
Abstract
Multiple sclerosis (MS) is most likely to adopt a progressive clinical course during middle age or beyond, and the number of older adults with MS is steadily increasing. Developing new strategies to manage progressive forms of MS, which do not respond to currently available disease-modifying therapies (DMTs), will require a deeper understanding of the mechanisms by which biological aging interacts with pathogenic pathways to propel disability accumulation. In experimental autoimmune encephalomyelitis (EAE), a widely used preclinical mouse model of MS, middle-aged animals experience a more severe and protracted clinical course than their younger counterparts. This exacerbated disease course is accompanied by persistent neuroinflammation. Clinical studies of age-related biomarkers, such as telomere length, senescence markers, and DNA methylation, suggest that biological aging is accelerated in people with MS compared with age- and sex-matched healthy controls. Furthermore, distinguishing biological age from chronological may afford more precision in determining aging effects in MS. Here we review the current literature on aging biology and its impact on MS pathogenesis. Future research on this topic may lead to the development of novel biomarkers and senotherapy agents that slow neurological decline in people with progressive MS by targeting relevant aging-related pathways.
Multiple sclerosis through the lens of geroscience
Multiple sclerosis (MS) is commonly considered a disease of young adulthood. However, the number of older adults with MS has increased significantly in recent years. In the United States, the prevalence of MS currently peaks between 55 and 64 years of age. 1 A relationship between age and the clinical course of MS has long been established through epidemiological studies that show advanced chronological age carries a higher risk of developing progressive disease phenotypes, which are relatively refractory to currently available disease-modifying therapies (DMTs). Although the association between aging and progressive MS is highly reproducible at the population level, there is considerable variability in disease progression among individuals with MS of the same chronological age. Biological age, which reflects the cumulative damage experienced by cells and tissues over time, may more accurately predict the severity of MS outcomes. Some patients transition to secondary progressive MS (SPMS) at a relatively young chronological age, while others do not experience obvious disease progression through middle age and beyond. These observations suggest a possible discordance between chronological and biological age. Although a number of demographic factors have been identified that predict a more aggressive MS course and greater disability, 2 the majority of disease variability is still unexplained. Prior work has demonstrated components of biological aging as a major driver of MS disease progression, and that interpersonal differences in the rate of biological aging may account for some of the clinical variability observed in patients. A growing body of translational studies employing cutting-edge techniques to measure biological aging supports this contention in MS.
Biological aging is driven by cellular, molecular, and epigenetic processes that contribute to cumulative tissue damage, loss of functional reserve, and diminution of regenerative potential. The exhaustion of compensatory damage–repair mechanisms ultimately leads to a decline in function, increasing frailty, and vulnerability to age-related diseases. Geroscience is the study of aging biology and its impact on disease. This rapidly growing field posits that curtailing aging mechanisms will reduce a broad spectrum of diseases in older adults. The pathophysiological features of MS evolve with age, and the application of geroscience to MS is in its nascency. This raises the question of whether markers of biological aging will correlate with clinical or radiological outcome measures in people with MS, or predict therapeutic responsiveness to DMTs. A deeper understanding of the interactions between pathways involved in aging and MS pathogenesis could ultimately lead to clinical trials of new classes of DMTs in progressive MS, such as senotherapeutics. In this review, we summarize the findings from preclinical and clinical studies of biological aging in MS and conclude with future directions for translating these findings into clinical practice.
Pathological characteristics of relapsing and progressive MS
A pathological hallmark of relapsing–remitting MS is the acute demyelination of white matter in the brain. Such plaques center around a leaky venule with perivascular inflammatory infiltration and can be visualized as a gadolinium-enhancing lesion on magnetic resonance imaging (MRI) consequent to the focal blood–brain barrier (BBB) breakdown. Infiltrating lymphocytes are largely confined to the perivascular space, whereas pro-inflammatory myeloid cells (monocyte-derived macrophages and dendritic cells) extend into the adjacent white matter where they release pro-inflammatory cytokines and phagocytose myelin.3,4 Clinical relapses closely correlate with the appearance of gadolinium-enhancing lesions. Hence, the acute white matter damage and neurological dysfunction that occurs during MS relapses are driven by an immune assault launched from the periphery against central nervous system (CNS) tissue. In contrast, progression independent of relapse activity is not associated with focal breaches of the BBB or new white matter lesion formation. In fact, clinical relapses and new T2 and gadolinium-enhancing lesions become less frequent as people with MS age.5,6 Rather, pathological features more characteristic of progressive MS include atrophy and neurodegeneration, and slowly expanding or “smoldering” lesions, with a gliotic core surrounded by a rim of classically polarized microglia actively phagocytosing myelin.4,7 There is also widespread microglial activation that extends throughout the parenchymal white matter, beyond the boundaries of the chronic lesions in “normal-appearing white matter.” 8
During the earliest stages of MS, low-grade meningeal inflammation is present and is associated with cortical subpial lesions. The meningeal infiltrates become more organized during progressive MS and form lymphoid follicle-like structures in the depth of sulci. 9 Cortical demyelination is more profuse in progressive than relapsing MS. Collectively, these observations indicate that the dominant neuroinflammatory pathways that drive tissue damage in progressive MS are contained behind the BBB and fundamentally different from those in relapsing MS. This likely explains why DMTs that profoundly suppress MS relapses by depleting circulating lymphocytes or curtailing their passage across the BBB, such as natalizumab, alemtuzumab, anti-CD20 monoclonal antibodies, and sphingosine-1-phosphate receptor modulators, are relatively ineffective in non-active progressive MS.
