system (CNS) disorders. The advent of increased incidence (mostly due to improved recognition) prompts the question: who should be managing autoimmune encephalitis? These patients are generally first encountered in the hospital, as well as general neurology and subspecialty clinics, such as epilepsy. Autoimmune neurology is a specialty which gives focus to evaluation and treatment of patients with autoimmune encephalitis, among other disorders, and trains neurologists accordingly. Some of those experts are dual trained in both MS and non-MS inflammatory/autoimmune CNS disorders. Many other autoimmune specialists are trained in non-MS care, such as hospital neurology, movement disorders, and epilepsy. General and other subspecialty providers increasingly find the need to be versed in management of autoimmune encephalitis.
Introduction
Until relatively recently, few neurological phenotypes were thought to have an autoimmune cause as a differential diagnostic consideration, save for some peripheral neuropathies (such as chronic inflammatory demyelinating polyneuropathy), neuromuscular junction disorders (myasthenia gravis and Lambert-Eaton syndrome) and multiple sclerosis (MS), and few beyond those diagnoses were empirically treated. Historically, neurologists had limited prescribing experience with immune therapies, beyond corticosteroids, intravenous immune globulin (IVIg), oral immune suppressants, and parenterally administered MS drugs.
Starting in the 1980s, neural antigen-specific antibody markers of diverse autoimmune neurological diseases, including encephalitis, began to be discovered. 1 This was slow at first, and then occurred almost exponentially since 2004.2,3 These IgG biomarkers are detected in serum, cerebrospinal fluid (CSF), or both. Some antibodies are accompanied by neurological phenotypes restricted to 1 or 2 syndromes, while others are associated with diverse or multifocal findings. 4 Every anatomical level of the nervous system has at least one described autoimmune cause to be considered in patients presenting with rapid onset and progression. Examples include encephalitis, epilepsies, myelitis, ataxias, stiff-person syndrome, MS mimics (neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein (MOG) autoimmunity), neuro-degenerative movement disorder mimics (e.g. chorea), as well as a large variety of autoimmune peripheral neuropathies.4–9 Some disorders are triggered in the context of occult systemic cancer (paraneoplastic), others in the course of infection (parainfectious), while others (like most systemic autoimmune disorders) are of unknown cause (idiopathic).10–12 Many affected patients have personal and family histories of more common autoimmune diseases, such as Hashimoto thyroiditis and rheumatoid arthritis. 13
This increase in disease recognition has resulted in a change in approach toward patients with rapidly progressive neurological disorders of unknown cause. Diagnostic criteria for autoimmune encephalitis have been developed. 14 In parallel, a diverse new generation of immune molecule-targeted intravenous monoclonal therapies has become mainstream. However, because of the rarity of these disorders, randomized-controlled trials of treatment are few.15–18 The message regarding the importance of early and sustained immune therapy has been repeated often and is now established practice. Indeed, seronegative autoimmune neurological cases are sometimes encountered and empiric trials of immunotherapy may be undertaken.19,20
MS doctors and rheumatologists have historically been the “go to” specialists for immunotherapy recommendations and implementation for non-MS autoimmune or other inflammatory central nervous system (CNS) disorders. The increased recognition of autoantibody-defined encephalitic disorders in general neurology practice, and across all subspecialties, triggers organizational and logistical questions. Herein, we will explore the questions of “who, when and where” for management of autoimmune encephalitis.
The spectrum of disorders
Discrete forms of autoimmune encephalitis, limited to the brain and brainstem, are outlined in Table 1. Common treatable examples include N-methyl-D-aspartate receptor (NMDA-R) encephalitis, leucine-rich glioma inactivated 1 (LGI1 encephalitis) and autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy.21,22 Though this article specifically addresses the questions of autoimmune encephalitis treatment, in reality, autoimmunity can affect every location in the nervous system, and sometimes can be multifocal, with both central and peripheral components.
Classic Antibody Biomarkers of Autoimmune Encephalitis.
