
Abstract
Vascular aging represents the common pathological basis of aging-related diseases, with vascular smooth muscle cells (VSMCs) senescence playing a pivotal role. P300/CBP-associated factor (PCAF), a transcriptional coactivator linked to diverse physiological processes, has an undefined role in VSMCs aging. This study investigated PCAF's function in VSMC senescence and its underlying mechanisms. Using siRNA adenovirus to downregulate PCAF and angiotensin II (AngII) to induce senescence, we assessed β-galactosidase activity, senescence-associated secretory phenotypes (SASP), and cell cycle proteins (p53, p21, p16). Transcriptomic analysis revealed PCAF's association with oxidative stress and the Nrf2-ARE pathway. Results demonstrated that PCAF downregulation significantly attenuated VSMCs senescence and AngII-induced oxidative stress by enhancing Nrf2-ARE pathway activity. Crucially, Nrf2 silencing reversed these protective effects. In conclusion, PCAF inhibition mitigates VSMCs senescence via Nrf2-ARE-mediated antioxidant activation, identifying PCAF as a promising therapeutic target for vascular aging.
Keywords
Introduction
As the global aging problem becomes more and more critical, the incidence of the accompanying aging-related diseases is also increasing year by year, which seriously threatens the life and health of the elderly people. 1 Many studies have shown that vascular aging is a common pathological basis for the various aging-related diseases. 2 Although vascular aging is a combined effect of vascular cells and vascular extracellular matrix senescence, the latest research showed that the senescence of vascular endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), the main components of the blood vessels, play the leading role in the pathogenesis of vascular aging. 3 Therefore, it is of great significance to clarify the mechanism of vascular cells senescence and find effective targets for inhibiting vascular cells senescence for improving vascular aging.
The senescence of vascular cells, as most senescent cells, is mainly characterized by irreversible cell cycle arrest and an active senescence associated secretory phenotype (SASP).4,5 The senescence of vascular cells could result in cell cycle arrest by initiating two cell cycle-related signaling pathways, p53-p21-RB (retinoblastoma protein, RB) and p16INK4A-RB.6–8 In addition, it could mediate the formation of SASP by affecting the activity of NF-κB, p38 MAPK and mTOR signaling pathways. 9 The recent research showed that senescent vascular cells were usually accompanied by the activation of p53-p21-RB and p16INK4A-RB pathways, and inhibiting the activity of these two signaling pathways could effectively delay the aging process of vascular cells. 10
P300/CBP-associated factor (PCAF) is a transcriptional coactivator, which could participate in the transcriptional regulation of genes by acetylating histones and non-histone proteins, and is closely related to various physiological activities such as cell growth, differentiation, and apoptosis. 11 Previous studies have shown that PCAF could participate in the modification of cell cycle-related protein molecules and has a regulatory effect on the cell cycle. 12 Downregulation of PCAF could reduce the activity of p53 protein, attenuate p53-dependent transcription of p21 protein, increase the expression of cyclin D1 and phosphorylation of RB, and ultimately drive the cell cycle. 13 Fei et al. 14 found that enforced expression of PCAF could upregulate the expression of the cell cycle negative regulator p16, resulting in cell cycle arrest. The latest research also found that downregulation of PCAF in vascular cells could remarkably inhibit the activation of NF-κB, p38 MAPK and mTOR, which mediate the formation of SASP, and could reduce the secretion of various SASP components, including TNF-α and IL-6.11,15 Nevertheless, as far as we know, there is no relevant report on the role and mechanism of PCAF in vascular cells senescence.
Although chronological aging is a key risk factor for vascular dysfunction, the pathological premature senescence induced by cardiovascular risk factors, such as hypertension, represents a distinct and clinically relevant process. 16 Angiotensin II (AngII), a critical mediator of the renin-angiotensin system, is not only pivotal in the pathogenesis of hypertension but also directly promotes vascular senescence and remodeling.17–19 Compared with aged rat models, which encompass complex, cumulative effects of chronological aging, the AngII-induced model offers a well-established, controllable, and clinically relevant system to dissect the molecular mechanisms underlying pathological vascular senescence. 20 This model allows us to specifically interrogate how acute or subacute elevation of vasoactive peptides drives premature senescence in vascular smooth muscle cells (VSMCs), thereby minimizing confounding factors associated with the aging process itself. Therefore, in this study, we employed AngII stimulation to investigate the role of PCAF in vascular senescence and explore its potential molecular mechanisms.
