
Abstract
Introduction:
Cilostazol is a phosphodiesterase 3 (PDE3) inhibitor and is one of the only approved medications shown to improve walking performance in patients with peripheral artery disease (PAD). However, its effects on skeletal muscle pathophysiology are poorly understood. Because skeletal muscle dysfunction contributes to mobility impairment in PAD, this study aimed to evaluate whether patients taking cilostazol exhibit differences in skeletal muscle pathophysiology compared to those not taking cilostazol.
Methods:
We conducted a cross-sectional analysis of 50 patients with PAD, including 15 patients taking cilostazol and 35 not taking cilostazol. Calf muscle strength was assessed via isometric dynamometry. Gastrocnemius muscle biopsies were analyzed for myofiber morphology, mitochondrial function using high-resolution respirometry, and gene expression via RNA sequencing.
Results:
No significant differences were observed in calf muscle strength (p = 0.49), myofiber cross-sectional area (Type I: p = 0.53; Type IIa: p = 0.59), or capillary density (p = 0.74) between groups. However, mitochondrial oxygen consumption under physiological energy demand was significantly higher in cilostazol-treated patients (p = 0.0137), although oxidative phosphorylation conductance (p = 0.38) and mitochondrial hydrogen peroxide emission were not different. RNA sequencing revealed transcriptomic overlap between groups, but gene set enrichment analysis identified upregulation of pathways related to mitochondrial gene expression and downregulation of inflammatory signaling in patients taking cilostazol.
Conclusion:
Cilostazol use in patients with PAD is associated with increased skeletal muscle mitochondrial oxygen consumption and modest transcriptomic changes, but it does not appear to alter muscle strength, fiber size, or capillarization. These findings suggest a limited impact of cilostazol on skeletal muscle structure, although potential metabolic effects warrant further investigation.
Keywords
Introduction
Peripheral artery disease (PAD) is caused by the atherosclerotic narrowing or blockage of the peripheral arteries, leading to lower-limb dysfunction, impaired walking performance, and increased risk of morbidity and mortality.1,2 Claudication is a hallmark symptom of mild to moderate PAD, characterized by exertional calf pain resulting from an inadequate blood supply to meet the metabolic demands of active muscle. The pain typically persists with continued activity and resolves with rest. Among patients with claudication about 25% experience worsening symptoms within five years and a smaller proportion progress to the most severe disease manifestation, chronic limb-threatening ischemia (CLTI), which is characterized by persistent rest pain and/or tissue loss. CLTI substantially increases the risk of limb amputation.3–5 Current medical management for PAD centers on reducing the risk of cardiovascular events. Unfortunately, there are currently only two available treatment options that have been shown to improve walking performance in patients with PAD5–7: (1) cilostazol, a phosphodiesterase 3 (PDE3) inhibitor that promotes vasodilation; and (2) supervised exercise training.
Skeletal muscle pathophysiology has emerged as an important contributor to the limb dysfunction and walking performance of patients with PAD.8–24 Although there is literature investigating how exercise impacts skeletal muscle biology, little is known about whether cilostazol affects skeletal muscle pathophysiology in patients with PAD. In the vasculature, inhibition of PDE3 leads to elevated intracellular cyclic adenosine monophosphate (AMP) levels and subsequent activation of protein kinase A, which promotes vasodilation, decreases platelet aggregation, suppresses vascular smooth muscle cell migration and proliferation, and exerts antiinflammatory effects. In addition, cilostazol has been shown to suppress reactive oxygen species generation through heme-oxygenase-1-mediated activation of AMP-activated protein kinase.25–27 However, despite cilostazol’s established vascular effects, its influence on skeletal muscle morphology, function, and mitochondrial bioenergetics has not been investigated in patients with PAD. This knowledge gap is particularly important given accumulating evidence that mitochondrial function, as well as muscle fiber size and type in the calf, are associated with walking performance in patients with PAD.8,10,12,28 Capillary density has also been reported to correlate well with treadmill peak walking time in people with PAD.29,30 To address this gap, we utilized a recently published cohort of patients with PAD 24 to examine whether patients taking cilostazol exhibited differences in skeletal muscle pathophysiology compared with patients not taking cilostazol.
Methods
Study population
This was a cross-sectional study involving patients with PAD recruited through UF Health and the Malcom Randall VA Medical Centers in Gainesville, FL, USA. This study was approved by the institutional review boards at the University of Florida and the Malcom Randall VA Medical Center (Protocol IRB201801553). All study procedures were carried out in accordance with the Declaration of Helsinki and patients provided informed consent. Patients with an abnormal ankle–brachial index (ABI) (< 0.9), indicating a diagnosis of PAD with a Rutherford Category between 3 and 4, met the inclusion criteria. Because toe vessels are less susceptible to stiffening, we also performed toe–brachial index (TBI) testing to confirm PAD diagnosis; a value < 0.7 was considered as a confirmation of PAD diagnosis. Patients with nonatherosclerotic occlusive disease (e.g., acute limb ischemia, vasculitis, Buerger’s disease, etc.) were excluded. Medical history, race, smoking history, and demographics were obtained by self-report. Co-existing medical conditions and medication usage were obtained through review of medical records.
Calf muscle strength testing
A maximal voluntary isometric strength test of the calf muscle was performed using a Lafayette dynamometer (Lafayette, IN, USA) mounted to a custom stand for stability. Patients were seated in the testing chair, and the leg was stabilized with the foot secured to the dynamometer. Next, patients placed their hands on their abdomen or chest to minimize trunk movements during the test. Following two to three practice trials, patients performed a maximal plantarflexion for 5 seconds, and continuous force analysis was observed to ensure a plateau was obtained. Patients repeated the test five times with 1.5-minute rest intervals between tests. Verbal encouragement was provided, and the maximal force (N) was used for analysis.