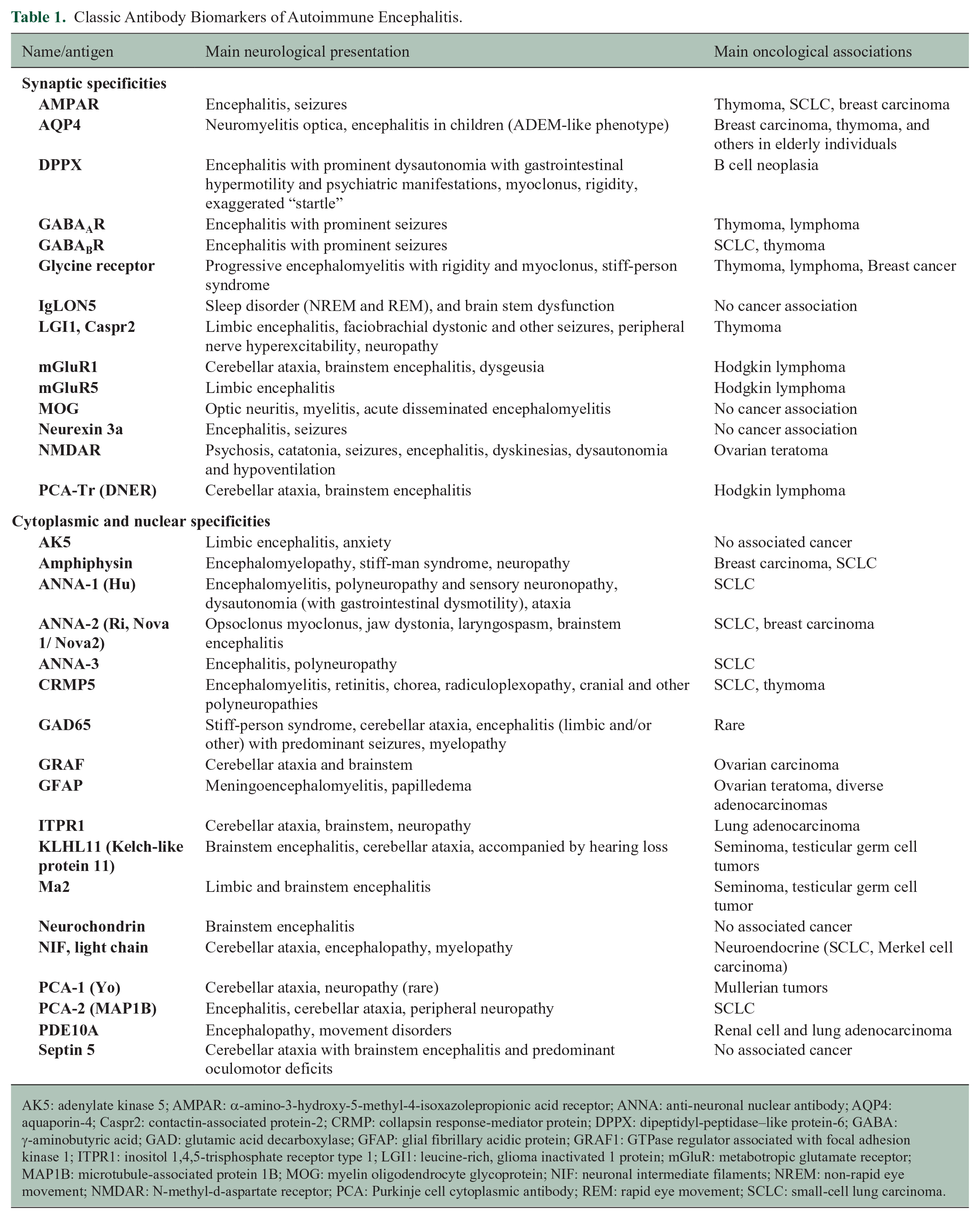
AK5: adenylate kinase 5; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ANNA: anti-neuronal nuclear antibody; AQP4: aquaporin-4; Caspr2: contactin-associated protein-2; CRMP: collapsin response-mediator protein; DPPX: dipeptidyl-peptidase–like protein-6; GABA: γ-aminobutyric acid; GAD: glutamic acid decarboxylase; GFAP: glial fibrillary acidic protein; GRAF1: GTPase regulator associated with focal adhesion kinase 1; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; LGI1: leucine-rich, glioma inactivated 1 protein; mGluR: metabotropic glutamate receptor; MAP1B: microtubule-associated protein 1B; MOG: myelin oligodendrocyte glycoprotein; NIF: neuronal intermediate filaments; NREM: non-rapid eye movement; NMDAR: N-methyl-d-aspartate receptor; PCA: Purkinje cell cytoplasmic antibody; REM: rapid eye movement; SCLC: small-cell lung carcinoma.
