
Abstract
Background and Objective:
Prior Epstein–Barr virus (EBV) infection is associated with an increased risk of pediatric-onset multiple sclerosis (POMS) and adult-onset multiple sclerosis (MS). It has been challenging to elucidate the biological mechanisms underlying this association. We examined the interactions between candidate human leukocyte antigen (HLA) and non-HLA variants and childhood EBV infection as it may provide mechanistic insights into EBV-associated MS.
Methods:
Cases and controls were enrolled in the Environmental and Genetic Risk Factors for Pediatric MS study of the US Network of Pediatric MS Centers. Participants were categorized as seropositive and seronegative for EBV-viral capsid antigen (VCA). The association between prior EBV infection and having POMS was estimated with logistic regression. Interactions between EBV serostatus, major HLA MS risk factors, and non-HLA POMS risk variants associated with response to EBV infection were also evaluated with logistic regression. Models were adjusted for sex, age, genetic ancestry, and the mother’s education. Additive interactions were calculated using relative risk due to interaction (RERI) and attributable proportions (APs).
Results:
A total of 473 POMS cases and 702 controls contributed to the analyses. Anti-VCA seropositivity was significantly higher in POMS cases compared to controls (94.6% vs 60.7%, p < 0.001). There was evidence for additive interaction between childhood EBV infection and the presence of the HLA-DRB1*15 allele (RERI = 10.25, 95% confidence interval (CI) = 3.78 to 16.72; AP = 0.61, 95% CI = 0.47 to 0.75). There was evidence for multiplicative interaction (p < 0.05) between childhood EBV infection and the presence of DRB1*15 alleles (odds ratio (OR) = 3.43, 95% CI = 1.06 to 11.07). Among the pediatric MS variants also associated with EBV infection, we detected evidence for additive interaction (p = 0.02) between prior EBV infection and the presence of the GG genotype in risk variant (rs2255214) within CD86 (AP = 0.30, 95% CI = 0.03 to 0.58).
Conclusion:
We report evidence for interactions between childhood EBV infection and DRB1*15 and the GG genotype of CD86 POMS risk variant. Our results suggest an important role of antigen-presenting cells (APCs) in EBV-associated POMS risk.
Introduction
Collective findings point to a role for both genetic variants 1 and environmental factors 2 in multiple sclerosis (MS) susceptibility. The identification of gene–environment (G × E) interactions in MS could help characterize the biological processes involved in disease pathoetiology and explain part of the reported missing heritability for MS. 3
Among the environmental risk factors for MS, prior infection with Epstein–Barr virus (EBV) has been consistently reported to be the strongest environmental risk factor associated with MS onset; 4 however, it has been challenging to elucidate the underlying biological mechanisms explaining this association 5 due to the long interval between EBV exposure and clinical onset in adults, as the peak incidence of EBV infection occurs in early childhood. 6
Consistent with adult MS studies,7–9 we and others have previously reported a strong association between EBV seropositivity and pediatric-onset MS (POMS).10,11 Evaluating environmental risk factors in POMS offers advantages over adult studies in terms of the temporality of exposure. 12 Although virtually all adults diagnosed with MS and more than 95% of healthy adults have evidence of a prior EBV infection, 4 only 0.1%–0.5% of adults develop MS. Therefore, EBV infection alone is not sufficient to cause the disease and other factors such as genetic traits may contribute substantially to EBV-associated MS risk. As a lower frequency of EBV seropositivity has been reported in POMS,11,13–15 it makes this age group ideal for assessing genetic susceptibility to EBV-associated MS.
A few reports suggest that genetic makeup modulates the immune response against EBV.16–18 Single-nucleotide polymorphisms (SNPs) associated with the antibody response to EBV infection have also been identified in MS genome-wide association studies (GWASs). 19 We sought to assess whether candidate HLA and non-HLA variants interact with EBV infection to influence the risk of developing POMS.
Methods
Study population
This multi-center case–control study enrolled POMS patients with a first clinical event before age 18 years and healthy controls from the same institutions who were frequency matched. Inclusion criteria have been previously described. 20 All cases were tested for myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG) with a cell-based assay. Pediatric MS experts ascertained MOG-IgG-positive children to confirm a final diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) and were excluded. Clinical and environmental data were gathered from cases and controls participating in the genetic and environmental risk factor study performed by the US Network of Pediatric MS Centers and collaborators, consisting of 16 pediatric MS centers. Participants and their families completed a detailed environmental questionnaire and provided blood samples for DNA and serum. The institutional review boards approved this study at the participating sites. Informed consent and assent (when appropriate) were obtained for each subject.