Preclinical and mechanistic studies of aging biology in autoimmune demyelinating disease
Studies have explored the effects of aging on CNS autoimmune disease by inducing experimental autoimmune encephalomyelitis (EAE), the most popular preclinical rodent model of MS. 10 In two recent studies, middle-aged EAE mice (aged 40–44 weeks) exhibited an exacerbated, protracted disease course with chronic disability, whereas younger adults (aged 8–12 weeks) experienced milder disease and frequent clinical remission.11,12 Using reciprocal bone marrow chimeras, wherein bone marrow cells from young adult or middle-aged EAE donors were transferred into lethally irradiated healthy young or middle-aged hosts, it was demonstrated that the exacerbated, chronic course of EAE is dependent on the presence of aged non-hematopoietic, radio-resistant cells, including glia. 11 Reminiscent of progressive MS, middle-aged mice with EAE had widespread microglial activation. Those microglia expressed a distinct set of genes involved in antigen presentation, cytokine and chemokine production, proteasome assembly, and Type I interferon responses. Moreover, microglia isolated from middle-aged mice expressed reduced levels of genes involved in homeostasis and transcripts encoding heat shock proteins, which have been implicated in neuroprotection. This suggests that aged microglia might contribute to chronic neurological deficits in EAE by driving persistent, smoldering neuroinflammation and producing pro-inflammatory factors that are toxic to oligodendrocytes and neurons, while relinquishing neuroprotective functions.
Age-dependent changes in microglia
The microglial transcriptome changes with aging, acquiring a signature consistent with a constitutively activated, as opposed to a homeostatic, state.13,14 Aged microglia also acquire an ameboid morphology and cell surface phenotype suggestive of activation. Consequently, compared with young microglia, aged microglia may be more predisposed to pathogenesis in the context of neurodegenerative diseases, including progressive MS. Age-related alterations in microglial properties are likely induced by a combination of cell intrinsic events, such as telomere shortening, increased lysosomal β-galactosidase activity, cell cycle arrest, epigenetic remodeling, DNA damage, and mitochondrial dysfunction.4–10 In addition, extrinsic factors such as chronic systemic inflammation and the loss of paracrine regulatory signals that actively maintain homeostasis are likely lost in middle-aged animals.
Microglial activation by cytokines and gut dysbiosis
Aging is characterized by a state of chronic, sterile, and low-grade inflammation. Aged myeloid cells, distributed in tissues throughout the body, assume a senescence-associated secretory phenotype (SASP), whereby they continually release pro-inflammatory cytokines, such as IL-6, IL-1, and TNFα, which diffuse into the bloodstream. In addition, age-related gut dysbiosis combined with increased intestinal permeability releases microbial-derived pathogen-associated molecular patterns (PAMPs) into circulation. Many of these cytokines and PAMPs are CNS-penetrant and capable of stimulating microglia in a senescent-accelerated prone-aged mouse model. 15 Elevations in plasma levels of myeloid-associated cytokines and chemokines, such as CCL11, CXCL1, and G-CSF, have been documented in progressive MS, and in some cases correlate with clinical measures of disability and radiological measures of neurodegeneration or lesion burden.16,17 The impact of such pro-inflammatory stimuli is exacerbated by a loss of regulatory signals that normally restrain microglial activation and stabilize a homeostatic phenotype. Neuronal-derived Fractalkine and CD200 interact with their cognate receptors on microglia to stabilize the homeostatic microglial phenotype. With advancing age, senescent neurons become dysfunctional, or drop out, decreasing these regulatory interactions and thereby lowering the threshold for microglial activation. In addition, there are age-related decreases in the production of gut microbiome-derived short chain and tryptophan metabolites which cross the BBB from the circulation and suppress microglial activation.13,18–21 Collectively, age-related changes in microglia might contribute to the transition from the relapsing–remitting to the progressive phase of MS.