While some classic autoimmune encephalitides are brain-restricted, others are not, particularly paraneoplastic disorders. For example, patients with collapsin response-mediator protein 5 (CRMP5) paraneoplastic autoimmunity may have a variety of disorders in addition to encephalitis, namely ophthalmitis, ataxia, myelopathy, and neuropathy.23–26 At the other end of the spectrum, some patients have monosymptomatic disorders masquerading as non-encephalitic disorders. The best described example of this is autoimmune seizures, the most treatable of which is LGI1 encephalitis. These patients typically present with one of several diverse temporal seizure types or classically faciobrachial dystonic seizures. 27 Antibody biomarkers of more chronic and less treatable disorders known as autoimmune epilepsies are specific for glutamic acid decarboxylase 65-kilodalton isoform (GAD65) and Ma1/Ma2 (anti-Ma and anti-Ta). Up to 50% of patients with clinically defined autoimmune encephalitis are antibody negative.28,29 In those patients, the “puzzle pieces” may include an encephalitic syndrome, absence of infectious biomarkers, the presence of other clues to autoimmunity from history, imaging, electroencephalogram, testing for non-neural antigen specific biomarkers in serum (e.g. thyroid peroxidase antibodies) and CSF (oligoclonal bands not detected in serum) as well as the presence or personal history of cancer, especially if one classically associated with paraneoplastic neurological syndromes (e.g. small-cell lung cancer or thymoma). A response to immunotherapy in those patients may also be diagnostic, though this remains controversial. 19 In recent years, immune checkpoint inhibitors (i.e. monoclonal autoantibodies that enhance immune responses by targeting negative regulatory molecules of immune activation, such as CTLA-4 and PD-1) are increasingly used in cancer therapy. 30 Autoimmune complications from those treatments can also affect the nervous system.10,31 Suspicions is raised in patients who present with neurological symptoms (often in the first weeks or months after treatment initiation) regardless of the underlying malignancy, as they can occur also with malignancies not previously associated with neurological autoimmunity (e.g. melanoma or renal-cell carcinoma). 10
Which specialists should treat autoimmune encephalitis? What can MS centers offer?
Overall, patients with autoimmune encephalitis needing initial care and continuity after hospitalization seem underserved. The pathways of referral and care vary from country-to-country and within countries, according to health care model (national insurance, private or mixed), resource availability, and geographical factors (urban, rural, or remote rural). Thus, the opinions herein should be interpreted in the context of the experiences of these U.S.-based authors, though the general principles should be broadly applicable.
Historically, and to some extent presently, many neurologists have limited prescribing immunotherapeutic experience, and may, as reflex, defer to MS colleagues who may or may not have the interest or training themselves. Non-neurologists, such as rheumatologists and oncologists (generally comfortable prescribing rituximab and cyclophosphamide) are sometimes approached to manage treatment despite limited experience in evaluating patients with autoimmune neurological disorders. Because of inherent immune therapy experience, MS centers (also known as MS clinics) are, in some respects, better equipped than most to treat, monitor, and follow-up patients with autoimmune encephalitis post-hospitalization.