Calf muscle biopsy
Gastrocnemius muscle specimens were collected via percutaneous muscle biopsy using sterile procedures, as previously described. 24 This procedure involved a small (< 0.5 cm) incision through the skin and placement of a suction-controlled sterile 14-gauge biopsy needle (BD Elevation Breast Biopsy System (Franklin Lakes, NJ, USA)) into the gastrocnemius muscle. A portion of the muscle was rapidly trimmed of fat/connective tissue and snap-frozen in liquid nitrogen for RNA sequencing analysis. A portion of muscle was frozen in isopentane (liquid-nitrogen-cooled) for histological analysis, and another portion was placed in ice-cold buffer X (50 mM K-MES, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 20 mM taurine, 5.7 mM ATP, 14.3 mM phosphocreatine (PCr), and 6.56 mM MgCl2-6H2O, pH 7.1) for the preparation of permeabilized fiber bundles.
Analysis of mitochondrial function
Fiber bundles were mechanically separated using fine forceps under a dissecting scope and then immediately permeabilized with saponin (30 μg/mL) for 30 minutes at 4°C on a mixer. Following permeabilization, fiber bundles were washed in ice-cold buffer Z (105 mM K-MES, 30 mM KCl, 1 mM EGTA, 10 mM K2HPO4, 5 mM MgCl2-6H2O, and 0.5 mg/mL bovine serum albumin [BSA], pH 7.1) for 15 minutes. High-resolution O2 consumption measurements were conducted at 37°C using the Oroboros O2k Oxygraph (Innsbruck, Austria) in buffer Z supplemented with creatine monohydrate (5 mM). To assess mitochondrial function in physiologically relevant conditions, this study utilized a creatine-kinase clamp system to set the level of cellular energy demand to which the fiber bundles were exposed. First, bundles were energized with 5 mM pyruvate, 2.5 mM malate, and 0.2 mM octanoylcarnitine, and measurements of State 2 oxygen consumption were collected. Next, the creatine-kinase clamp was added including 20 U/mL creatine kinase, 5 mM ATP, and 1 mM PCr to mimic a near-maximal exercise condition. Subsequent additions of PCr were added stepwise to bring the cellular energy demand down to resting conditions. The slope of the relationship between cellular energy demand (∆GATP) and oxygen consumption (JO2), termed OXPHOS conductance, was calculated. The rate of respiration was expressed as pmol/s/mg fiber wet weight. All respiration measurements were conducted with a working range [O2] of ~350 to 200 μM and were normalized to the bundle wet weight obtained using a Mettler Toledo MX5 microbalance (Columbus, OH, USA). Mitochondrial H2O2 emission was measured fluorometrically in a separate muscle bundle via the Amplex UltraRed/horseradish peroxidase detection system (excitation:emission 530:590) (ThermoFisher Scientific, Waltham, MA, USA), as previously described.24,31 All mitochondrial analyses were performed by researchers that were blinded to the participant group.
Muscle histopathology
All histopathology analysis and quantification were performed by researchers who were blinded to the participant group. The skeletal muscle fiber cross-sectional area (CSA) and myosin heavy chain isoform (fiber type) were assessed using immunofluorescence microscopy. Frozen sections (5 µm) of the muscle specimen were cut using a Leica CM3050 cryostat (Deer Park, IL, USA), mounted on microscope slides, and blocked in animal-free blocking solution (Vector Laboratories, Newark, CA, USA; Cat. No. SP-5035-100) for 1.5 hours. To label the muscle fiber membrane, sections were incubated with a primary antibody against laminin (Developmental Studies Hybridoma Bank, University of Iowa; Cat. No. 2E8; 1:100 dilution). To label myofiber types and capillaries, the sections were incubated with primary antibodies against myosin heavy chain (MyHC) isoforms MyHC I (Cat. no. BA-D5; 1:100 dilution), MyHC IIa (Cat. no. SC-71; 1:500 dilution) (both from the Developmental Studies Hybridoma Bank, University of Iowa), and CD31/PECAM1 (Abcam, Waltham, MA, USA; Cat. no. ab28364; 1:50 dilution) overnight at 4°C. The following morning, slides were washed four times for 5 minutes with phosphate-buffered saline (PBS) and then incubated with appropriate Alexa-Fluor-conjugated secondary antibodies (ThermoFisher Scientific; Cat. no. A21140, A21121, A21137, A32733; all 1:300 dilution) for 1 hour at room temperature. Following another round of 5-minute washes (four times) with 1× PBS, the coverslips were mounted with Vectashield hardmount (Vector Laboratories; Cat. no. H-1400). Slides were imaged at 20× magnification with an EVOS FL2 Auto microscope (Thermo Fisher Scientific), and tiled images of the entire muscle section were used for analysis. The proportion of myofiber types (myosin heavy chain) and quantification of myofiber CSA were performed using the automated analysis platform Myosight (Baylor College of Medicine, Houston, TX, USA). 32 To quantify the density of capillaries, the tiled images were thresholded in ImageJ/Fiji (National Institutes of Health, Bethesda, MD, USA) and counted by a blinded investigator, and the vessel density was normalized to the area of the muscle section.
Succinate dehydrogenase (SDH) activity staining was performed by allowing unfixed muscle sections to air-dry for 5 minutes before placing the slides in a Coplin jar containing 130 mM sodium succinate, 0.2 mM phenazine methosulfate, 1 mM sodium azide, and 1.5 mM nitrotetrazolium blue chloride (in 0.2 M phosphate buffer, pH = 7.0) for 60 minutes at 37°C. Slides were then washed twice in 1 × PBS at room temperature before being dehydrated by dipping in increasing ethanol concentrations and cleared with xylenes. Coverslips were mounted using toluene. Transmitted light images were obtained at ×20 magnification with an EVOS FL2 Auto microscope (Thermo Fisher Scientific), and the mean intensity was used to quantify SDH abundance/activity.
Bulk RNA sequencing
Total RNA was extracted from gastrocnemius muscle using the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA, USA; Cat. No. R2052). Library preparation and mRNA sequencing via Poly-A selection was performed by Genewiz (Azenta Life Science, South Plainfield, NJ, USA). The 150-bp reads (paired-end) were sequenced on an Illumina HiSeq 4000 (San Diego, CA, USA). Reads were trimmed to remove adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36 (Heinrich Heine University, Germany), and then mapped to the Homo sapiens GRCh38 reference genome using the STAR Aligner v.2.5.2b (Alex Dobin, Cold Springs Harbor Laboratory, USA). Unique gene hit counts were calculated using featureCounts from the Subread package v.1.5.2. The hit counts were summarized and reported using the gene_id feature. Only unique reads that fell within exon regions were counted. After extracting hit counts, the gene hit counts table was used for downstream differential expression analysis. DESeq2 was used to perform a comparison of gene expression between the groups (cilostazol vs no cilostazol). The Wald test was used to generate p-values and log2 fold changes. Gene set enrichment analysis (GSEA) was performed using GSEAPY.