Adaptive immunity and aging
Age-associated changes in lymphocyte populations and functions lead to immunosenescence, a systemic phenotype hallmarked by an increase in pro-inflammatory cytokines. Senescence also occurs on a cellular level, referred to as cellular senescence, which is characterized by irreversible cell cycle arrest. Senescent memory T cells and B cells secrete SASP factors.22–24 SASP factors, released by lymphocytes located in the perivascular infiltrates of chronic, active MS lesions could potentially diffuse to the lesion rim and activate microglia. Granzyme K-expressing CD8T cells accumulate during aging 24 and are enriched in progressive MS brain tissues. 25 Extracellular human Granzyme K has been shown to stimulate the expression and release of SASP factors, such as CCL2, IL-6, IL-8, and from nearby cells, causing a pro-inflammatory cytokine response. 26 In a recent paper, it was reported that older adults with MS have increased frequencies of circulating CD4T cells with activated and cytotoxic profiles compared with normal controls. 27 Furthermore, expression of the inhibitory receptor, CTLA-4, was decreased on the T cells from aging adults with MS, while expression of co-stimulatory molecules was elevated on B cells, which together could drive enhanced and chronic CD4T cell activation.
Remyelination failure and aging
Senescent microglia and infiltrating myeloid cells are less efficient at clearing cellular and myelin debris within white matter lesions. Buildup of debris has been shown to impair oligodendrocyte progenitor cell (OPC) recruitment and remyelination. 28 The senescence of OPCs themselves may also contribute to diminished remyelinating capacity. Myelin repair has been shown to decline with age in numerous animal models and is attributed, in part, to a decreased capacity of OPCs to migrate to sites of injury and to differentiate into mature pro-myelinating oligodendrocytes. 29 These OPC deficits correlate with age-associated epigenetic modulation of oligodendrocyte differentiation, loss of responsiveness to pro-differentiation signals, mitochondrial dysfunction, increased DNA damage, and dysregulated mTOR activity.30,31 In EAE, OPCs located in chronic demyelinating lesions upregulate Sirtuin 1 (SIRT1), which is indicative of OPC cell cycle arrest. 32 Such observations suggest that the pharmacologic reversal age-associated changes in OPCs might facilitate myelin repair in the context of MS and other demyelinating disorders.
Despite studies in EAE providing insights into aging pathophysiology and CNS autoimmune disease, challenges exist in their translation in human studies. There is increasing interest in measuring biological aging in people with MS to determine the associations of aging mechanisms with MS outcomes.
Biological aging in people with MS
The utility of biological age in predicting susceptibility to adverse outcomes has been demonstrated across multiple clinical contexts. However, there are still ongoing challenges to identify a single, or set of, aging biomarkers that can reliably and accurately determine the rate of aging in both humans and laboratory animals. 33 Nevertheless, candidate biomarkers have been derived from hallmarks of aging processes, and some are beginning to shed new light on MS disease outcomes and progression.
Telomere length is a well-recognized aging biomarker that has been linked to the development of age-associated diseases, such as cardiovascular disease and dementia. 34 Telomere length is also the most studied marker of biological aging in MS. The attrition of telomeres as the protective endcaps of genomic DNA predicts the onset of cellular senescence. 35 Peripheral blood leukocytes from individuals with MS typically have shorter telomere lengths compared to healthy controls. 36 Furthermore, a large cohort study by Krysko et al. 37 found that, independent of chronological age, shorter leukocyte telomere length was associated with higher disability and lower brain volume in people with MS. These findings suggest that there may be accelerated biological aging as a downstream effect of replicative senescence of telomere shortening in people with MS. Some limitations in telomere length, such as measurement variability, hinder its utility, and different approaches to measuring telomere length complicate comparisons between studies and possibly among longitudinal samples of the same study.