Materials and methods
Materials and agents
Angiotensin II (Ang II) and the senescence β-galactosidase (SA-β-Gal) staining kit were procured from Sigma-Aldrich (#05-23-0101) and Beyotime Biotechnology (#C0602), respectively. Dihydroethidium was from Beyotime Biotechnology (#S0063, China). Assay kits for superoxide dismutase (SOD) (#A001-3) and catalase (CAT) (#A007-1-1) were procured from Nanjing Jiancheng Bioengineering Institute (China). Primary antibodies were commercially sourced as follows: glyceraldehyde-phosphate dehydrogenase (GAPDH) (#2118), nuclear factor-erythroid 2-related factor 2 (Nrf2) (#12721), PCAF (#3378), tumor suppressor protein p53 (p53) (#2524), and heme oxygenase-1 (HO-1) (#43966) from Cell Signaling Technology (USA). PCAF (#sc-13124), NAD[P]H quinone oxidoreductase-1 (NQO1) (#sc-376023), cyclin-dependent kinase inhibitor 1A (p21) (#sc-6246), and cyclin-dependent kinase inhibitor 2A (p16) (#sc-1661) from Santa Cruz Biotechnology (USA). Intercellular cell adhesion molecule-1 (ICAM-1) (#sc-8439) and vascular cell adhesion molecule 1 (VCAM-1) (#sc-13160) were obtained from Santa Cruz Biotechnology (USA). The rat Nrf2 small interfering RNA (siRNA) and control siRNA were purchased from GenePharma (China).
Construction of adenoviral vectors
Adenovirus construction followed a previously established protocol. 11 GeneChem (Shanghai, China) designed and synthesized a specific siRNA sequence targeting the rat PCAF gene (Kat2b, GenBank Accession NC_005108.4), along with a scrambled siRNA negative control. The sense sequences were 5′-GACAAACTGCCTCTTGAGAAA-3′ (PCAF siRNA) and 5′-TTCTCCGAACGTGTCACGT-3′ (control siRNA). Subsequently, adenoviruses encoding PCAF siRNA (Ad-PCAF-RNAi) or a control (Ad-GFP) were generated by co-transfecting HEK293 cells using standard calcium phosphate methods.
Primary cells culture and adenoviral infection of cells
Animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and ARRIVE guidelines and received approval from the Animal Care and Use Committee of Renmin Hospital of Wuhan University (Approval No. WDRM20210104). Thoracic aortic vascular smooth muscle cells (VSMCs) were harvested from 5-6 week old male Sprague-Dawley (SD) rats (150–200 g, provided by the Hubei Experimental Animal Centre) as described previously. 21 Briefly, SD rats were housed in a specific pathogen-free (SPF) environment and acclimated for one week with SPF maintenance feed. The rats were then anesthetized and placed in the supine position. Following thoracotomy, the thoracic aorta was excised and transferred to a culture dish containing cold (4 °C) DMEM/F12 medium. After removal of the perivascular adipose tissue, the aorta was longitudinally opened and placed in another culture dish with DMEM/F12 medium. The endothelial cells were gently removed by scraping the intimal surface with a pair of curved ophthalmic forceps, and the vascular media was subsequently separated. The medial layer was then cut into approximately 1 mm2 explants and transferred to cell culture plates. The plates were placed in a cell culture incubator for about 30 minutes to allow the explants to adhere. Next, DMEM/F12 medium supplemented with 20% fetal bovine serum (FBS) was carefully added, and the explants were maintained undisturbed in the incubator. The medium was replaced every other day until VSMCs migrated from the edges of the explants. Cells between passages 3 and 5 were maintained in DMEM/F12 + 10% FBS.
Transduction utilized adenoviral vectors (Ad-PCAF-RNAi or Ad-GFP; MOI = 30) applied to VSMCs at approximately 50% confluence for 4 h. After the transduction period, fresh medium was applied prior to further experimentation. Twenty-four hours after successful viral transfection, VSMCs were then stimulated with 2 μM AngII for 24 h to induce senescence.
SA-β-Gal staining
After processed as required, the cells were stained using a senescence-β-galactosidase staining kit. The specific steps were implemented according to the kit instructions. After staining, five representative pictures were randomly taken from each group, and the ratio of the number of positive cells to total cells was calculated.
Western blot
Total cellular proteins were extracted with RIPA lysis buffer (Beyotime Biotechnology, #P0013B), while cytosolic and nuclear fractions were isolated using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, #P0027). Following quantification via BCA assay, equal protein aliquots (30 µg) underwent separation on 10% SDS-PAGE gels and electrophoretic transfer onto polyvinylidene fluoride (PVDF) membranes at 200 mA constant current for 2 h. Membranes were then blocked for 2 h at room temperature with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBST). After TBST washes, membranes were probed overnight at 4°C with primary antibodies. Subsequent incubation with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies proceeded for 1 h at room temperature. Finally, protein bands were visualized using enhanced chemiluminescence (ECL) reagent and imaged with a Bio-Rad ChemiDoc Touch Imaging System.
Immunofluorescence staining
VSMCs plated on coverslips within 6-well plates underwent sequential processing: fixation in 4% paraformaldehyde (15 min), permeabilization with 0.3% Triton X-100 (10 min), and blocking in 5% goat serum (1 h). Primary antibody incubation proceeded overnight at 4°C, followed by 1-h exposure to fluorophore-conjugated secondary antibodies at room temperature. Nuclei were counterstained with DAPI prior to mounting with anti-fade reagent. Fluorescence images were acquired using a microscope, with all experiments independently repeated ≥3 times.