Statistical analysis
The normality of data was tested with the Shapiro–Wilk test and inspection of Q-Q plots. Data involving comparisons of normally distributed data from groups were analyzed using an unpaired, two-tailed Student’s t-test. Data involving comparisons of nonnormally distributed data from groups were analyzed using a Kruskal–Wallis test. Chi-squared analysis was used to determine differences in population proportions for relevant clinical characteristics. Pearson correlation analyses were run as two-tailed tests to determine associations between outcome variables. Two-way ANOVA was used for data involving group comparisons across conditions for mitochondrial analyses. For bulk RNAseq, the Benjamini–Hochberg method was used to calculate false discovery rate-corrected p-values. In all cases, p < 0.05 was considered statistically significant. All statistical testing was conducted using software from Python (Python Software Foundation) or GraphPad Prism (GraphPad Software, LLC; version 10.0). Data are presented as the mean ± SD unless otherwise specified.
Results
Fifty patients with PAD were included in this study: 35 patients not taking cilostazol and 15 patients taking cilostazol. The participant characteristics for these groups are shown in Table 1. Patients taking cilostazol had similar physical and medical characteristics compared to those not taking cilostazol; however, those taking cilostazol had a lower prevalence of heart failure (p = 0.0438), which is expected because heart failure is a contraindication for cilostazol. The median duration of cilostazol usage was 50 months (minimum = 3 months, maximum = 178 months). Six patients were taking 100 mg twice daily and the remaining nine patients were taking 50 mg twice daily. Information about medication adherence was not collected or available. None of the patients were actively enrolled in a supervised exercise training program at the time of testing or in the 3 months preceding testing. Moreover, none of the patients had a history of surgical intervention on the index limb used for testing. No patients had undergone a catheter-based intervention (i.e., angiography) in the 3 months prior to testing.
Patient characteristics.
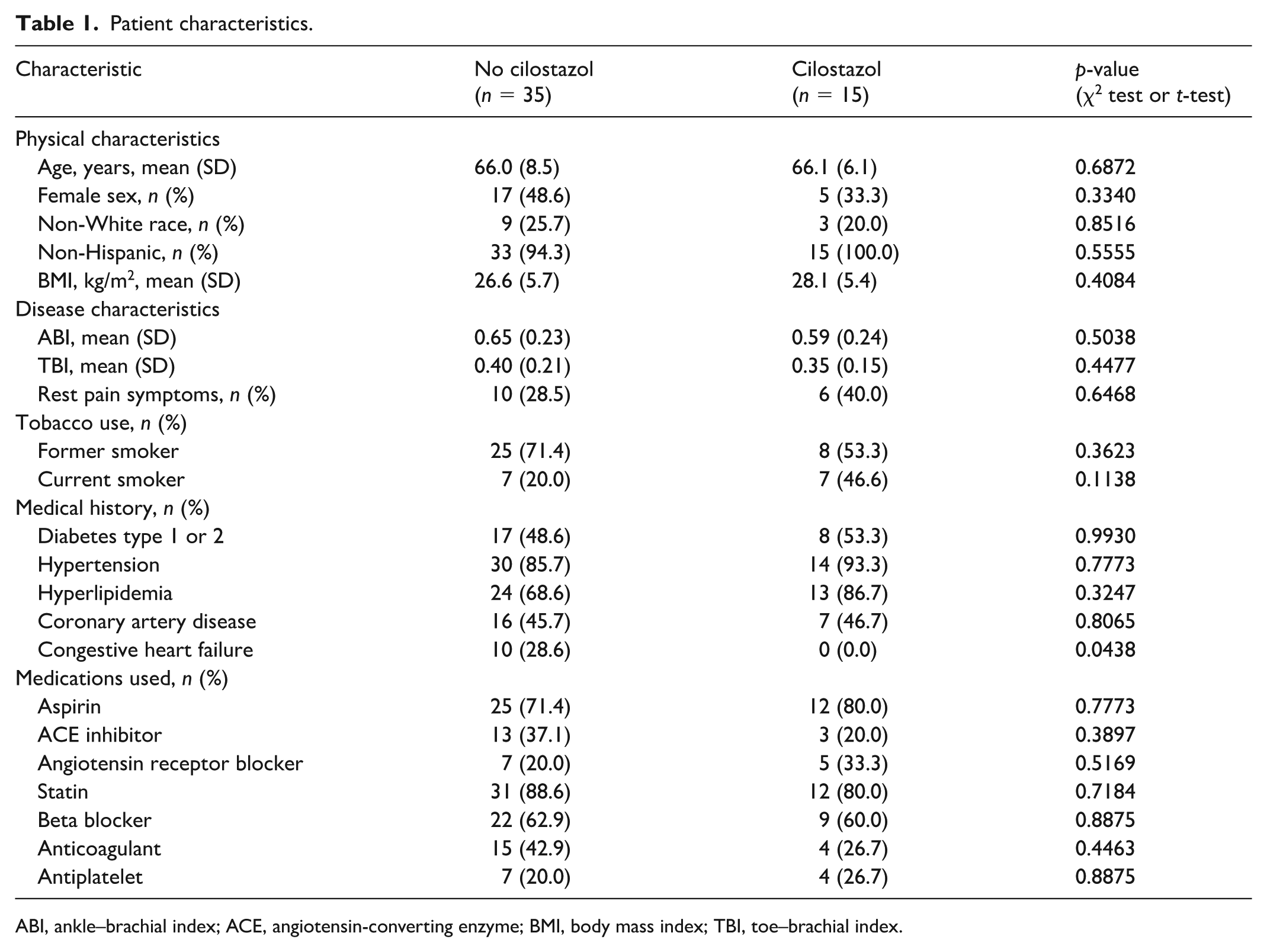
ABI, ankle–brachial index; ACE, angiotensin-converting enzyme; BMI, body mass index; TBI, toe–brachial index.