Understandably, some MS centers only wish to evaluate and treat MS patients, and there are generalists, or otherwise specialized neurologists, who have gained additional training or experience in the management of autoimmune encephalitis. Dependency on MS centers as the exclusive providers of therapeutic care for autoimmune encephalitis patients is problematic also for logistical reasons beyond provider interest and training. First, the referral pathway to an MS specialist, or rheumatologist, may not be sufficiently expedient for treatment and follow-up of autoimmune encephalitis. In the experience of the authors, specialty referral often occurs late, at the point of, or after, hospital discharge. In that scenario, the patient may have been undertreated as an inpatient, sent to a rehabilitation environment prematurely, without a clear 9- to 12-month plan for outpatient care. Second, MS centers were developed to treat MS, the evaluation, treatment and surveillance of which diverges to some extent from that of autoimmune encephalitis. Thus, there are opportunities for other providers willing to acquire the knowledge and experience of treating autoimmune encephalitis. In addition, health care networks could develop their own systems of managing patients with autoimmune encephalitis. Ideally, all neurology providers would have a working knowledge to effectively diagnose, manage and, where appropriate, judiciously transition care of encephalitis cases to an autoimmune expert, with or without MS background.
At Mayo Clinic, and elsewhere nationally and internationally, specialty clinics (often called Neuroim-munology or Autoimmune Neurology Clinics) have been developed for management of diverse non-MS autoimmune disorders, including autoimmune encephalitis. Such services can permit timely referral, evaluation, treatment, and surveillance of suspected and confirmed autoimmune encephalitis cases. Patients might be identified in the hospital setting and evaluated by an autoimmune neurology-trained neuro-hospitalist. Those patients could then be transitioned to an outpatient autoimmune specialty clinic. Clinics may receive referrals from diagnostic laboratory practice, in addition to those from outside neurologists, patients, and their primary care providers. As well as a 1-year non-MS inflammatory CNS disease training, providers often have trained in another subspecialty (MS, epilepsy, behavioral neurology, movement disorders, or neuromuscular medicine).
While this “hub and spoke” model might be ideal for communities in the catchment of large academic centers, this is not practical for many others (too many other priorities, and too few patients with autoimmune encephalitis). General neurologists or other subspecialists (including MS providers) with interest and experience could provide this care also. In some circumstances, additional guidance could be provided from an autoimmune neurology-trained specialist, potentially by phone or electronic consultation. Those providers could also receive a period of training, education, and updates as outlined below.
While many patients receive intravenous treatments in autoimmune neurology clinics as outpatient (including plasma exchange, corticosteroids, cyclophosphamide, IVIg, rituximab and other monoclonal antibody therapies), most return to non-expert providers closer to home for treatment and surveillance of side effects and efficacy. It is preferable for those patients to return for expert surveillance care for objective neurological follow-up to determine outcome. For the prescription of complex immunotherapy regimens, collaboration with non-neurology community-based specialists (such as rheumatologists, oncologists and hematologists) may be beneficial. In addition, a neurologist (preferably with interest and experience, or some training) could provide follow-up post-treatment to determine outcome, and the necessity of treatment plan changes (drug, dose, and schedule).
Educational considerations
In addition to providing training, autoimmune neurology fellowships also foster program-development post-fellowship, beyond the few centers offering such services at present. There are several advantages to this model of training and care: ease of access for patients, some uniformity of training, establishment of evaluative and treatment protocols, and ultimately multicenter clinical trials, some of which will start enrolling in the next 1–2 years.
Specialized programs could be blended with existing programmatic educational infrastructure. Training in the diagnosis and management of these eminently treatable disorders should start during residency. Too often, the authors come across under-management (e.g. no spinal fluid evaluation nor electroencephalography (EEG) completed, and “5 days” of IV methylprednisolone only as treatment) and over-management (attention difficulties, thyroid antibodies and im-proved mood after 5 days of IV methylprednisolone over-interpreted as resolving encephalopathy).
There are some inherent challenges to making the correct diagnosis. There is the potential for normality on one or more standard diagnostic tests (such as standard CSF protein and white cell values, EEG, and magnetic resonance imaging (MRI)). In addition, antibody test under-utilization (serum without CSF antibody testing ordered, or vice versa), low (but not negligible) positive predictive value of some serum-based antibody tests (e.g. calcium channel antibodies), and nuanced interpretation of others (e.g. GAD65 antibody) contribute to the diagnostic challenges.
Diagnosis and management of autoimmune neurological disorders could form a module of any 2-year neuroimmunology training program, which might consist of 1 year of MS training, and 1 year of other miscellaneous autoimmune or other inflammatory CNS disorders. Expertise in the treatment of other inflammatory CNS disorders, such as sarcoidosis, histiocytosis, and Susac syndrome is also required for any neuroimmunologist.