SOD, CAT and ROS detection
Cells were collected, and the commercial kits were used to analyze activity levels of SOD (Nanjing Jiancheng Bioengineering Institute, China, #A001-3), CAT (Nanjing Jiancheng Bioengineering Institute, China, #A007-1-1) and ROS (Mainly measures superoxide anion levels; Beyotime Biotechnology, China, #S0063). All procedures were performed in accordance with the manufacturers’ instructions. Three independently repeated experiments were performed.
Co-Immunoprecipitation (IP)
Cell lysates were collected using IP lysis buffer supplemented with protease inhibitors, incubated on ice for 30 min, and then centrifuged at 12,000 g for 30 min. After collecting the supernatant, an aliquot was reserved as the total lysate for Input control, and the remaining sample was used for subsequent co-immunoprecipitation steps. Detailed procedures can be found in reference. 22 Briefly, under 4°C conditions, 5 μg of anti-PCAF antibody (Cell Signaling Technology, USA, #3378) was added to the experimental group, while the negative control group received the same concentration of IgG antibody (Cell Signaling Technology, USA, #3900) from the same species as the PCAF antibody. The samples were incubated overnight at 4°C with rotation to allow the PCAF antibody to fully bind to its corresponding antigen, forming PCAF-bound immune complexes. Subsequently, 50 μL of Protein A/G agarose bead slurry (pre-equilibrated with lysis buffer) was added to each tube and incubated overnight at 4°C with rotation to precipitate the immune complexes. The samples were then centrifuged at 2500 g for 3 min at 4°C, and the supernatant was carefully removed. Next, the agarose beads were washed 3–4 times with pre-chilled IP lysis buffer, each time rotating at 4°C for 5–10 min, and the final wash supernatant was removed as completely as possible. After washing, 2× SDS loading buffer was added to the beads, followed by boiling for 5–10 min. After a brief centrifugation, the supernatant (IP sample) was transferred to a new EP tube. The IP samples and Input controls were then subjected to SDS-PAGE electrophoresis. After transfer, immunoblotting was performed using anti-Nrf2 antibody (Cell Signaling Technology, USA, #12721), anti-PCAF antibody (Cell Signaling Technology, USA, #3378), and anti-GAPDH antibody (Cell Signaling Technology, USA, #2118). Finally, the expression levels of Nrf2, PCAF, and GAPDH were detected using a gel imaging system (Bio-Rad ChemiDoc Touch Imaging System).
Transcriptome sequencing analysis
Following total RNA extraction, qualified samples were processed for library construction. Post-library preparation, initial quantification via Qubit 3.0 fluorometry confirmed concentrations ≥1 ng/μL. Insert fragment size distribution was validated using the Qsep400 bioanalyzer prior to PE150 sequencing on Illumina NovaSeq6000. Raw sequencing reads underwent quality filtering (generating Clean Data) and reference genome alignment (yielding Mapped Data). Subsequent bioinformatic analyses included: library quality assessment, structural variant calling, differential expression profiling, gene functional annotation, and pathway enrichment.
Statistical analysis
Experimental results were presented as mean ± standard deviation (SD) and the statistical analysis was performed with Statistical Product and Service Solutions 22.0 software. Analysis of variance (ANOVA) was used for multiple comparisons and least significant difference t test (LSD) for post hoc tests. A P value less than 0.05 was considered statistically significant.
Results
PCAF downregulation attenuates AngII-induced VSMCs senescence
VSMCs were stratified into four experimental cohorts: Control, AngII-induced senescence (AngII), AngII + control adenovirus (AngII + Ad-GFP), and AngII + PCAF-targeting adenovirus (AngII + Ad-PCAF RNAi). Initial immunoblot analysis revealed PCAF upregulation in AngII-stimulated VSMCs versus controls (P < 0.05, Figure 1A-B). No significant PCAF expression alteration occurred in AngII + Ad-GFP relative to AngII alone (P > 0.05), whereas Ad-PCAF RNAi transfection substantially suppressed PCAF levels compared to Ad-GFP controls (P < 0.05).