Environmental data
Batched EBV-viral capsid antigen (VCA)-IgG testing was performed at the Oklahoma Medical Research Foundation using commercially available, standardized enzyme-linked immunosorbent assays (ELISA) (Wampole Laboratories, Princeton, NJ, USA) as described previously. 21 Quality control (QC) requirements included having positive, negative, and calibration controls that met predefined measures.
Genetic data
We selected candidate HLA and non-HLA variants for assessing G × E interactions in POMS. The major HLA MS risk factors included the presence of HLA-DRB1*15 alleles and the absence of HLA-A*02 alleles. Non-HLA candidates, on the contrary, included only two POMS risk variants in CD86 and IL2RA (rs2104286 and rs2255214, respectively), 22 which have been associated with response to EBV infection, as well. 19
All participants’ DNA samples were genotyped for DRB1 status, as previously reported. 23 DRB1 status was classified as either carrying one or more HLA-DRB1*15 alleles or none. Each study participant was genotyped using the Infinium 660K BeadChip or HumanOmniExpress BeadChip. Using PLINK v.1.9, strict QC checks and genotype comparisons between samples on two Illumina platforms were carried out. As previously described, 22 alignment, QC, phasing, imputation, and variant filtering were carried out. The SNP weighting method 24 was used to estimate the percentage of genetic ancestry related to four major populations (European, East Asian, West African, and Native American). The HLA-A*02 (rs2975033) tagging SNP was imputed. It has been reported that the HLA-A*02 allele and the allele “A” of rs2975033 is in perfect linkage disequilibrium (r2 = 0.97). 25 Therefore, it has been assumed that persons with the GG genotype of rs2975033 do not have HLA-A*02. The subjects were split into two groups based on whether they carried any HLA-A*02 alleles or none. In addition, the subjects were classified according to the presence of the GG genotype of CD86 SNP (rs2255214) and the presence of the TT genotype of IL2RA SNP (rs2104286).
Statistical analysis
Controls and POMS cases were frequency matched for sex and race. The odds of having POMS associated with prior EBV infection variables (dichotomized variable based on serostatus (positive vs negative) and quantitative variable based on anti-VCA-IgG serum levels), presence of HLA-DRB1*15, absence of HLA-A*02, and non-HLA candidate SNPs were assessed separately using logistic regression models adjusted for age, sex, genetic ancestry and mother’s education as a proxy of socioeconomic status (SES). Additive interaction between prior EBV infection and genotype was assessed in the adjusted logistic regression model. The relative excess risk due to interaction (RERI) and the attributable proportion (AP) of disease due to interaction were calculated using published methods.26,27 To calculate RERI and AP and their confidence intervals, we used adjusted odds ratio (OR) from our logistic regression models. We first ran our adjusted logistic regression models in Stata and then used the “nlcom” command according to the formula of RERI and AP. The output included the adjusted estimates of RERI and AP, their 95% confidence intervals (CIs), and p-values.
In order to determine whether multiplicative interaction existed, a multiplicative interaction term was also included in logistic regression models. Stata 15.0 was used for all statistical analyses (Stata Corp, College Station, TX, USA).
Result
Participant characteristics
This analysis included 473 POMS cases and 702 pediatric controls with available anti-VCA-IgG level data. The average age of onset of POMS cases was 14.65 + 2.6 years.
The characteristics of the study participants are shown in Table 1. Controls and POMS cases were frequency matched for sex and race. However, the POMS cases had a higher proportion of Hispanics and mothers with lower levels of education. POMS cases were significantly more likely to have evidence of prior EBV infection (94% of POMS cases vs 60% of controls) (Table 1). The OR based on logistic regression models adjusted for age, sex, genetic ancestry, and mother’s education was calculated. Childhood EBV infection was associated with higher odds of POMS (OR = 9.89; 95% CI = 6.21 to 15.73).
Characteristics of study participants at the time of enrollment.