Absolute calf muscle (plantar flexor) strength did not differ significantly between patients taking and not taking cilostazol (p = 0.49), nor did the calf strength normalized to body weight (p = 0.69) (Figure 1A). The mean myofiber CSA showed no differences between groups for either Type I (p = 0.53) or Type IIa myofibers (p = 0.59) (Figures 1B and 1C). The proportion of Type I fibers trended toward being higher in the cilostazol-treated group, although this did not reach statistical significance (p = 0.11) (Figure 1D). Next, we quantified capillary density in the gastrocnemius muscle biopsy specimens by immunolabeling for CD31/PECAM1, with representative images shown in Figure 2A. Quantitative analysis showed no difference in the capillary density between patients taking and not taking cilostazol (p = 0.74) (Figure 2B), indicating that cilostazol has minimal impact on skeletal muscle capillarization.
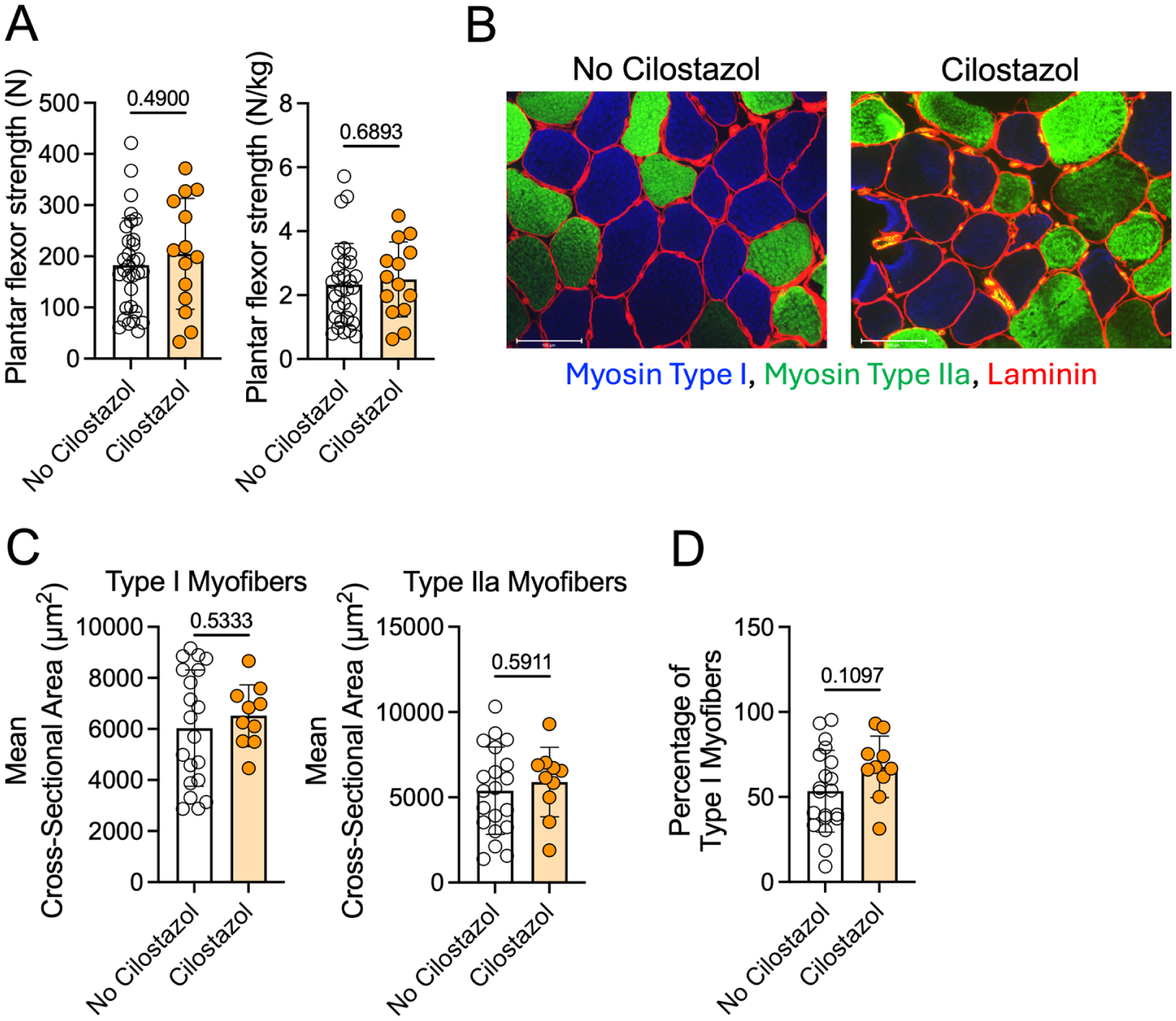
Patients with PAD taking cilostazol have similar calf muscle strength and myofiber area compared to patients not taking cilostazol.