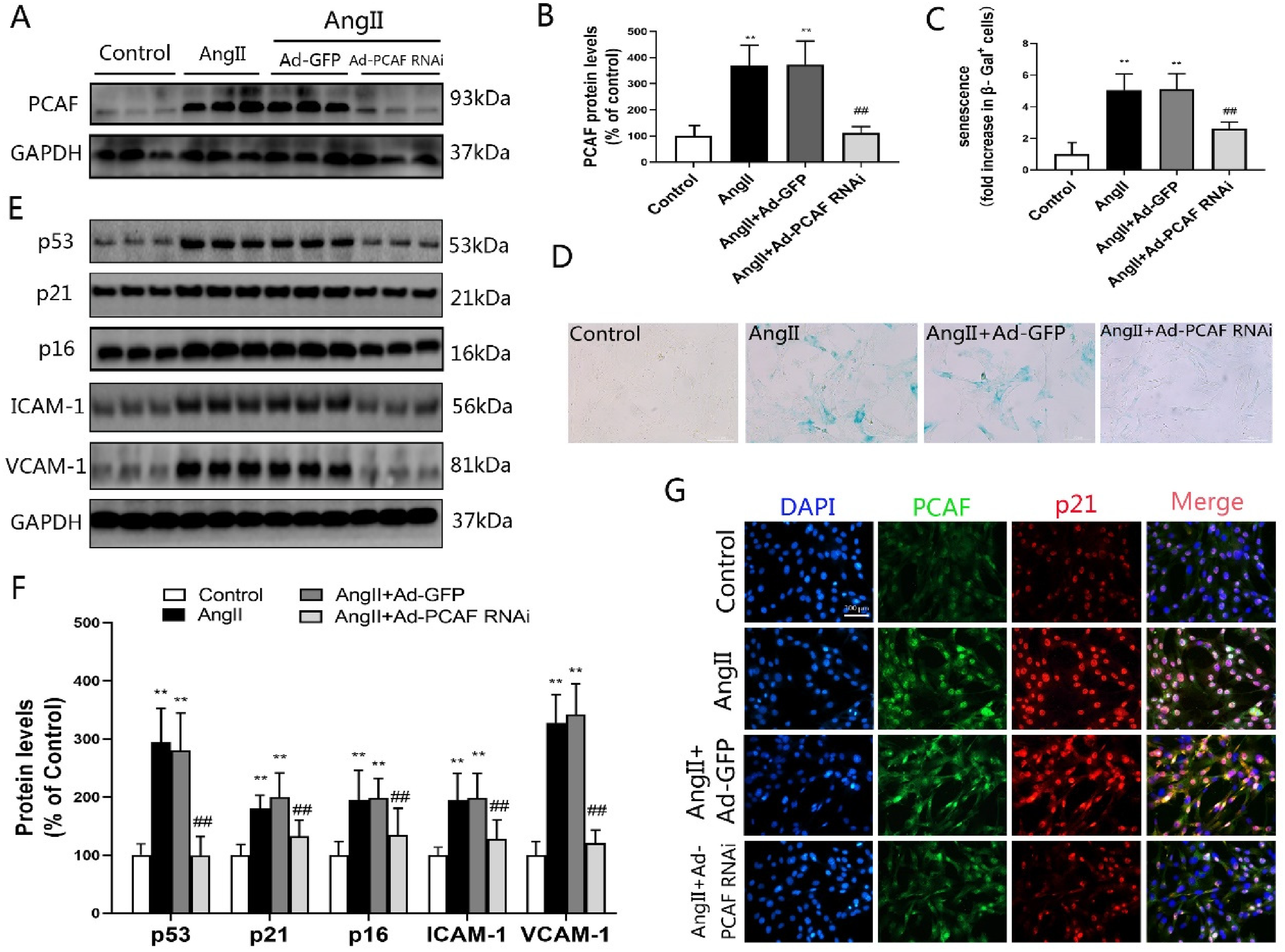
PCAF downregulation attenuates AngII-induced VSMCs senescence. (A-B) PCAF expression in VSMCs assessed by Western blotting (n=6). (C-D) VSMCs senescence measured via β-galactosidase activity (scale bar=100 μm, 400×; n=5). (E-F) Protein levels of p53, p21, p16, ICAM-1, and VCAM-1 in VSMCs by Western blot (n=6). (G) Immunofluorescence co-localization of PCAF, p21 in VSMCs (scale bar=100 μm, 400×; n=5). **P<0.01 vs. the control group; ##P<0.01 vs. the AngII induction with Ad-GFP group (AngII+Ad-GFP group).
Senescence phenotypes were assessed through β-galactosidase activity, immunoblotting and immunofluorescence. β-Galactosidase staining demonstrated enhanced activity in AngII and AngII + Ad-GFP groups relative to controls (P < 0.05, Figure 1C-D). Conversely, Ad-PCAF RNAi treatment significantly reduced enzymatic activity versus Ad-GFP counterparts (P < 0.05). Western blotting confirmed coordinated upregulation of senescence markers (p53, p21, p16) and adhesion molecules (ICAM-1, VCAM-1) in AngII and AngII + Ad-GFP groups (P < 0.05, Figure 1E-F). This effect was reversed in Ad-PCAF RNAi-treated cells, showing marked downregulation of all targets.
Immunofluorescence analysis corroborated AngII-mediated induction of both PCAF and p21 (Figure 1G). Critically, PCAF knockdown via Ad-PCAF RNAi substantially attenuated p21 expression. Collectively, these data demonstrate that PCAF suppression inhibits AngII-driven VSMCs senescence.