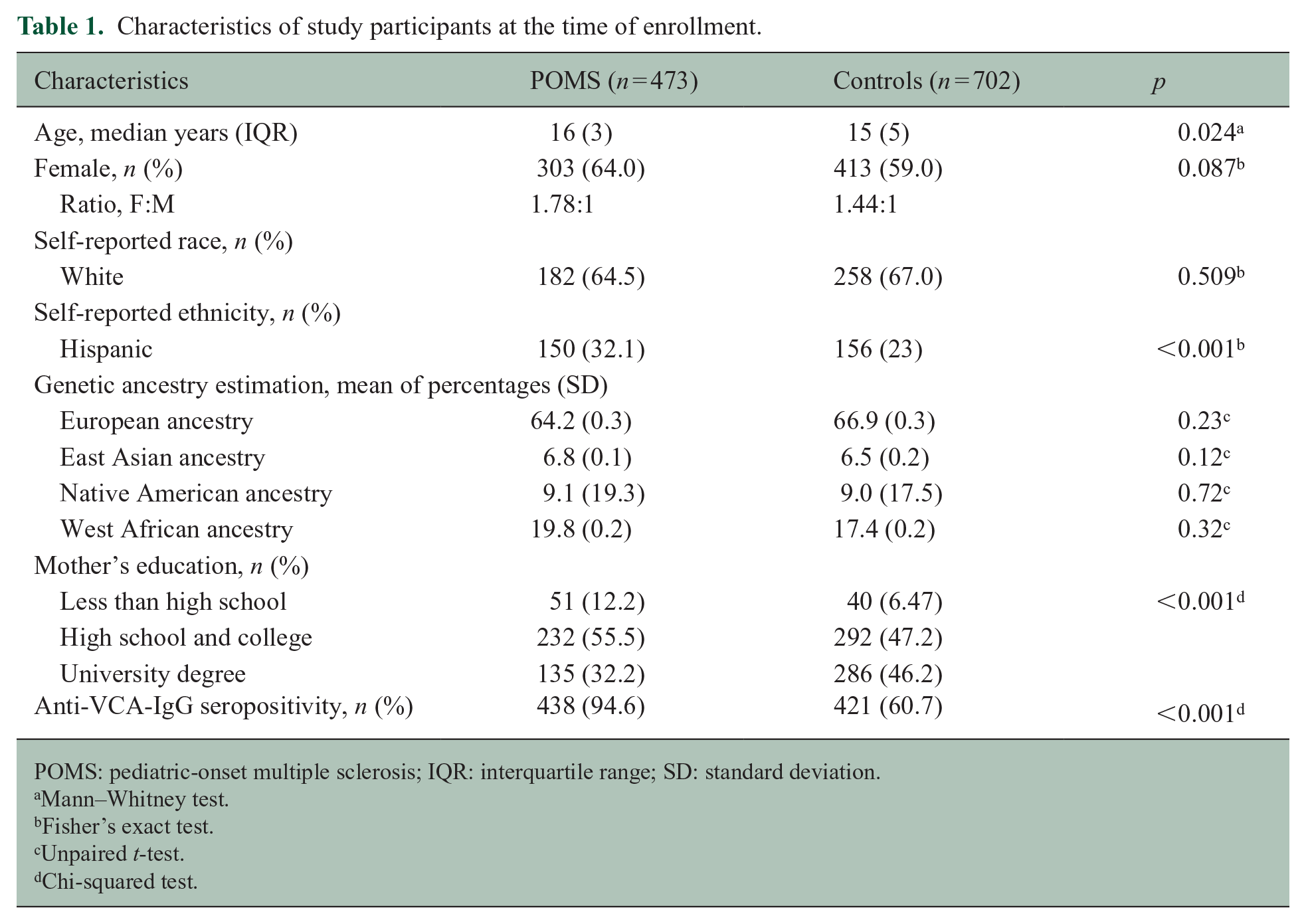
POMS: pediatric-onset multiple sclerosis; IQR: interquartile range; SD: standard deviation.
Mann–Whitney test.
Fisher’s exact test.
Unpaired t-test.
Chi-squared test.
Genetic characteristics
POMS cases were more likely to carry at least one HLA-DRB1*15 allele compared to controls (Table 2). Based on the imputed genotype for HLA-A*02, the proportion of participants without any HLA-A*02 alleles was higher in the POMS group (61%) compared to controls (56%). In addition, the proportion of children who carry the risk genotype for rs2255214 and rs2104286 was higher in POMS cases compared to controls.
Distribution of genetic covariates between POMS cases and controls.

POMS: pediatric-onset multiple sclerosis; SNP: single-nucleotide polymorphism.