For post-fellowship providers, there is a plethora of educational opportunities available. Courses and dedicated update sessions are offered at academic national and international meetings both general (e.g. American Academy of Neurology (AAN), and American Neurological Association) and subspecialty (e.g. Consortium of MS centers, International League Against Epilepsy, Movement Disorders Society). There are also Continuing Medical Education (CME)-approved publications and eLearning opportunities through the AAN. There is also the opportunity for case presentation and discussion through the monthly AAN Autoimmune Neurology Section Webinar.
Thus, there are a variety of ways in which neurology providers with an interest in autoimmune neurology could become proficient in the management of autoimmune encephalitis; further expertise can be gained through standard neuroimmunology training programs and specialized autoimmune neurology programs. Future requirements may include neuroim-munology subspecialty board examinations, though such endeavors in neurology are frequently hard to accomplish because of financial and time-resource difficulties.
Treatment principles
Similar to MS relapses, attacks of encephalitis need to be promptly treated, and corticosteroids and plasma exchange are the mainstays of treatment. In addition, there is overlap in medium to long-term preventive treatment principles. For example, B cell-expressed CD20 molecules are a target for monoclonal antibody therapies in both MS (ocrelizumab and rituximab) and autoimmune encephalitis (rituximab). Treatments for MS, a common disease, were often studied initially in animal models, were subject to rigorous randomized-controlled trials, and are almost all approved by regulatory agencies, such as the Food and Drug Administration (FDA). In contrast, “standard” treatments for autoimmune encephalitis were initially extrapolated from established treatment approaches to more common autoimmune diseases, such as myasthenia gravis and MS. Over the last decade, there has been increasing class IV evidence for the treatment of autoimmune encephalitis (uncontrolled retrospective case series or expert opinion). Thus, the contemporary use of some drugs for MS treatment and not autoimmune encephalitis, or vice versa, may not always be derived from pathophysiologic insights.
Acute treatment of autoimmune encephalitis
Immunotherapy is the cornerstone of autoimmune encephalitis treatment (Table 2), and an overview is presented here. Further details may be found in various review articles.21,32 Steroids are often the first line of treatment, either high-dose intravenous or oral. The authors typically use intravenous methylprednisolone for 3–5 days, followed by weekly infusions for 5–11 weeks with osteoporosis and Pneumocystis jirovecii prophylaxis. Plasma exchange treatments are also effective and are used before any monoclonal antibody treatments that they can deplete. Those exchanges can be accomplished early, even alternate day with steroid infusions, or right after the completion of the initial 5 days of steroid treatment. IVIg is often utilized in patients in whom corticosteroids might not be tolerated (monthly or weekly). The prolonged initial treatment can also serve as a trial to evaluate objective immunotherapy responses in case of a diagnostic dilemma, or in cases where autoimmune encephalitis has been previously diagnosed and the current clinical manifestations might represent sequelae. In cases of severe autoimmune encephalitis in the hospitalized patient (e.g. patients with NMDAR-IgG), treatment escalation (such as rituximab or cyclophosphamide) is increasingly considered early on in the illness.
Common immunotherapies used for autoimmune encephalitis.
