
Abstract
Seronegative neuromyelitis optica spectrum disorder (NMOSD) is a diagnostic challenge. This study aimed to characterize misapplication of the 2015 international criteria for seronegative NMOSD. We reviewed 35 consecutive seronegative NMOSD referrals to a tertiary center. Two (6%) met formal criteria, and 2 (6%) were clinically suspected of NMOSD without fulfilling radiologic supportive criteria. Alternative diagnoses were common including multiple sclerosis (37%), optic neuritis (14%), functional neurologic disorder (11%), and other (38%). Immunosuppression with rituximab was used in 3 of 4 patients with functional neurologic disorder. Misapplication of the 2015 criteria is common, and alternative explanations in seronegative NMOSD patients should be strongly considered.
Keywords
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune disease characterized by relapsing demyelinating attacks, commonly manifesting with optic neuritis (ON) and longitudinally extensive transverse myelitis (LETM). The disorder is strongly associated with AQP4-IgG, which has over 99% specificity for NMOSD diagnosis using cell-based assays. 1 In 2015, the international NMOSD criteria were proposed and divided patients into seronegative and seropositive groups based on AQP4 status. 2 Since then, myelin oligodendrocyte glycoprotein antibodies (MOG-IgG) have emerged, accounting for another subset of NMOSD-like cases. 3 However, some patients remain negative for both antibodies. 4 In these cases, diagnosis relies heavily on clinical and radiological criteria.
Misapplication of the seronegative NMOSD diagnosis can lead to inappropriate treatment and unnecessary referrals. Despite this, data on diagnostic errors in seronegative NMOSD remain limited. This study aimed to assess the accuracy of such diagnoses among patients referred to a tertiary care center.
Methods
This retrospective study was approved by the Mayo Clinic (IRB 21-001492) and identified consecutive patients seen by the Mayo Clinic from January 1st, 2018– December 31st, 2023 for a consultation regarding seronegative NMOSD. Forty-five patients were identified using text search. Medical records were reviewed to include those meeting the following criteria:
An outside diagnosis/referral of seronegative NMOSD.
Serum testing for AQP4-IgG via cell-based assay.
Cerebrospinal fluid analysis.
Ten patients were consequently excluded from analysis (Figure 1). All patients underwent MOG-IgG testing, except for one patient ultimately diagnosed with multiple sclerosis (MS). This case was included because according to the international diagnostic criteria, a diagnosis of MOG antibody-associated disease (MOGAD) would have been excluded even in the presence of high-titer MOG-IgG, due to identification of a better alternative diagnosis. Two blinded evaluators independently assessed whether patients met the 2015 NMOSD criteria, which included exclusion of alternative diagnoses (Supplemental Table S1). Discrepancies were reviewed by the principal investigator. Evaluators were blinded to the other’s determination until the analysis portion of the study. There was a 94% consensus on the initial evaluation.
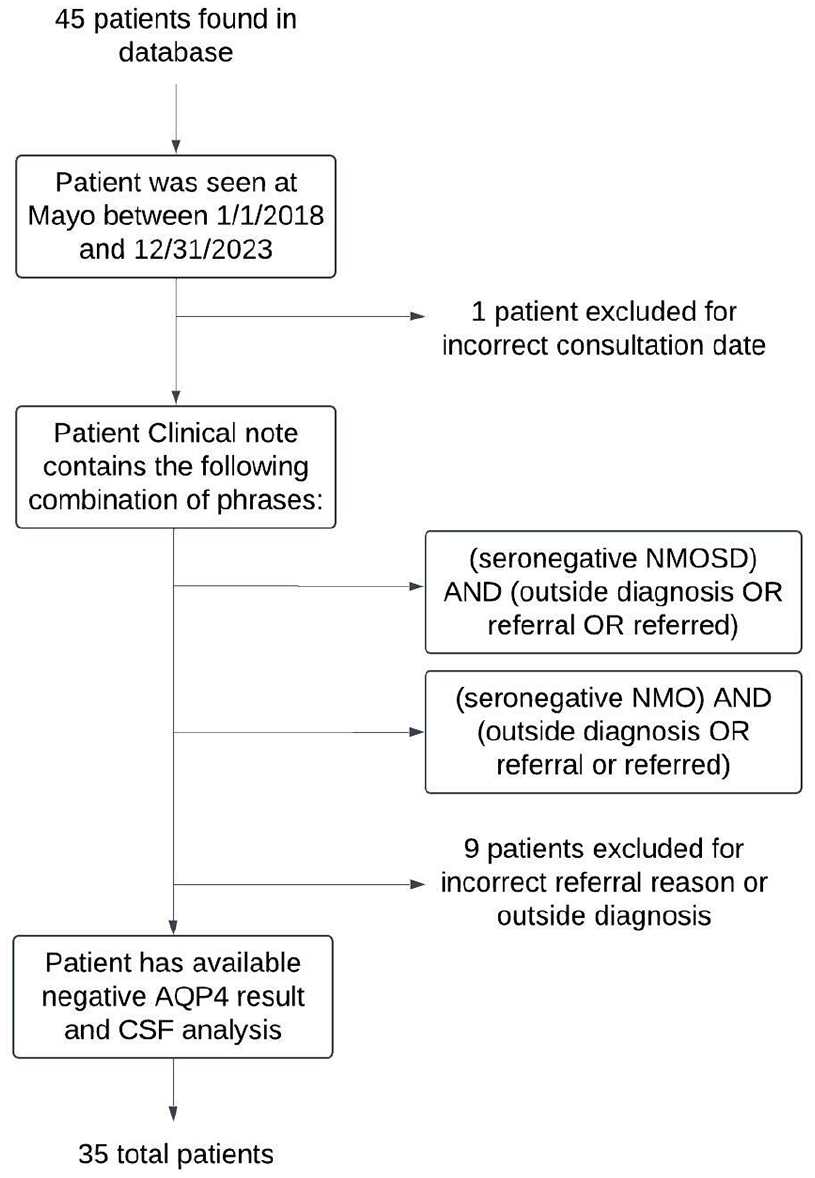
Methodology for patient inclusion.