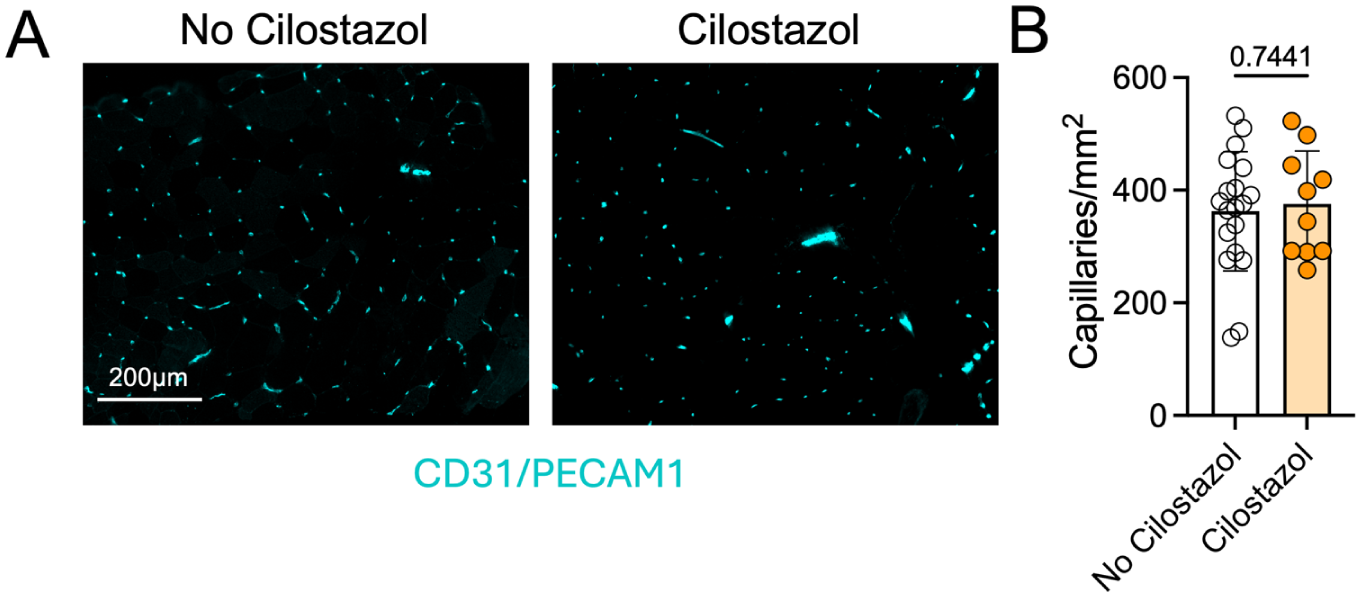
Patients with PAD taking cilostazol have similar calf muscle capillary density compared to patients not taking cilostazol.
Mitochondrial respiration was assessed in permeabilized gastrocnemius muscle fibers from patients with PAD treated with or without cilostazol using a creatine-kinase clamp to simulate physiological energy demands. Oxygen consumption (JO2) was significantly higher in cilostazol-treated patients when compared to patients not taking cilostazol (p = 0.0137) (Figure 3A). OXPHOS conductance, the slope of the relationship between JO2 and energy demand (∆GATP), was not different between groups (p = 0.38), even after controlling for statin usage, diabetes, and heart failure as covariates (p = 0.078) (Figure 3B). This finding suggests that whereas patients taking cilostazol had higher rates of oxygen consumption than those not taking cilostazol, the ability of the mitochondria to increase oxygen consumption in response to changes in energy demand was not impacted differently between groups. Using traditional respirometry involving saturating adenosine diphosphate (ADP)-stimulating conditions, there was no difference in maximal State 3 respiration between groups (p = 0.78), even after controlling for statin usage, diabetes, and heart failure as covariates (p = 0.27) (Figure 3C). Mitochondrial H2O2 emission was not different between groups across varying energy demand levels (Figure 3D) or under State 2 (no energy demand) conditions (p = 0.11) (Figure 3E). When fueled with succinate in the absence of energy demand, with or without inhibition of the matrix antioxidant systems, mitochondrial H2O2 emission and production levels showed no difference between groups (Figure 3F). SDH enzyme activity assessed histochemically in muscle sections also showed no difference between groups (p = 0.50) (Figure 3G). Correlation analyses in this modest-sized cohort identified significant positive correlations between calf muscle OXPHOS conductance and the percentage of Type I myofibers, capillary density, State 3 respiratory capacity, and SDH staining intensity (Table 2). Plantarflexor muscle strength was significantly correlated with the Type IIa myofiber CSA, percentage of Type I myofibers, capillary density, SDH staining intensity, and State 3 respiratory capacity (Table 2). OXPHOS conductance and plantarflexor strength were also positively correlated in this cohort (Pearson r = 0.38, p = 0.0223).
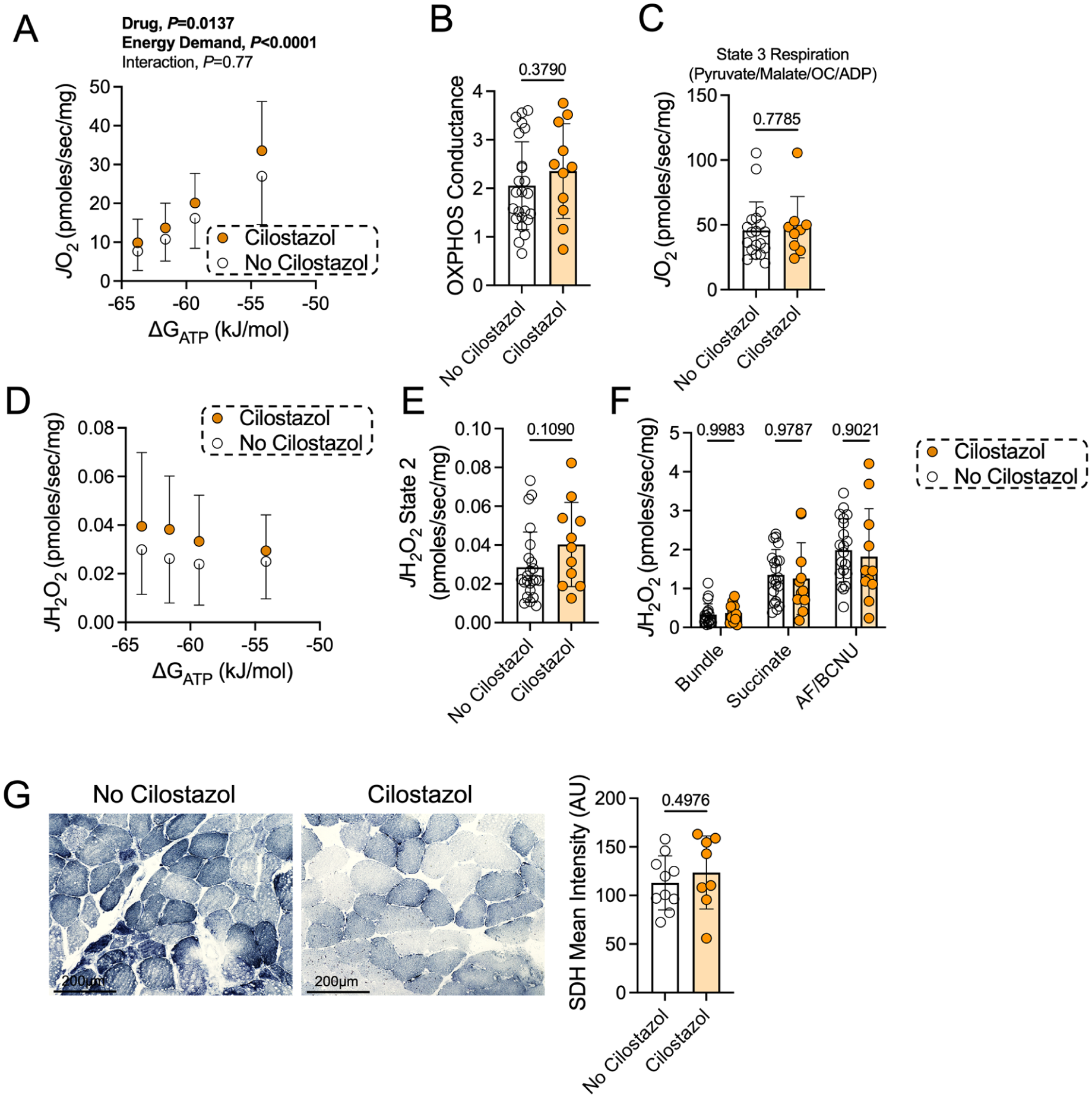
Patients taking cilostazol have modestly higher rates of mitochondrial oxygen consumption under physiological energy demand conditions.