RNA-seq analysis of VSMCs after PCAF downregulation
The study indicates that vascular aging generally accompanies biological changes in vascular smooth muscle cells such as oxidative stress and cell cycle. In order to elucidate the impact of PCAF on these processes, we extracted total RNA for transcriptome RNA-seq analysis. The experimental process is shown in Figure 2A. Unsupervised hierarchical clustering analysis clearly demonstrates a significant separation between the RNA data of VSMCs samples infected with Ad-PCAF RNAi and control samples, indicating a marked shift in the transcriptome of PCAF-knockdown VSMCs (Figure 2B). Gene ontology (GO) enrichment analysis of the RNA-seq data reveals significant enrichment in biological processes such as oxidative stress and cell cycle. Among the differentially expressed genes (DEGs), knocking down the PCAF gene in VSMCs not only induces downregulation of aging-related genes, but also significantly reduces levels of oxidative stress (Figure 2C–D).
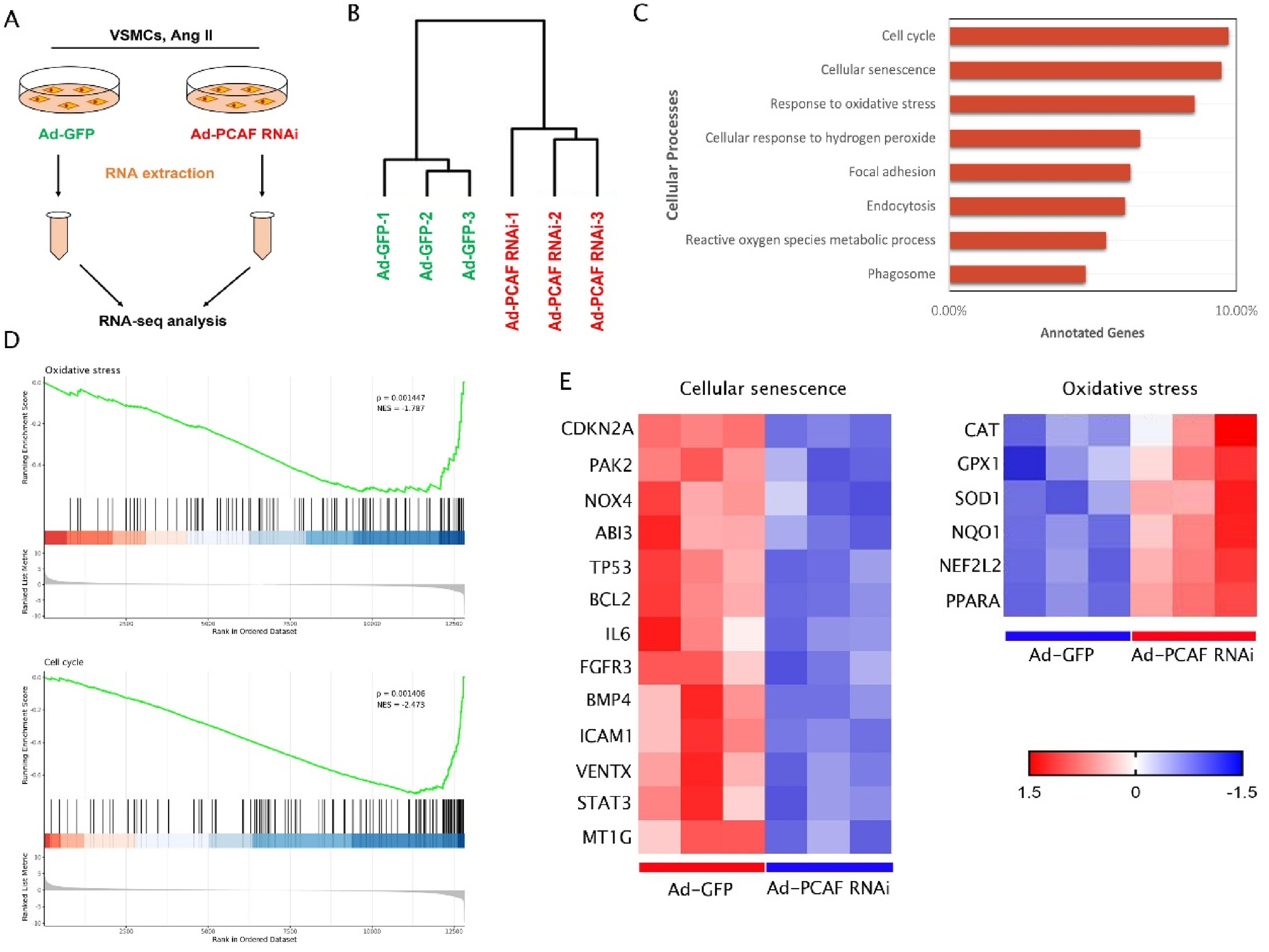
RNA-seq analysis of VSMCs in AngII induced control and PCAF knockdown groups. (A) Flow chart of RNA-seq detection and analysis experiment. (B) Hierarchical clustering image of the distribution and relationship between VSMCs samples infected with Ad-PCAF RNAi and control samples. (C) Go (gene ontology) enrichment analysis of the impact of PCAF knockdown on cellular biological events based on RNA-seq data, and the biological events of PCAF knockdown group are shown in the figure. (D-E) Heatmap of mRNA expression of differentially expressed genes (DEGs) in the control group and PCAF downregulation group detected by RNA-seq.