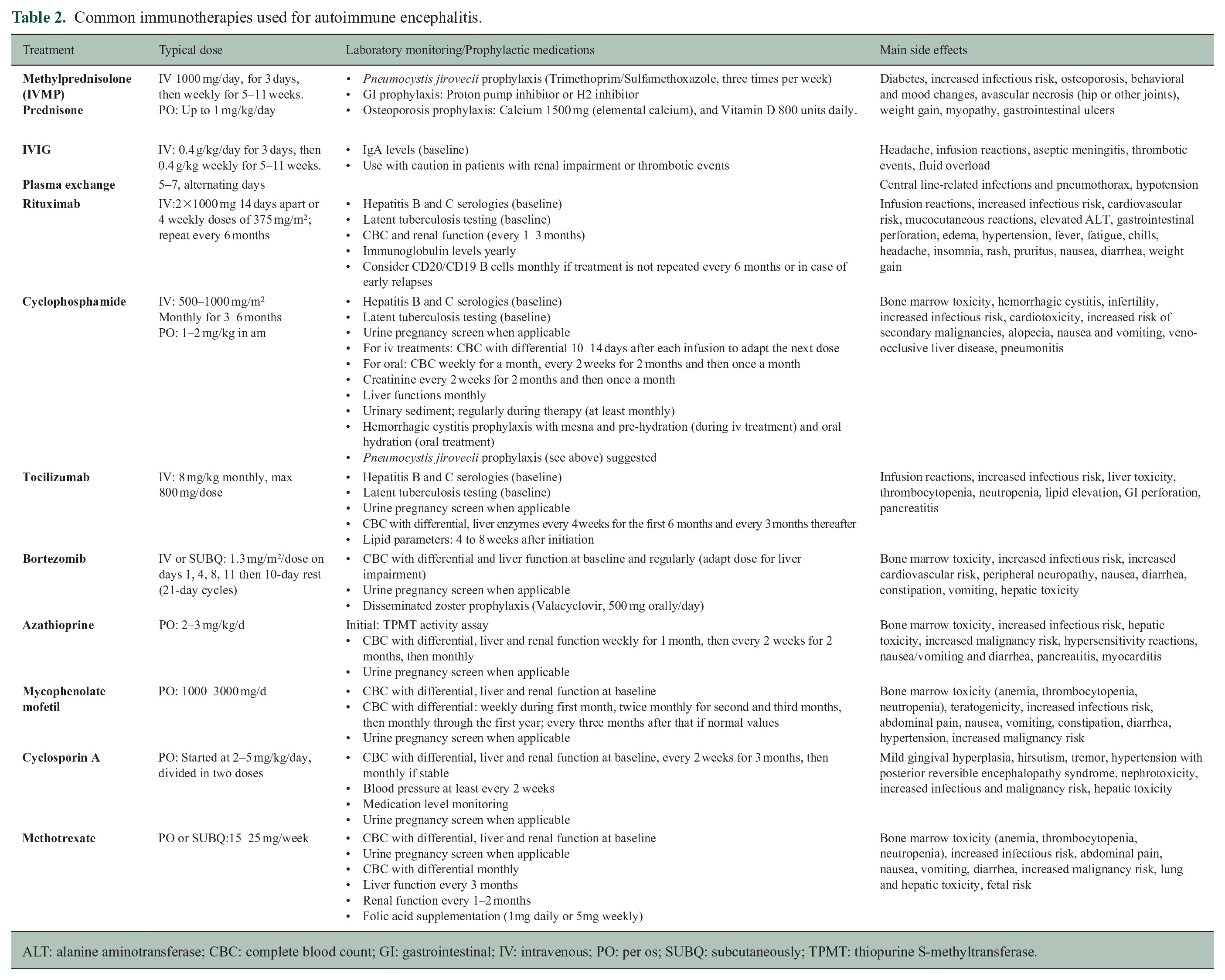
ALT: alanine aminotransferase; CBC: complete blood count; GI: gastrointestinal; IV: intravenous; PO: per os; SUBQ: subcutaneously; TPMT: thiopurine S-methyltransferase.
Chronic treatment of autoimmune encephalitis
Some autoimmune encephalitides can be monophasic and a prolonged initial immunotherapy and a slow taper over several months to avoid early relapses can be sufficient.18,19,29 Others have a protracted course and relapses can occur, prompting consideration for maintenance immunotherapy with often initial overlap with steroids or IVIg. Different options exist depending on the disease and the patient’s profile. 33 Oral immunosuppressants such as azathioprine or mycophenolate mofetil can be used as steroid-sparing agents and long-term treatments for those with relapsing disease. Rituximab, a B-cell depleting therapy is considered in patients that need it as first line and have failed or not tolerated other treatments. Cyclophosphamide, either oral or monthly intravenous, is used in severe cases and in classic paraneoplastic T-cell mediated diseases (e.g. ANNA-1 or anti-Hu associated encephalitis). This treatment should be used early in the disease course for 3–6 months and if effective, reused in case of relapses. Bortezomib, a proteasome inhibitor, has also been successful in refractory NMDA-R encephalitis cases, 17 while inebilizumab (targeting plasmablasts) brought improvement in another CNS B-cell mediated disease, neuromyelitis optica. 15 Finally, treatments targeting interleukins and their receptors have been used in adjunction to more traditional immunotherapies in severe cases.34,35 The optimal duration of maintenance immunotherapy is not known but the authors suggest re-evaluating in 3–5 years in a stable patient.
