
Abstract
Background:
Area postrema syndrome (APS) is a core feature of neuromyelitis optica spectrum disorder (NMOSD). Charcot–Marie–Tooth disease type 1X (CMT1X), caused by gap junction protein beta-1 (GJB1) mutations, can rarely involve the central nervous system (CNS).
Objectives:
To report a case of CMT1X presenting with APS mimicking NMOSD.
Methods:
Clinical, electrophysiological, radiological, and genetic evaluations were performed.
Results:
A 43-year-old woman experienced intractable hiccups, nausea, and a dorsal medullary lesion. Genetic testing revealed a pathogenic GJB1 p.Arg22Gln variant. The lesion resolved, and she remained relapse-free without maintenance immunotherapy.
Conclusion:
GJB1-related disorders should be considered in patients with episodic CNS symptoms, particularly when accompanied by features of hereditary neuropathy, to avoid misdiagnosis.
Keywords
Introduction
Area postrema syndrome (APS), characterized by intractable hiccups, nausea, and vomiting, is a hallmark of neuromyelitis optica spectrum disorder (NMOSD). Charcot–Marie–Tooth disease type 1X (CMT1X), caused by mutations in the GJB1 gene (gap junction protein, beta-1), encodes connexin 32 (Cx32), which is essential for gap junction formation in the central nervous system (CNS) and peripheral nervous system (PNS). 1 CMT1X is the second most common subtype of Charcot–Marie–Tooth disease. While the classical presentation involves sensorimotor polyneuropathy, patients may also exhibit stroke-like CNS manifestations. 2 This report discusses a case initially misdiagnosed as NMOSD but later identified as an acute CNS episode related to a pathogenic GJB1 mutation.
Case report
A 43-year-old woman presented with a 4-day history of acute dizziness, nausea, persistent hiccups, unsteadiness, and sensory disturbances involving both legs with a sensory level at the inguinal region. Neurological examination revealed rightward gaze-evoked nystagmus, upper limb hyperreflexia, lower limb spasticity, and an extensor plantar response on the left. Sensory examination indicated a T12 sensory level with diminished light touch and vibration. The sensory symptoms reached nadir within hours and did not fluctuate over 4 days. No sphincter involvement was found.
Brain magnetic resonance imaging (MRI) showed a T2-weighted fluid-attenuated inversion recovery (FLAIR) hyperintense lesion at the dorsal medulla extending to the cervicomedullary junction, without contrast enhancement or diffusion restriction (Figure 1(a) and (b)). A whole-spine MRI revealed no additional lesions. Cerebrospinal fluid analysis showed 0 white blood cells (WBCs)/μL, no oligoclonal bands, and an immunoglobulin G (IgG) index of 0.46. Visual evoked potentials were unremarkable. Serum fixed cell-based assays for anti-aquaporin-4 (AQP4) and anti-myelin oligodendrocyte glycoprotein (MOG) antibodies, which were conducted before any immunotherapy, returned negative results. Although the sensory level persisted, symptoms of hiccups, unsteadiness, and numbness improved in severity following a 5-day course of intravenous methylprednisolone based on the initial NMOSD diagnosis.
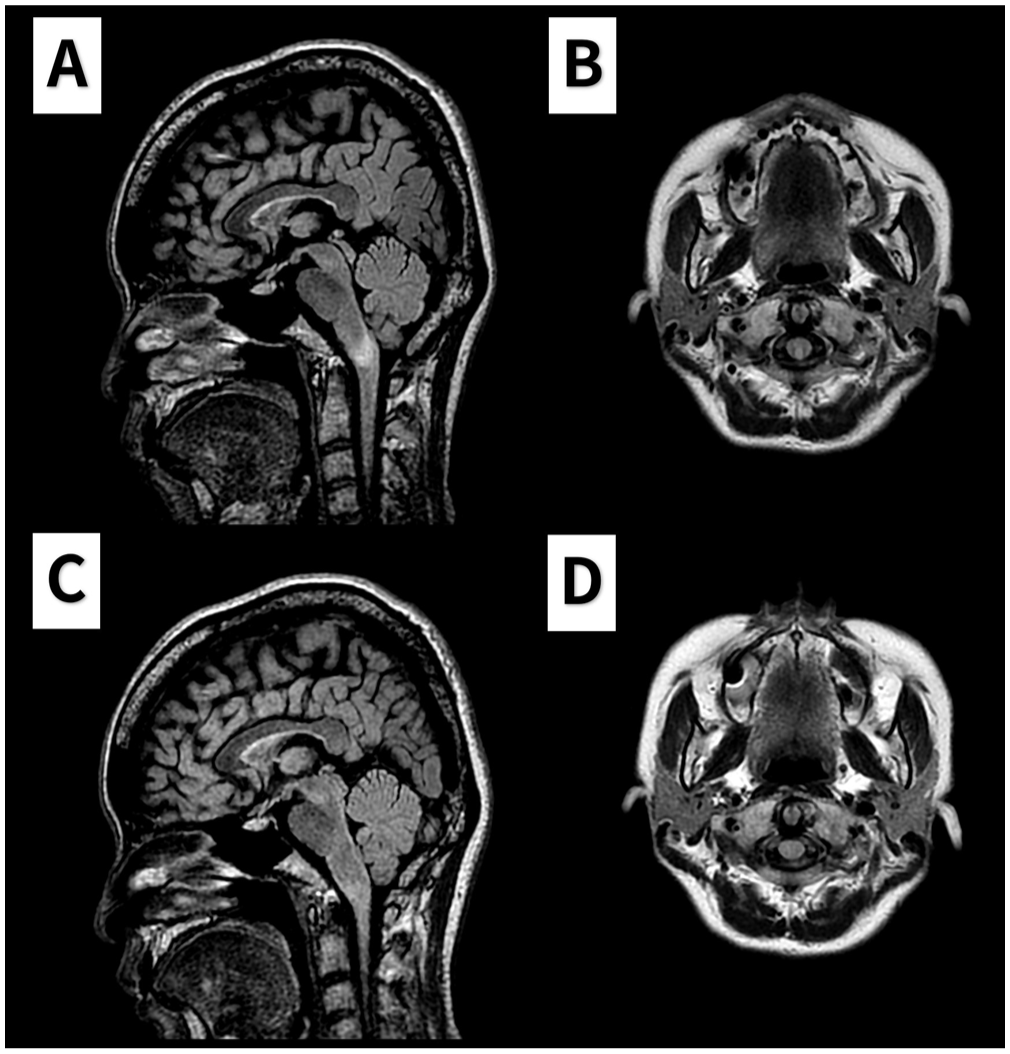
The brain MRI during the acute episode and 1-year follow-up. (a–b) T2-weighted fluid-attenuated inversion recovery (FLAIR) images obtained during the acute episode show a hyperintense lesion at the cervicomedullary junction in the sagittal (a) and axial (b) views. (c–d) Follow-up T2 FLAIR images acquired 1 year later demonstrate resolution of the previously noted lesion.