PCAF suppression mitigates AngII-induced oxidative stress in VSMCs
Oxidative stress is a well-established driver of vascular aging. To investigate how PCAF downregulation modulates this stress in VSMCs, we assessed key oxidative stress markers: SOD, CAT, and ROS. AngII stimulation significantly compromised SOD and CAT activities (P < 0.05, Figure 3A-B) but markedly elevated ROS levels (P < 0.05, Figure 3C-D). Crucially, suppressing PCAF expression counteracted these AngII-induced effects: both SOD and CAT activities were significantly restored compared to the AngII + Ad-GFP group (P < 0.05), and ROS generation was substantially reduced (P < 0.05). These findings demonstrate that PCAF downregulation mitigates oxidative stress injury in VSMCs.
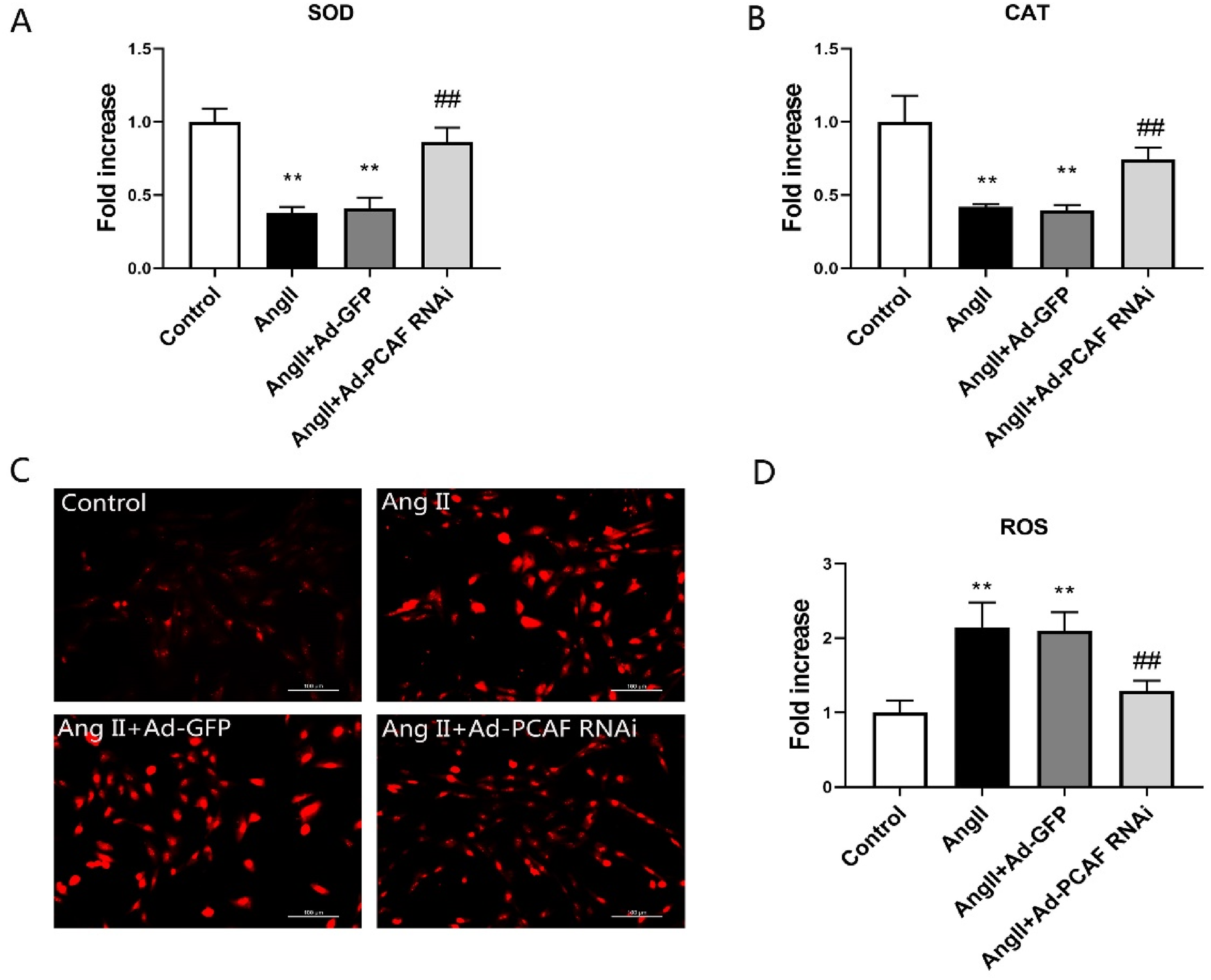
PCAF suppression mitigates AngII-induced oxidative stress in VSMCs. (A) SOD activity in VSMCs (n = 5). (B) CAT activity in VSMCs (n = 5). (C-D) ROS levels detected by dihydroethidium (DHE) staining in VSMCs (scale bar=100 μm, 400×; n = 5). **P < 0.01 vs. the control group; ##P < 0.01 vs. AngII + Ad-GFP group.