Results
Median age at the initial attack for the referred seronegative NMOSD patients was 40 years (interquartile range = 33–48 years). Twenty-four of 35 patients (69%) were female. Twenty-five patients in the cohort were White, 4 were other/mixed race, 2 were Asian, 1 was Black, and 3 were unknown. Three patients identified as Hispanic or Latino.
Of the 35 patients referred for seronegative NMOSD, only 2 (6%) met the 2015 diagnostic criteria. Both presented with ON and LETM with appropriate magnetic resonance imaging (MRI) findings, dissemination in space, and no alternative explanation. The categories into which these patients fell are denoted using the asterisk symbol (*) in the following results. Two additional patients were felt to have suspected NMOSD not meeting criteria, both having ON and transverse myelitis (TM) without longitudinally extensive lesions at the time of TM. One patient was found to have atypical MOGAD who presented with area postrema syndrome, bilateral leg weakness, and hypersomnolence. The most common mimicking conditions included MS in 13 of 35 cases (37%), idiopathic ON in 5 of 35 cases (14%), and non-neurologic pathologies such as functional vision loss in 4 of 35 cases (11%). Seven of 13 (54%) patients ultimately diagnosed with MS were found to have ⩾2 unique oligoclonal bands in their cerbrospinal fluid (CSF) analysis.
The most common patient presentations were isolated ON (11/35, 31%), combined patient presentations (10/35, 29%*), and isolated TM (6/35, 17%) (Figure 2). Combined patient presentations included patients with combinations of ON, TM, area postrema syndrome, and/or acute brainstem syndrome. Of the 10 patients with combined presentations, five were diagnosed with MS, one had a glioma, two met the 2015 seronegative NMOSD diagnostic criteria, and two were suspected of NMOSD not meeting criteria (discussed earlier). Four of 10 (40%) patients with a combined presentation had ⩾2 unique oligoclonal bands in their CSF analysis; of these four, two were seronegative NMOSD patients.
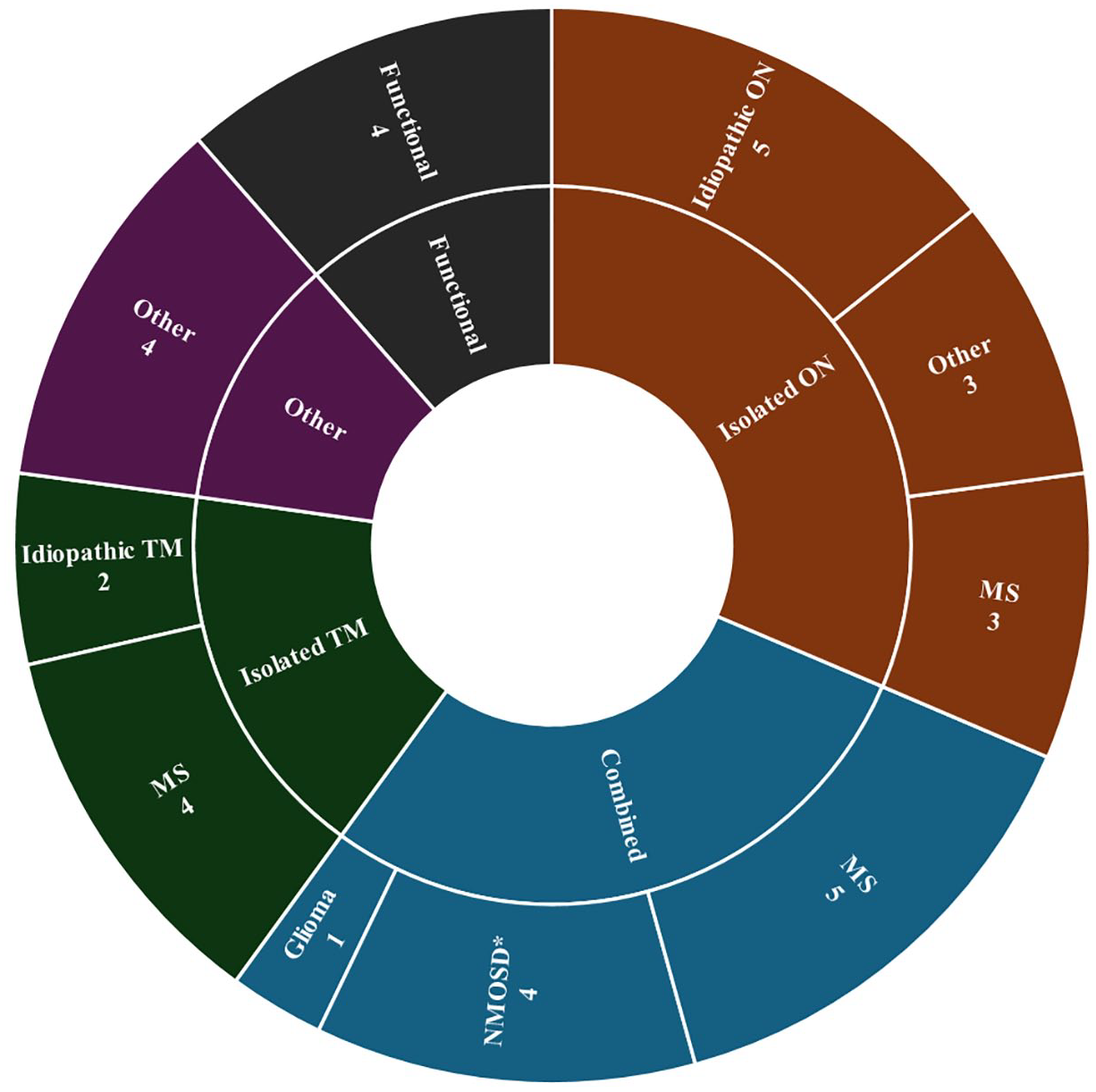
Final diagnoses of referrals for seronegative NMOSD.
Rituximab was frequently used in the cohort (25/35, 71%) including 9 of 13 MS cases (69%), three of four with functional neurologic disorder (75%), and all four patients felt to have seronegative NMOSD.
Discussion
Our study found that 88% of patients referred for seronegative NMOSD had an alternative condition. Previous studies have explored the implications of misdiagnosing NMOSD, focusing on the impact of missed diagnoses.4,5 However, overdiagnosis of NMOSD and improper treatment can be similarly harmful. We found inappropriate treatment with rituximab in three of four (75%) patients with functional disease. Sequelae of overuse of rituximab are multifaceted and serious, including infections and unnecessary cost burden to patients and the healthcare system. 6