Correlation analyses.
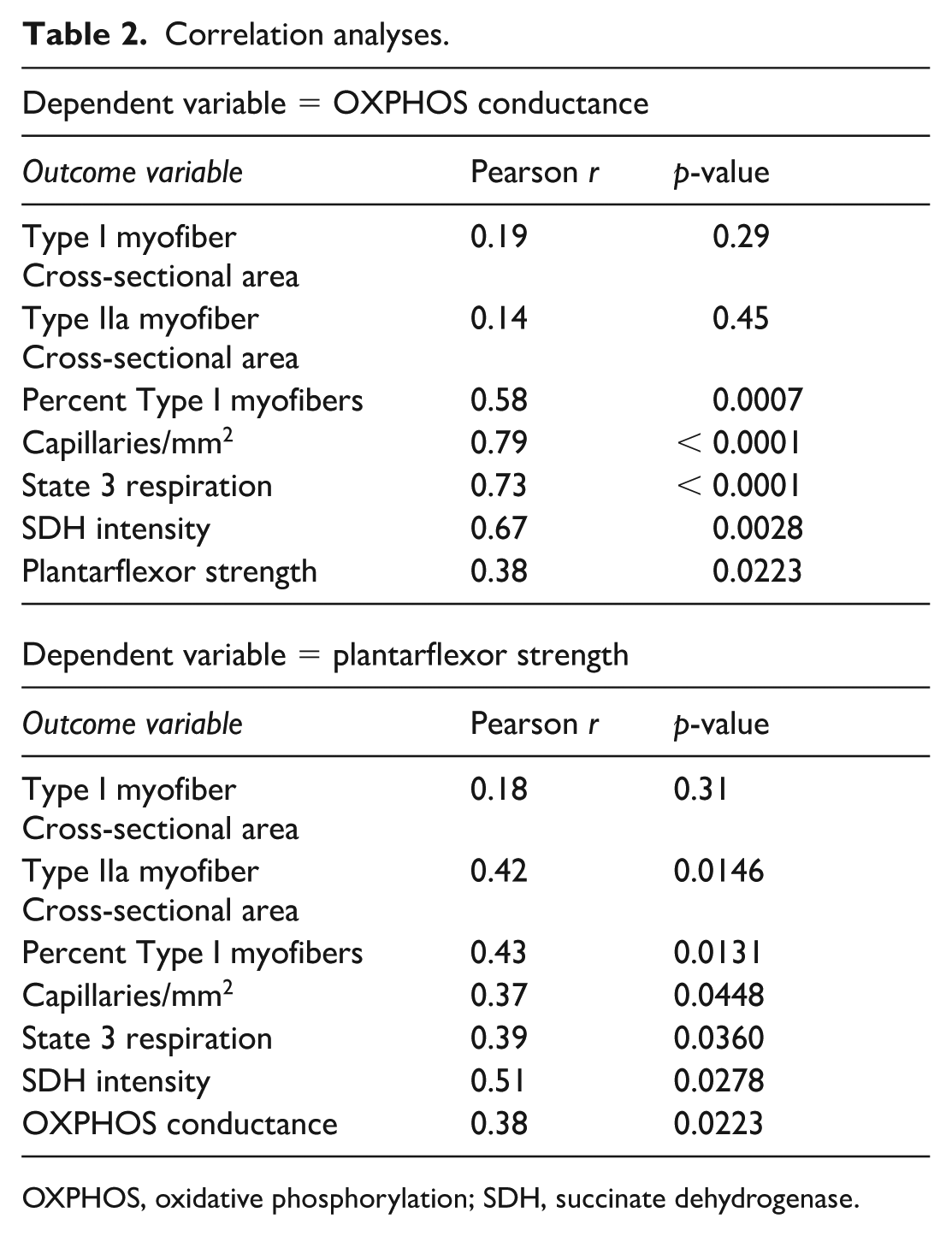
OXPHOS, oxidative phosphorylation; SDH, succinate dehydrogenase.
We analyzed bulk RNA sequencing data collected from the gastrocnemius muscle specimens of patients with PAD taking (n = 15) and not taking (n = 28) cilostazol. Expression was quantified for 25,149 genes. Principal component analysis of this transcriptome revealed substantial overlap between gene expression profiles of muscle from patients with PAD taking and not taking cilostazol (Figure 4A). However, partial least squares discriminant analysis (PLS-DA) was able to identify some distinct clustering between these groups (Figure 4A). Using an unadjusted p-value of less than 0.05, patients with PAD taking cilostazol had 1315 differentially expressed genes (DEGs), with 674 genes downregulated and 641 upregulated (Figure 4B) compared to patients with PAD not taking cilostazol.