Nrf2-ARE signaling pathway was significantly enriched in VSMCs with PCAF downregulation
To further clarify the mechanism of PCAF in vascular aging, we performed pathway enrichment analysis of differentially expressed molecules from sample RNA-seq data and found that Nrf2-ARE signaling pathway was significantly enriched in VSMCs with PCAF downregulation (Figure 4).
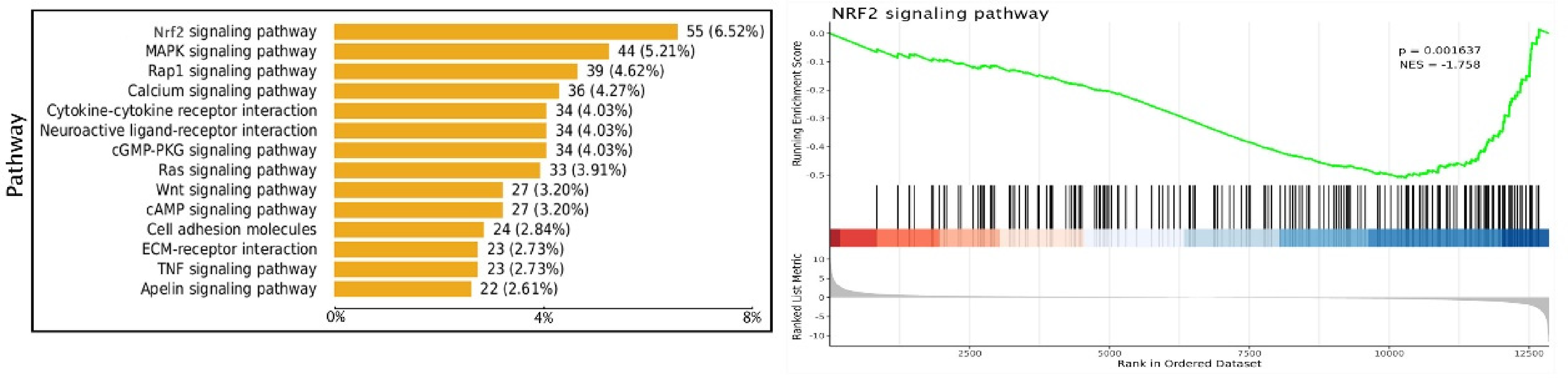
Pathway enrichment analysis.
PCAF downregulation activates the Nrf2-ARE pathway in AngII-stimulated VSMCs
To investigate the PCAF-Nrf2 relationship, co-immunoprecipitation (Co-IP) was performed. This revealed an interaction between PCAF and Nrf2 in AngII-stimulated VSMCs, which intensified following PCAF knockdown (Figure 5A). Subsequent Western blot analysis demonstrated that AngII induction significantly decreased both total and nuclear Nrf2 protein levels compared to the Control group (Figure 5B, C; P < 0.05). Conversely, PCAF downregulation reversed this effect, elevating Nrf2 expression in both compartments relative to the AngII + Ad-GFP group (P < 0.05). Immunofluorescence staining corroborated these Nrf2 localization findings (Figure 5D).