During hospitalization, the examination revealed pes cavus and atrophy in the intrinsic hand and lower leg muscles. The patient had previously attributed her poor exercise performance to personal limitations. Nerve conduction studies demonstrated diffuse sensorimotor polyneuropathy (conduction velocities: 39–44 m/s in the upper limbs and 27–30 m/s in the lower limbs). Whole exome sequencing identified a heterozygous missense mutation in GJB1 (NM_000166.6:c.65G > A; p.Arg22Gln), which was subsequently confirmed in her father, who had slowly progressive gait disturbance and sensorimotor polyneuropathy.
Three months post-event, the T12 sensory level resolved, leaving only residual numbness in both soles. One year later, repeat MRI confirmed complete resolution of the cervicomedullary lesion with no new abnormalities (Figure 1(c) and (d)). The patient remained relapse-free after 3 years without maintenance immunotherapy.
Discussion
APS is a key feature of NMOSD, as per the 2015 International Panel for NMO Diagnosis (IPND) criteria. The clinical presentation, combined with radiological findings, is particularly specific for NMOSD. 3 Our patient initially presented with APS with a T2-hyperintense lesion in the dorsal medulla. This led to an initial diagnosis of NMOSD, prompting the initial treatment with pulse steroid therapy. Although a T12 sensory level was noted, no spinal cord lesion was found on MRI. We postulated that the sensory symptoms were caused by the dorsomedial medullary lesion due to involvement of the dorsal column system, particularly the gracile nuclei.
However, several factors prompted a reevaluation of the diagnosis: the absence of anti-AQP4 and anti-MOG antibodies, lack of other core NMOSD features such as myelitis or optic neuritis, and the patient’s absence of clinical or radiological features during a 3-year follow-up without maintenance immunotherapy. Because patients with area postrema-onset NMOSD often exhibit a higher annual relapse rate and shorter time to recurrence, particularly in the absence of immunotherapy, 4 the patient’s stable 3-year course following a single episode without immunosuppressive treatment is highly atypical for NMOSD. Furthermore, the patient does not meet the 2015 IPND criteria after more than 3 years of follow-up, warranting consideration of an alternative diagnosis. The atypical presentation of our case suggests that this episode may be more consistent with transient CNS involvement in CMT1X, although a seronegative NMOSD cannot be entirely excluded and continuous follow-up remains necessary.
CMT1X, caused by a pathogenic GJB1 variant, primarily affects sensorimotor function but can also present with transient CNS manifestations, particularly with certain mutations like p.Arg22Gln.2,5,6 These episodes may present with upper motor neuron signs and are often associated with hyperintensities on brain imaging.5,6 Although most commonly involving the centrum semiovale and corpus callosum, infratentorial and periventricular white matter lesions were reported.7,8 Potential triggers include systemic inflammation, fever, or high-altitude exposure, although spontaneous episodes can also occur. 5 Male CMT1X patients exhibit CNS manifestations more commonly, while clinically significant CNS involvement in female carriers has also been reported.4,9
The mechanisms underlying CNS involvement in GJB1-related disorders are not fully understood. One proposed mechanism involves impaired glial gap junction coupling in which connexin 32 is expressed.5,10 Reduced expression of connexins has been observed in multiple sclerosis lesions, suggesting a broader role of gap junction dysregulation in demyelinating disorders. 11
In conclusion, this case underscores the necessity of screening for subtle features of hereditary neuropathy in patients with episodic CNS dysfunction, as recognizing GJB1-related pathology can prevent lifelong immunosuppression for a misdiagnosed inflammatory disorder.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical considerations
The report was approved by the Institutional Review Board of National Cheng Kung University Hospital (No. B-EC-113-026).
Consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for anonymized patient information and images published in this article.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request, subject to approval by the institutional review board and with the patient’s informed consent.