Recent research has identified key areas of errors in the application of the 2015 NMOSD diagnostic criteria. Contentti et al. 7 found that 10%–49% of neurologists inaccurately identified whether case scenarios demonstrated dissemination in space, and only 32% accurately identified the non-cardinal syndromes for seronegative patients. These findings indicate that lack of familiarity with diagnostic criteria could be a contributor in misapplication. Our data indicate that establishing an accurate diagnosis can be challenging in patients with combined presentations including ON or TM. Juryńczyk et al. 8 noted that in expert review of patients with overlapping NMOSD and MS features, NMOSD characteristics tended to dictate diagnosis. We postulate this may relate to indeterminate MRI findings and nonspecificity of oligoclonal bands for a diagnosis of MS. Patients with a combined presentation are most likely to ultimately have NMOSD, and only 40% of our cohort with a combined presentation had ⩾2 unique oligoclonal bands. In the absence of oligoclonal bands, interpretation of MRI findings becomes essential in determining the correct diagnosis.
Prior studies showed that MOGAD explained up to 40% of AQP4-IgG seronegative NMOSD. 9 We identified only one MOGAD patient, demonstrating that providers are now appropriately screening for both AQP4-IgG and MOG-IgG in patients with an NMOSD phenotype prior to referral.
This study highlights misapplication of the 2015 NMOSD diagnostic criteria in seronegative cases and the need to consider alternative explanations in seronegative patients not meeting criteria. Future revisions of the NMOSD diagnostic criteria should strive to better delineate cases of seronegative demyelinating disease; however, our findings suggest that most diagnostic errors stemmed from providers’ misapplication or lack of familiarity with the existing criteria, rather than shortcomings of the criteria themselves.
Supplemental Material
sj-docx-1-msj-10.1177_13524585251360648 – Supplemental material for Unveiling Misdiagnosis: Rethinking Seronegative NMOSD
Supplemental material, sj-docx-1-msj-10.1177_13524585251360648 for Unveiling Misdiagnosis: Rethinking Seronegative NMOSD by Mary V. Lang, Grant M. Welk, Deena A. Tajfirouz, Kevin D. Chodnicki, Sean J. Pittock, Eoin P. Flanagan and John J. Chen in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors thank Mayo Clinic for their institutional support.
Data availability statement
The data supporting the findings of this study are available upon reasonable request of the corresponding author.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Mayo Clinic.
Ethical statement
This study was approved by the Institutional Review Board of Mayo Clinic (IRB: 21-001492) and conducted in accordance with the Declaration of Helsinki.
Informed consent/patient consent
Written consent was obtained from all individual participants included in the study.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.