Surveillance and follow-up of autoimmune encephalitis
In MS practice, patients are generally followed-up during an attack or in the context of medication failure or intolerance, or for routine yearly imaging and assessment. Follow-up measures include symptoms, examination, MRI changes, and disability scores. In contrast, autoimmune encephalitis patients tend to have a severe, prolonged monophasic illness requiring increased care and visits initially (3–6 months), which reduce in number over time. Follow-up measures vary, depending on the tests demonstrating abnormalities at presentation, but should be objective (neurological or paraclinical), particularly given that corticosteroids can produce non-specific feelings of well-being. These include bedside examinations (particularly the mini-mental status examination or MOCA), brain imaging (MRI or PET-CT), seizure diary, EEG (interictal or monitoring) or neuropsychometric testing. Antibody values, either quantitative (such as IU/mL or nmol/L) or semi-quantitative (such as end-point dilutions (titers)), are controversial as surrogates of neurological outcome. 36 In the experience of the authors, these values tend to drift down as the illness resolves and with treatment, regardless of outcome. Seronegativity may occur, though immunological memory predicts ongoing detection of antibody, albeit at low values. Though not useful for predicting disease course or outcome of any given attack, a substantial rise in antibody values at a remote time from initial attack with new neurological symptoms is consistent with relapse of neurological autoimmunity, and sometimes cancer. 37
Patients with LGI1 or MOG encephalitis generally respond quickly to steroids, but should be tapered slowly over 9 months with reducing need for 6–12 weekly surveillance appointments over the course of 12–18 months. IVIg could be utilized instead if steroids are poorly tolerated.18,38,39 In contrast, patients with NMDA-R encephalitis recover very slowly and are usually significantly disabled post-hospital discharge. 33 Patients with GAD65 neurological autoimmunity may present with limbic encephalitis or autoimmune epilepsy. 40 Save for those patients with coexisting gamma aminobutyric acid A (GABA-A) receptor autoimmunity, most GAD65 autoimmune patients have disappointing responses to immune therapies, but may stabilize.
Visits every 3–6 months for up to 2 years are often required to ensure steady (though slow) recovery. Topics for discussion with patients include judicious tapering of immunotherapies and anti-seizure drugs, and screening for chronic complications of brain injury associated with encephalitis. These include headaches, seizures, behavioral changes, mood disorders, sleep disorders, and autonomic dysfunction (e.g. orthostatic hypotension, dysrhythmias, and bowel, bladder and erectile dysfunction). Referral to appropriate specialists may be required to address more complex or refractory symptoms. Multidisciplinary encephalitis clinics to cater for these chronic patient needs seem important for future service development.
Conclusion
Increasingly, neurologists require fundamentals of evaluation and treatment of autoimmune CNS disorders, including encephalitis, particularly for the early illness stages. Efficient pathways of referral to specialists with expertise in autoimmune neurology are important to ensure timely treatment and continuity of care. MS centers have a role to play in the management of patients with autoimmune encephalitis, so long as individual providers have sufficient interest and expertise through formal training or interest, continuing education, and experience. MS centers are but one site at which current and future autoimmune encephalitis patients will be managed. The hospital, the clinic, the local infusion center all have a role to play. Ultimately, the care of these patients should be directed by neurologists, as the outcomes measured are neurological, rather than immunological.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. McKeon reports grants from Alexion, grants from Grifols, grants from Euroimmun, outside the submitted work; in addition, Dr. McKeon has a patent Septin-5-IgG pending, a patent PDE10A-IgG pending, a patent MAP1B-IgG pending, and a patent GFAP-IgG pending. Dr. Zekeridou has a patent on PDE10A-IgG as a biomarker of paraneoplastic neurological autoimmunity pending.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.