Assessment of G × E interactions
In Table 3, we stratified analyses based on evidence of prior EBV infection in childhood compared to genetic risk factors (presence of HLA-DRB1*15, absence of HLA-A*02, and presence of other candidates SNPs genotype) in POMS cases and controls. Without evidence of prior EBV infection, the presence of at least one HLA-DRB1*15 allele did not significantly increase the odds of POMS. When both HLA-DRB1*15 and evidence of EBV infection were present, the odds of having POMS increased to 16.64 (95% CI = 9.56 to 28.94). The RERI for prior EBV infection and the presence of HLA-DRB1*15 alleles (10.25, 95% CI = 3.78 to 16.72) suggested an additive interaction and indicated the risk fraction in the presence of both prior EBV infection and HLA-DRB1*15 exceeds the addition of risks. The AP revealed that approximately 60% (AP = 0.61, 95% CI = 0.47 to 0.75) of the POMS risk in those with HLA-DRB1*15 and childhood EBV infection was attributable to the interaction between these two risk factors. Without prior EBV infection in childhood, the absence of HLA-A*02 alleles did not significantly increase the odds of having POMS. In the individuals with a lack of HLA-A*02 alleles and prior EBV infection in childhood, the OR of having POMS increased to 9.04 (95% CI = 4.68 to 17.44); however, RERI and AP were not significant. Furthermore, we detected evidence for additive interaction between a prior childhood EBV infection and the presence of the GG genotype in rs2255214, risk variants of CD86 (AP = 0.30, 95% CI = 0.03 to 0.58).
Additive interactions between prior EBV infection and genetic risk factors.
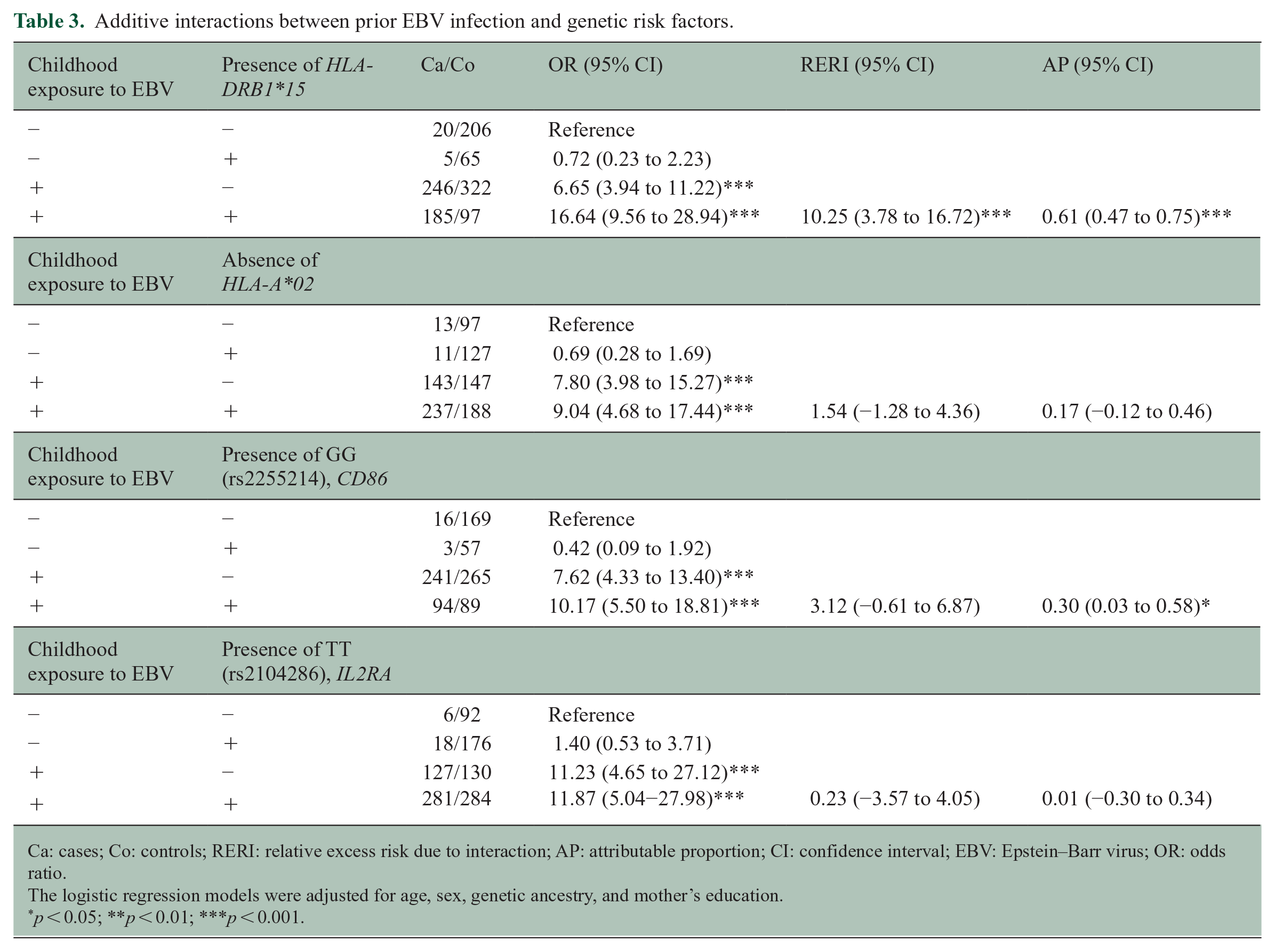
Ca: cases; Co: controls; RERI: relative excess risk due to interaction; AP: attributable proportion; CI: confidence interval; EBV: Epstein–Barr virus; OR: odds ratio.
