
Abstract
We describe a case of tumefactive demyelinating lesion (TDL) that occurred during B-cell depletion therapy by ofatumumab and discuss its pathogenesis through serial magnetic resonance imaging and biopsied histopathology. Ten months after initiating subcutaneous ofatumumab following two episodes of transverse myelitis, the patient developed a large tumefactive lesion with closed-ring contrast-enhancement and intralesional microhemorrhages and was diagnosed as having a TDL by brain biopsy. After repeated intravenous methylprednisolone and plasma exchanges, the TDL gradually shrank, although a perilesional hypointense ring remained on susceptibility-weighted images. TDLs can occur even during B-cell depletion therapy; however, the underlying mechanism is not fully elucidated.
Introduction
Tumefactive demyelinating lesions (TDLs) are rare central nervous system manifestations that mimic brain tumors and occasionally require brain biopsy.1,2 They typically occur as initial events but can appear during the relapsing-remitting course of multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD), and myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease.1,3 TDLs can be triggered by initiating or discontinuing disease-modifying therapies for MS, such as natalizumab, alemtuzumab, and fingolimod. 2 However, the emergence of TDLs under B-cell depletion therapy has only been described in one patient treated with ocrelizumab. 4
Case report
A 35-year-old Japanese patient developed left hemiparesis 21 months after initiating subcutaneous ofatumumab and 2 months after discontinuing oral prednisolone. Approximately 2.5 years previously, the patient developed a longitudinally extensive spinal cord lesion (Th1–Th12) on magnetic resonance imaging (MRI) (Figure 1(a)). Laboratory tests were negative for infectious diseases (syphilis, tuberculosis, influenza, COVID-19), tumor markers (carcinoembryonic antigen, carbohydrate antigen-19-9, carbohydrate antigen-125, soluble interleukin-2 receptor), and autoantibodies (anti-nuclear, anti-SS-A, anti-SS-B, anti-dsDNA, anti-neutrophil cytoplasmic antibodies). Serum and cerebrospinal fluid were negative for aquaporin-4 (AQP4)-immunoglobulin G (IgG) and MOG-IgG in cell-based assays. Cerebrospinal fluid showed mild hypercellularity (22/µL) and increased total protein (59 mg/dL) and myelin basic protein (1110 pg/mL), with no oligoclonal bands. Brain MRI revealed several periventricular lesions without contrast enhancement (Figure 1(b)). The patient received three courses of intravenous methylprednisolone (IVMP, 1000 mg for 3 days), followed by plasma exchanges, which gradually alleviated her symptoms. Four months later, the patient reported the subacute worsening of leg weakness and sensory loss. Despite no new or enlarging lesions on MRI, a relapse was diagnosed based on her clinical symptoms, and subcutaneous ofatumumab was started after IVMP. Rituximab was not prescribed due to off-label treatment limitations with National Health Insurance. The patient remained stable on ofatumumab for 21 months until readmission for left hemiparesis and sensory loss. A large (5.3 × 7.1 × 6.1 cm) hyperintense lesion on fluid-attenuated inversion recovery sequences with a mass effect and complete ring enhancement was observed in the right hemisphere (Figure 1(c)). Blood and cerebrospinal fluid tests were again negative for infections and autoantibodies. A stereotactic-guided brain biopsy was performed on day 6 and the patient underwent three courses of IVMP and seven plasma exchange sessions, which partially improved her symptoms. Fluid-attenuated inversion recovery hyperintensity and ring enhancement enlarged on day 14, then shrank and lost contrast enhancement by day 101. Intriguingly, grouped small hypointense signals were observed inside the contrast-enhanced ring on day 1, and small hypointense signals then gradually formed ring-like patterns on susceptibility-weighted imaging (SWI). Histopathological examination revealed multiple microhemorrhages; aggressive monocyte/macrophage infiltration; perivascular T-cell infiltration; absence of CD20+ cells; mild loss of myelin basic protein, glial fibrillary acidic protein, and AQP4; and disrupted α-smooth muscle actin (Figure 1(d)). C9neo deposition was absent (data not shown). The patient was finally diagnosed with NMOSD without AQP4-IgG and started on tacrolimus and prednisolone maintenance therapy. No further relapses occurred for 1.5 years.