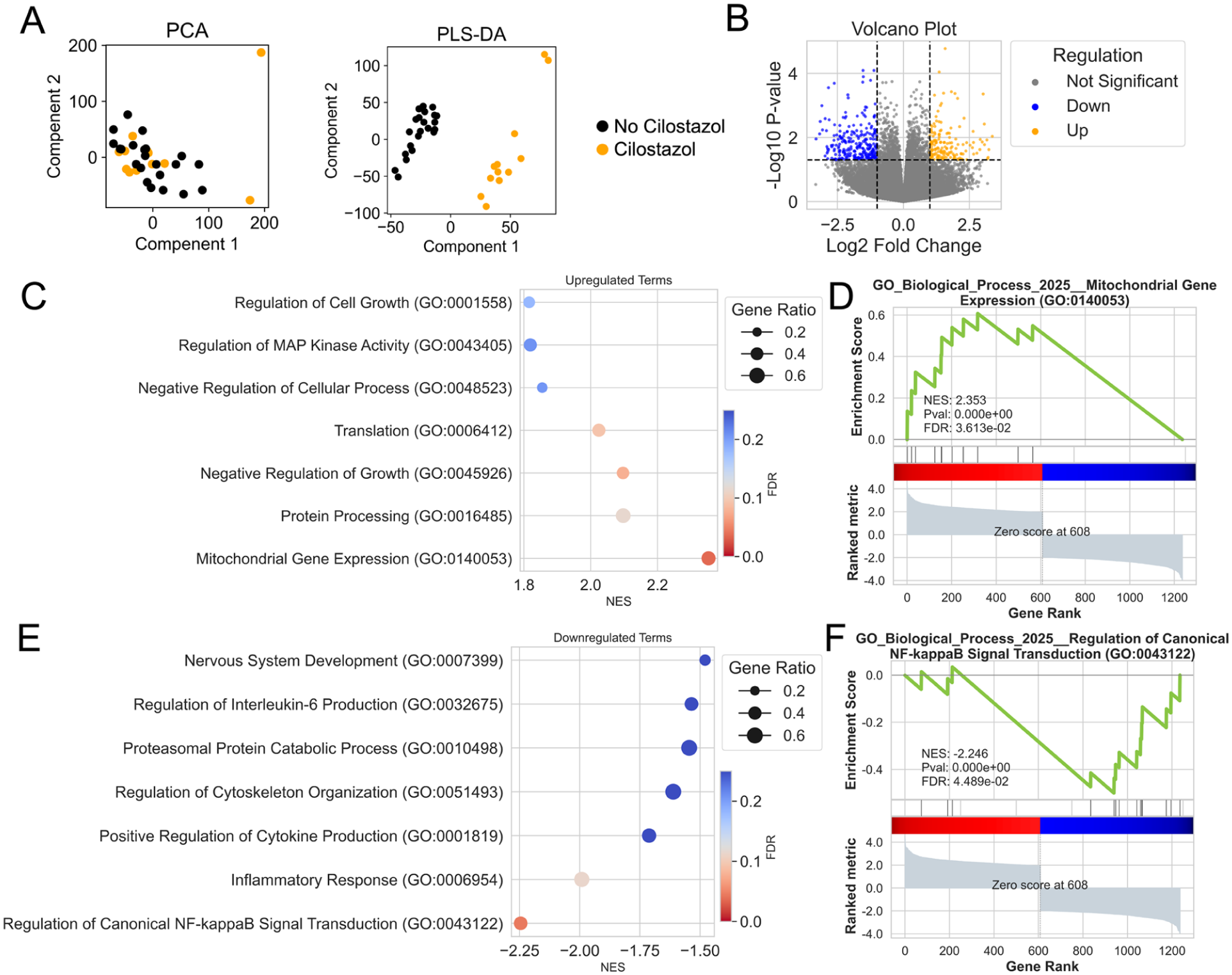
Muscle transcriptome differences in patients with PAD taking cilostazol and patients not taking cilostazol.
Gene set enrichment analysis (GSEA) revealed that the term ‘mitochondrial gene expression’ was significantly upregulated in patients with PAD taking cilostazol (Figures 4C and 4D). This finding is consistent with the observed increases in muscle mitochondrial respiration at varying levels of energy demand (shown in Figure 3A). GSEA also revealed that patients with PAD taking cilostazol had downregulation of inflammatory signaling pathways such as ‘regulation of canonical NF-κB signal transduction’ and ‘inflammatory response’ (Figures 4E and 4F), suggesting that cilostazol may exert antiinflammatory effects on skeletal muscle. In addition, upregulation was observed in pathways related to ‘protein processing’ and ‘translation’, indicating potential enhancement of protein turnover and synthesis machinery in skeletal muscle from patients taking cilostazol, although these effects did not increase myofiber areas (Figure 1). Full DEG and GSEA results from the RNA sequencing are available in Supplemental Dataset 1.
Discussion
Although there is a wealth of clinical evidence supporting the efficacy of cilostazol in improving walking performance in patients with PAD experiencing claudication symptoms, 7 to our knowledge, there are no studies that have examined whether taking cilostazol alters calf muscle pathophysiology in these patients. Herein, we conducted a cross-sectional analysis to compare muscle pathophysiology in patients with PAD grouped by whether or not they were taking cilostazol. The results of this analysis found no differences in calf muscle strength, myofiber area, or capillary densities between patients taking and not taking cilostazol. However, patients taking cilostazol display increased skeletal muscle mitochondrial respiration under physiologically relevant energy demand conditions, yet show no differences in maximal ADP-stimulated respiration or hydrogen peroxide production. Skeletal muscle RNA sequencing revealed substantial overlap between the transcriptomes of patients taking and not taking cilostazol. GSEA analysis of DEGs indicated that patients taking cilostazol have increased expression of genes related to mitochondria and decreased expression of inflammation-related genes.