The logistic regression models were adjusted for age, sex, genetic ancestry, and mother’s education.
p < 0.05; **p < 0.01; ***p < 0.001.
Table 4 shows evidence for multiplicative interactions between prior EBV infection and genetic risk factors in logistic regression models adjusted for age, sex, genetic ancestry, and mother’s education. Although there was evidence for multiplicative interaction between prior EBV infection and the presence of HLA-DRB1*15 (OR = 3.43, 95% CI = 1.06 to 11.07), the interaction terms in the adjusted logistic regression models for the absence of HLA-A*02 alleles and other non-HLA-POMS-risk-variants were not statistically significant.
Models assessing multiplicative interactions for odds of POMS.
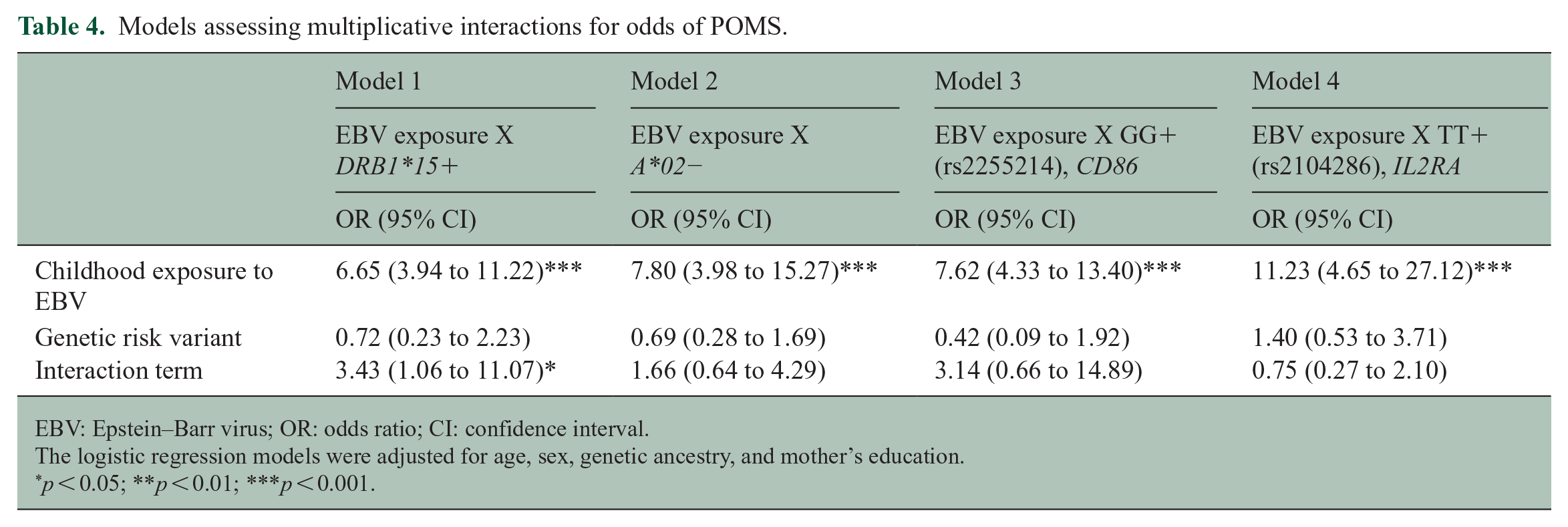
EBV: Epstein–Barr virus; OR: odds ratio; CI: confidence interval.