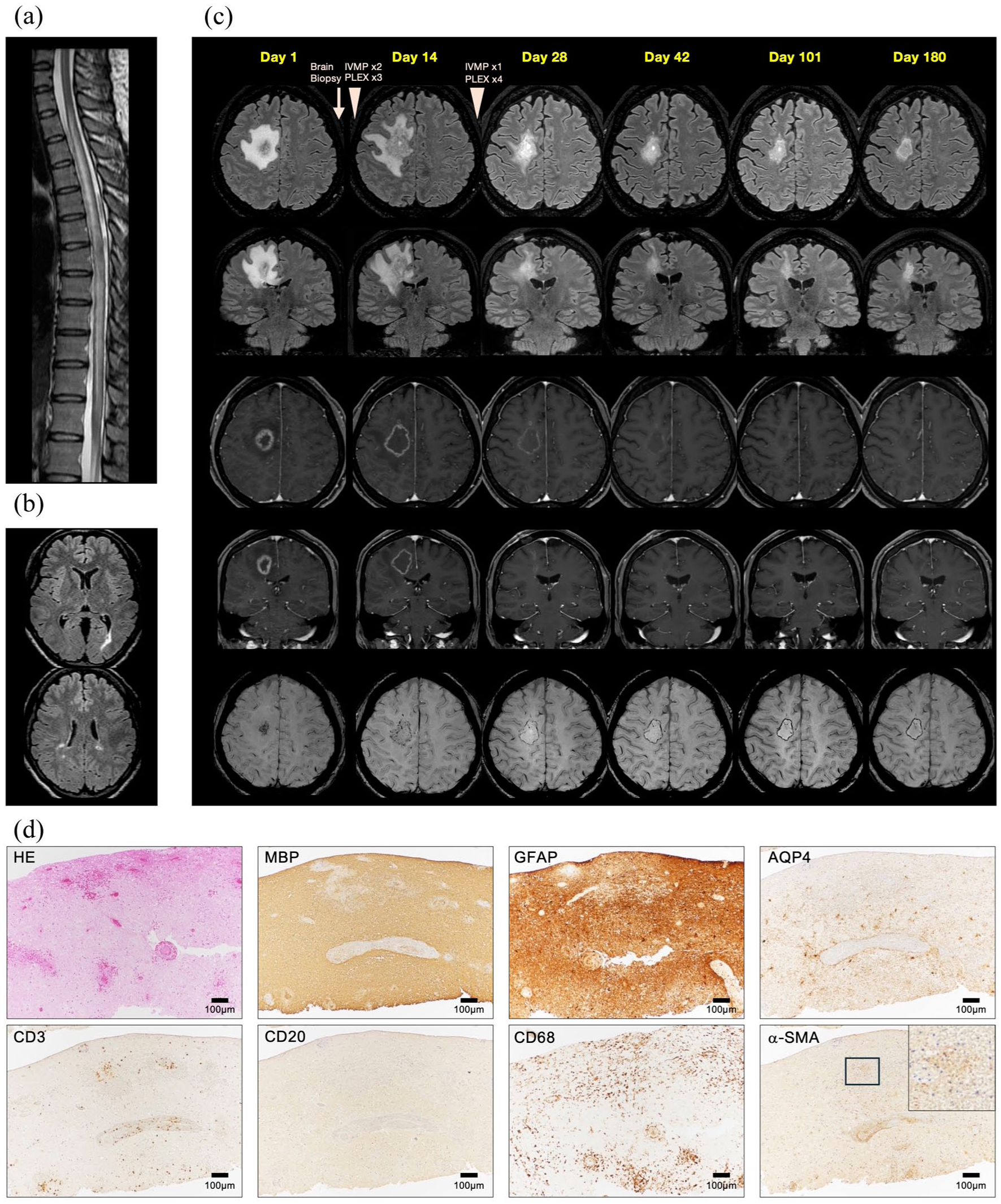
Serial magnetic resonance imaging (MRI) and biopsied histopathology of the tumefactive demyelinating lesion: (a) Sagittal T2-weighted image of the thoracic and lumbar spinal cord, and (b) axial fluid-attenuated inversion recovery (FLAIR) images of the brain at the first event. (c) Axial and coronal FLAIR, axial and coronal gadolinium-enhanced T1-weighted imaging, and axial susceptibility-weighted imaging on days 1, 14, 28, 42, 101, and 180 after admission. (d) Hematoxylin-eosin (HE) staining and immunostaining of myelin basic protein (MBP), glial fibrillary acid protein (GFAP), CD3, CD20, CD68, aquaporin-4 (AQP4), and α-smooth muscle actin (α-SMA) are shown. The upper right panel for α-SMA represents a magnified view of the area within the square. IVMP, intravenous methylprednisolone; PLEX, plasma exchange.