There are only a few studies that have reported how phosphodiesterase inhibitors, particularly PDE3 inhibitors, impact skeletal muscle biology. These studies primarily involved in vitro studies of muscle contractility, preclinical animal models, or cell culture systems. In isolated muscles, treatment with milrinone or amrinone was found to improve isometric force levels and fatigue resistance, although these drugs may have reduced shortening velocity.33,34 As these direct exposures are vastly different from oral consumption of cilostazol, it is difficult to relate these observations to the lack of calf muscle strength differences observed in the current patient cohort. One study in cultured muscle cells suggested that cilostazol increases fatty acid oxidation, 35 a finding which is potentially congruent with our observed increases in respiration with muscle bundles energized with a mixture of pyruvate, malate, and the medium chain fatty acid octanoylcarnitine (Figure 3A). Beyond PDE3 inhibitors, a few other studies focused on muscle biology reported that PDE4 inhibitors could reduce muscle atrophy and proteolysis.36,37 These observations are also potentially congruent with the transcriptome results in our study, as patients taking cilostazol had modest upregulations of genes in the pathways ‘protein processing’ and ‘translation’.
Cilostazol is currently one of the only FDA-approved pharmacological agents indicated specifically to improve walking distance in patients with claudication due to PAD. Its use is supported by multiple randomized controlled trials and meta-analyses showing modest but consistent improvements in treadmill walking time and quality of life.5,7 However, the real-world utilization of cilostazol remains limited for several reasons. First, cilostazol is contraindicated in patients with PAD who also have heart failure due to concerns about increased mortality seen with PDE3 inhibitors in this population. 5 Given the high prevalence of heart failure among patients with PAD, this contraindication reduces the proportion of patients with PAD taking cilostazol. Second, although cilostazol improves walking performance, it does not reduce the risk of cardiovascular events or disease progression, limiting its utility as a disease-modifying therapy. Furthermore, side effects such as headache, palpitations, and diarrhea often lead to poor adherence. Our findings support the idea that cilostazol may modestly influence skeletal muscle mitochondrial respiration but do not suggest a strong effect on broader muscle pathology. These nuanced effects may help explain why the clinical benefits of cilostazol are modest and variable across patients. As such, prescribing cilostazol requires careful patient selection, weighing potential benefits in walking ability against contraindications and tolerability concerns.
This study has several limitations. First, this was a cross-sectional comparison of muscle pathology in patients with PAD who were and were not taking cilostazol. Therefore, the results shown should not be used to draw conclusions about how cilostazol may or may not change muscle pathophysiology in patients with PAD. Future longitudinal studies involving analyses of the same patients before and after taking cilostazol are needed to form such conclusions. Second, we only assessed calf muscle strength in this cohort of patients; measures of walking performance or physical activity levels were not quantitatively performed. Because regular exercise training and increasing physical activity levels can positively influence muscle pathophysiology, we cannot rule out effects due to differences in physical activity levels; however, none of the patients were actively enrolled in an exercise training program at the time of testing or in the 3 months preceding the testing. Third, as this was a cross-sectional analysis, analyses of how chronic cilostazol treatment might alter the trajectory of changes in muscle pathophysiology across time in patients with PAD cannot be performed. More studies are needed to determine if cilostazol might alter the progression of muscle pathology in patients with PAD. Fourth, comorbid conditions such as congestive heart failure, diabetes, and statin use can influence skeletal muscle and mitochondrial function. However, these comorbidities are highly prevalent among patients with PAD and we did not control for their individual contributions within the current study. Therefore, their potential influence on the observed outcomes cannot be fully excluded. Finally, some patients (n = 10 not taking cilostazol and n = 6 taking cilostazol) in our cohort presented with rest pain symptoms at the time of the muscle biopsy procedure. However, guidelines for the management of more severe forms of PAD are much less clear regarding the benefits of cilostazol. Although there were no differences in the proportion of these more severe cases between groups, this may have potential to bias the data reported.
Conclusion
This study provides a cross-sectional comparison of muscle pathophysiology in patients with PAD who were and were not taking cilostazol. Although patients taking cilostazol had no differences in calf muscle strength, myofiber area, or capillary density, they exhibited greater levels of mitochondrial oxygen consumption compared to patients not taking cilostazol. However, the higher rates of oxygen consumption did not confer changes in the performance of the oxidative phosphorylation system or differences in mitochondrial H2O2 production. RNA sequencing revealed a similar transcriptome profile of the calf muscle in these groups. These data suggest a similar muscle pathophysiology exists in patients with PAD taking or not taking cilostazol. Future longitudinal studies are needed to clarify whether the initiation of cilostazol treatment changes muscle pathophysiology in patients with PAD.
Supplemental Material
sj-xlsx-1-vmj-10.1177_1358863X261429979 – Supplemental material for Cilostazol use and calf muscle pathophysiology in people with peripheral artery disease
Supplemental material, sj-xlsx-1-vmj-10.1177_1358863X261429979 for Cilostazol use and calf muscle pathophysiology in people with peripheral artery disease by Jaewon Choi, Kyoungrae Kim, Trace Thome, Lauren Stone, Nicholas Vugman, Caroline G Pass, Victoria R Palzkill, Qingping Yang, Erik M Anderson, Brian Fazzone, Scott A Berceli, Salvatore T Scali and Terence E Ryan in Vascular Medicine
Footnotes
Data Availability Statement
Sequencing data are available from the NCBI Gene Expression Omnibus (GEO) using accession number GSE287118.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Terence E Ryan was supported by National Institutes of Health (NIH) grants R01-HL149704 and HL171050 and the American Heart Association grant 25EIA1369187. Salvatore T Scali was supported by NIH grant R01-HL148597. Scott A Berceli was supported by NIH grant R01-DK119274. Kyoungrae Kim was supported by the American Heart Association grant POST903198. Trace Thome was supported by NIH grant F31-DK128920. Victoria R Palzkill was supported by NIH grant F31-HL174156 and the American Heart Association grant 24PRE1193999. Caroline G Pass was supported by the American Heart Association grant 24PRE1196311.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.