Epigenetic alterations are a well-defined aspect of biological aging that has only recently been studied in people with MS. DNA methylation across a set of cysosine-phosphate-guanine (CpG) sites is a major epigenetic operator that changes with age and can be assessed in tissues and blood.Site-specific percent methylation at each CpG site can be quantified and used to derive an “epigenetic clock” that tracks closely with chronological aging.38,39 Different clock algorithms can be created from methylation data across specific subset of cells or tissues and trained to identify distinct physiological events or outcomes. The residual between epigenetic age and chronological age identifies individuals who are biologically older or younger. Those with accelerated epigenetic aging have been shown to have higher risk of age-related diseases, such as cardiovascular disease, diabetes, neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease, and premature mortality.40,41
Early findings suggest that epigenetic age is accelerated in people with MS. DNAm PhenoAge, a second-generation epigenetic clock with improved accuracy in predicting aging outcomes, was accelerated in whole blood from a cohort of individuals with MS compared to healthy controls. 42 Similarly, evidence of epigenetic age acceleration was observed in glial cells from the post-mortem brain tissue of patients with progressive compared to controls. 43 However, these studies were limited by lack of longitudinal follow-up and analysis of MS outcomes. Further studies are needed to confirm these preliminary findings in MS and determine associations of epigenetic age with MS outcomes.
A few other markers of biological aging have been studied in people with MS. Thymic involution plays an important role in immunosenescence causing decreased output of T cells from the thymus. A surrogate of thymic output is the measurement of T cell receptor excision circles (TRECs), which are small circular DNAs created during rearrangement of T cell receptor genes. Several studies show that TREC numbers are lower in people with MS compared to age-matched controls, suggesting premature immunosenescence in MS.22,23,44 Reproductive aging has also been studied in MS based on the increased risk of developing progressive MS as women reach perimenopause. Decreases in anti-Müllerian hormone (AMH) mark ovarian aging and can predict > 80% of the variability in the age of menopause. In a longitudinal study of women with MS, AMH levels in women with MS were associated with disability progression and brain atrophy after adjusting for chronological age, disease duration, and body mass index (BMI). 45
Future directions
An increased understanding of the role of biological aging in MS pathogenesis and clinical manifestation could lead to advances in the prognostication and treatment of progressive MS. As discussed above, evidence from EAE mice and early human studies suggests that biological aging is accelerated in MS (Figure 1). Of clinical relevance, biological age may correlate more strongly with MS disease progression and outcomes than chronological age. The rate of biological aging can vary between individuals. Interventions targeting aging mechanisms have the potential to modify the pace of aging, thus representing a novel potential treatment strategy to mitigate disability and delay onset of progression in MS.
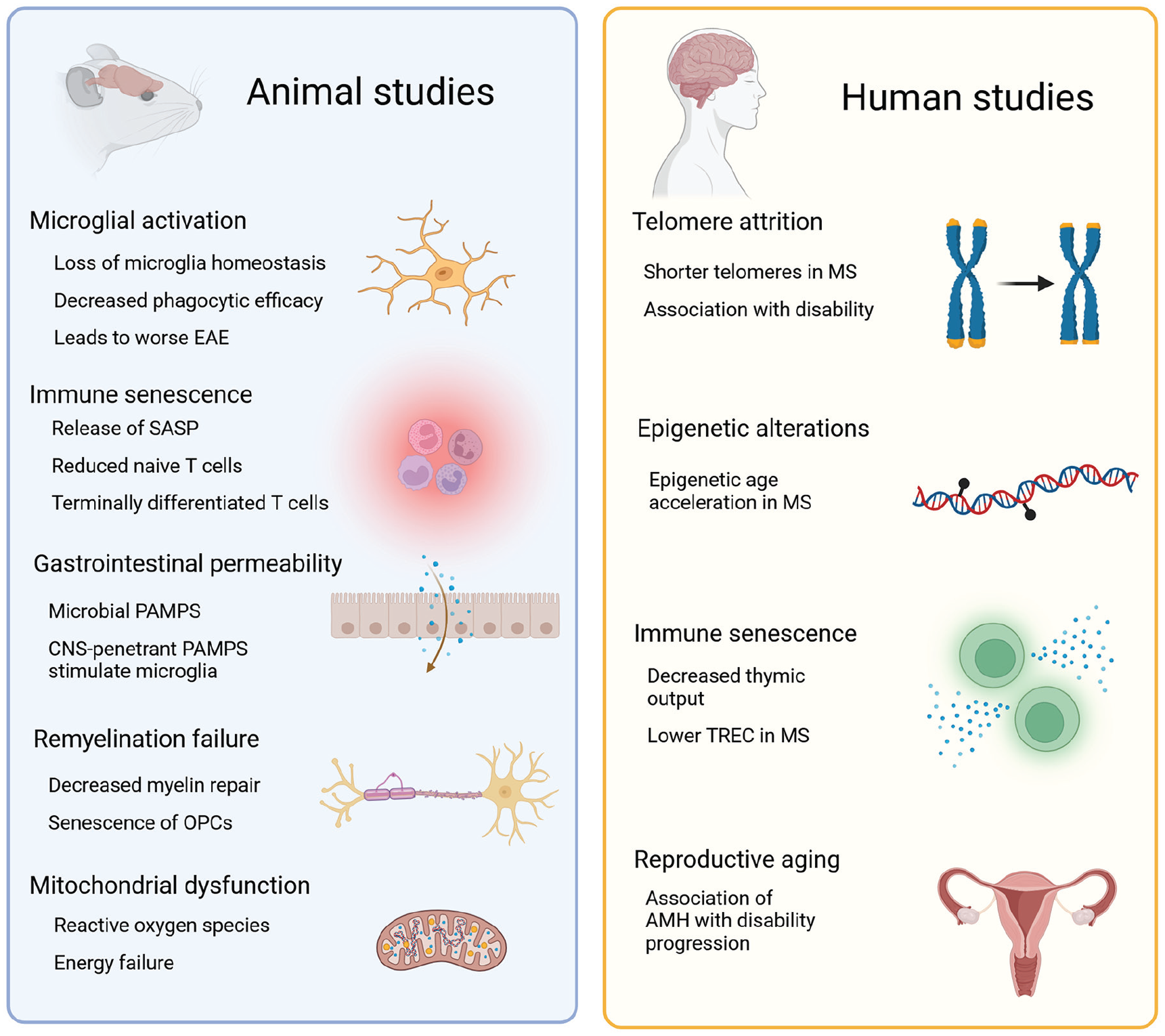
Observed forms of biological aging in animal models and human studies of MS. Left, a variety of postulated mechanisms contribute to the age-related dysregulation of the immune system in mouse models of MS. Right, in human studies, aging biomarkers show evidence of accelerated biological aging in people with MS.