Discussion
Anti-CD20 monoclonal antibodies effectively suppress disease activity of various autoimmune conditions, including MS, NMOSD, and MOG antibody-associated disease, despite the distinct roles of B cells in their pathogenesis. 5 Recent studies suggest that anti-CD20 therapy is effective as maintenance therapy for tumefactive demyelination;6,7 however, B-cell contributions to TDL pathogenesis remain unclear. Typical TDL histopathology shows active inflammatory demyelination with predominant macrophage infiltration.1,8,9 T cells are observed in many TDL cases, whereas B-cell infiltration is confirmed in 20% to 76% of cases.8,9 Although B cells were depleted in peripheral blood and biopsied tissue, and we consider it intriguing that this atypical TDL appeared during B-cell depletion therapy, we cannot exclude the possibility that B cells elsewhere contributed to TDL development.
Serial MRI scans of this patient revealed the longitudinal development of the TDL. Initially, the lesion was characterized by three layers: microhemorrhage-cluster in the center, closed ring-enhancement, and perilesional edema. Two weeks later, all layers had enlarged, and hypointense SWI signals were distributed in a ring-like pattern. Four weeks later, the TDL started shrinking, with reduced contrast enhancement and edema; the hypointense SWI ring remained. Ten weeks later, enhancement and perilesional edema had nearly disappeared, leaving the hypointense SWI ring. These findings suggest acute inflammation with predominant vascular damage or vasculitis in the early phase and the accumulation of iron-containing macrophages at the lesion’s edge in the chronic phase. Histopathological findings supported this speculation, demonstrating predominant microhemorrhages, vascular damage, inflammatory cell infiltration, and mild demyelination in the early phase. Hemorrhagic central nervous system inflammatory demyelinating lesions have been documented with histopathology showing demyelination and aggressive macrophage infiltration, similar to the present case. 10 However, B-cell involvement has not been documented.
Another case report described a patient with MS who experienced a TDL 13 months after starting ocrelizumab, with a similar size, location, and strong perilesional edema. 4 However, our patient presented a unique phenotype with microhemorrhages in the acute phase, mild demyelination, perilesional ring formation in the chronic phase, and a limited response to immunotherapies. Although it is unclear whether the present case shows a specific TDL phenotype or whether the lack of response to B-cell depletion relates to the pathology, neuroimmunologists should be aware that TDL can flare during B-cell depletion therapy.
Footnotes
Acknowledgements
We thank Professor Paul Morgan (Cardiff University, UK) for providing the anti-C9neo antibody.
Data Availability Statement
Data sharing is not applicable to this article, as no data sets were generated or analyzed during the current study.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: K.S. received honoraria from Alexion, Biogen, Chugai, Mitsubishi Tanabe, Novartis, and UCB. K.M. received honoraria from Alexion, Biogen, Chugai, Novartis, and Tanabe Mitsubishi. E.T. received honoraria from Novartis and UCB. M.W. received honoraria from Alexion, Argenx, Biogen, Chugai, Mitsubishi Tanabe, Novartis, UCB, and Viatris. A.M., Y.K., K.I., D.I., and K.T. have nothing to disclose. N.I. received honoraria from Alexion, Biogen, Chugai, Eisai, Mitsubishi Tanabe, Novartis, Takeda, and Teijin and research support from Chugai, Japan Blood Products Organization, and Sumitomo.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by JSPS KAKENHI (JP23K06964), the Rinsho Igaku Shinkou Zaidan, the Nakatomi Foundation, and the Takeda Science Foundation.
Informed Consent
Written consent was obtained from the patient.