The logistic regression models were adjusted for age, sex, genetic ancestry, and mother’s education.
p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
In this study, we identified evidence supporting G × E interactions between childhood EBV infection and POMS risk variants that modify POMS risk (presence of HLA-DRB1*15 alleles and GG genotype in rs2255214 POMS risk variant CD86). These observed G × E interactions provide new insight into the biological mechanisms underlying EBV infection–associated MS risk.
Among environmental risk factors associated with MS, an association between prior EBV infection and MS risk has been consistently reported.4,7,10,15,28–30 The peak incidence of EBV infection is in childhood, with another peak in adolescence associated with infectious mononucleosis (IM). 6 The risk of MS in those with adolescent EBV infection with a history of IM is two- to three-fold higher than in individuals who are EBV-positive with no history of mononucleosis; however, the risk of developing MS is around 15 times higher among those infected with EBV in early childhood compared with individuals who are EBV-negative, based on the results from meta-analysis.4,6,30 In line with previous findings, we also demonstrated that childhood EBV seropositivity increased the risk of POMS compared to EBV-seronegative children (OR = 9.89; 95% CI = 6.21 to 15.73), Around 70% of 10–14 years olds in the general population of a developed country are EBV-seropositive, 31 which is similar to our observation in healthy pediatrics (~60%).
We show evidence for additive and multiplicative interactions between childhood EBV infection and HLA-DRB1*15, the strongest genetic risk factor for POMS. 22 In those carrying HLA-DRB1*15, the POMS risk associated with childhood EBV infection exceeds the addition of the separate risks (RERI~10 and AP~0.6). Several studies have evaluated the interaction between prior EBV infection and HLA-DRB1 on MS.28,32–36 Similar to our results, a meta-analysis of pooled data indicated significant additive interaction between HLA-DRB1*15:01 and prior EBV infection. 37 Whether the interaction between EBV infection and HLA-DRB1*15:01 in MS is multiplicative remains controversial.28,32–37 Similar to our previous report, 10 we also observed interaction on the multiplicative scale. The foreign peptides derived from EBV have high avidity to DR2b heterodimer encoded by DRB1*15:01 38 which could provide functional evidence to support our observed results.
Only two POMS non-HLA risk variants, rs2104286, and rs2255214, 22 have been associated with response to EBV infection. 19 We observed an additive interaction between childhood EBV exposure and the presence of GG (rs2255214) in CD86. Antigen-presenting cells (APCs) express CD86, a co-stimulatory ligand that triggers the immune response. 25 Furthermore, in MS patients, higher expression of CD86 has been reported on B-cells serving as APCs,27,28 which highlighted the important role of CD86 expressed on B-cells in MS pathoetiology. 20 The G × E interactions we report emphasize the possible critical role of B-cells in EBV-associated MS risk.
The large cohort of POMS and frequency-matched controls, the diversity of geographic origin, the rigorous case ascertainment by pediatric MS specialists, and the exclusion of confirmed MOGAD are some of the strengths of our study. In addition, we adjusted our models for genetic ancestry and mothers’ education as a surrogate for SES. We used a robust marker of prior EBV infection, anti-VCA-IgG, rather than Epstein–Barr virus nuclear antigen 1 (EBNA-1) IgG which is not produced in some individuals with prior infection and can disappear over time. 39 Additional strengths include the study of children, allowing the use of prior EBV infection in early childhood rather than the level of VCA-IgG as done in adult studies for which most participants have been exposed to EBV.
While serological testing for EBV was performed after disease onset, disease duration at the time of sample collection was short. Although we adjusted analyses for several potential confounders, the results could have been affected by unmeasured or unknown confounders. Our sample size did not allow us to look at a large number of genetic variants. Future studies with larger sample sizes are needed to replicate our findings and further evaluate potential G × E interactions related to EBV infection.
In summary, childhood EBV infection is a stronger risk factor for POMS in individuals who carry specific genetic factors. These interactions between childhood EBV infection and the presence of HLA-DRB1*15 and the POMS risk variant in CD86 (rs2255214) suggest a possible role of B-cells serving as APCs for EBV infection–associated MS risk.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The funding for this work includes R01NS071463 (PI E.W.), NMSS HC-1509-06233 (PI T.C.C.), UM1AI144292 (PI J.A.J.), and McDonald fellowship award of MSIF (PI A.Z.).