It is currently unknown whether MS DMTs affect markers of biological aging and whether certain classes of DMTs, including those with neuroprotective effects, have differential effects on the pace of aging. Although DMTs are familiar treatments for MS, drugs specifically targeting processes of aging are new to neurologists caring for people with MS. For instance, senolytics are drugs that target the vulnerabilities of senescent cells and are in clinical trials for the treatment of various age-related diseases. 46 Preclinical studies highlight the potential role of cellular senescence and SASP-mediated pro-inflammatory processes in EAE progression. In people with MS, there is evidence of increased peripheral blood markers of senescence and increased inflammatory cytokines with aging in the cerebrospinal fluid.47,48 While SASP markers are one way to study senescence, another approach is to measure senescence-induced gene expression, such as p16Ink4a and p21, which are tumor suppressor genes that induce cell cycle arrest.49,50 Additional use of aggregate panels or cell-type-specific senescence markers may allow for more accurate identification of senescence patterns. Clinical studies on the impact of markers of cellular senescence on MS progression are still needed, and clinical trials of senolytics in MS remain a possibility.
An important step in advancing the study of biological aging in MS will be to provide further evidence of accelerated biological aging in individuals with worse disease outcomes and progressive MS phenotypes across other hallmark mechanisms of aging. Similarly, studies can be designed to determine whether biologically “older” people with MS are at increased risk of disease progression. This may translate into prognostic indicators of disease progression that can be used to identify people who may benefit from early aggressive therapies. Ultimately, longitudinal studies are needed to determine whether accelerated aging correlates with faster disease progression.
The design of studies measuring biological aging in MS requires robust aging biomarkers. These biomarkers should ideally represent common pathways affected by both aging processes and MS pathophysiological mechanisms. Frequently studied aging biomarkers center around the “hallmark” mechanisms of aging. 51 While no single biomarker exists that accurately reflects the entire spectrum of aging processes in humans, markers of epigenetic alterations and cellular senescence represent promising measures backed by epidemiological studies. 52 Interestingly, many of the hallmarks of aging contribute to an inflammatory state. Given the robust evidence for increased central and peripheral markers of inflammation in MS, there is a compelling need to investigate the link between biological aging and MS disease progression.
Interdisciplinary collaborations between MS clinicians, neuroimmunologists, and geroscience researchers will be vital to the design, implementation, and interpretation of studies examining the relationship between aging biomarkers and MS progression. The study of biological aging in MS has the potential to shift current treatment paradigms from focusing on symptom management in non-active progressive MS to disease modification. This approach embodies the geroscience hypothesis, which posits that therapeutically targeting aging physiology will prevent or delay the worsening of age-related diseases. 53 There may come a time when our repertoire of MS therapeutics includes agents that block aging pathways to lengthen years free of disability progression. Until then, a framework for measuring biological aging in MS must be established to validate aging mechanisms as a driver of disease progression in MS.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The manuscript is supported by funding from the National Institute on Aging (R03AG078946).