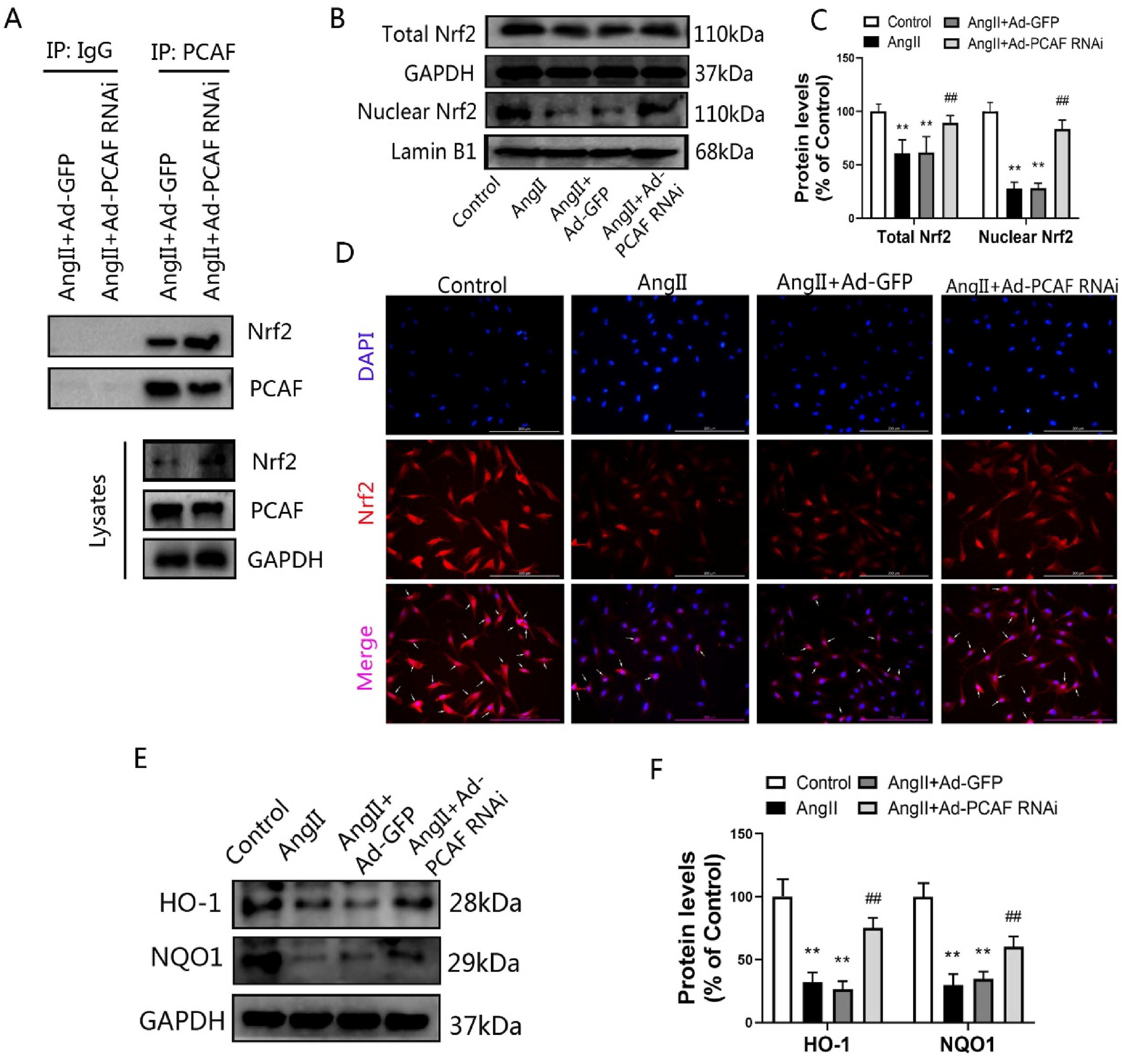
PCAF downregulation activates the Nrf2-ARE pathway in AngII-stimulated VSMCs. (A) PCAF-Nrf2 interaction in VSMCs by co-immunoprecipitation (n=3). (B-C) Total and nuclear Nrf2 expression in VSMCs by Western blot (n=5). (D) Subcellular localization of Nrf2 and nuclei via immunofluorescence (scale bar=200 μm, 400×; n=5). (E-F) HO-1 and NQO1 protein levels in VSMCs by Western blot (n=5). **P<0.01 vs. the control group; ##P<0.01 vs. AngII+Ad-GFP group.
Furthermore, assessment of key Nrf2 downstream effectors, HO-1 and NQO1, showed their expression was markedly suppressed by AngII versus Control (Figure 5E, F; P < 0.05). Similar to Nrf2, PCAF knockdown counteracted AngII's inhibition, significantly increasing HO-1 and NQO1 levels compared to AngII + Ad-GFP (P < 0.05). Collectively, these data indicate that PCAF downregulation enhances Nrf2-ARE signaling pathway activity.
Nrf2 silencing abrogates the protective effects of PCAF downregulation against oxidative stress and senescence in VSMCs
To determine whether Nrf2 activation mediates the protective effect of PCAF downregulation on VSMCs senescence, Nrf2 siRNA was employed. Successful Nrf2 silencing, confirmed by significantly reduced expression of Nrf2 and its downstream targets HO-1 and NQO1 (Figure S1), enabled subsequent analysis. Compared to the Ad-GFP group, PCAF knockdown (AngII + Ad-PCAF RNAi) significantly elevated SOD and CAT levels while reducing ROS (Figure 6A-D). However, Nrf2 silencing reversed these effects: SOD and CAT levels became significantly lower, and ROS levels significantly higher, than in the AngII + Ad-PCAF RNAi group (P < 0.05). This indicates that Nrf2 is essential for the antioxidant benefits conferred by PCAF downregulation. Similarly, assessment of cellular senescence revealed reduced senescence in the PCAF knockdown group versus Ad-GFP controls (P < 0.05) (Figure 6E-F). Critically, this protective effect was abolished upon Nrf2 silencing, as senescence significantly increased in the AngII + Ad-PCAF RNAi + Nrf2 siRNA group compared to AngII + Ad-PCAF RNAi alone (P < 0.05).
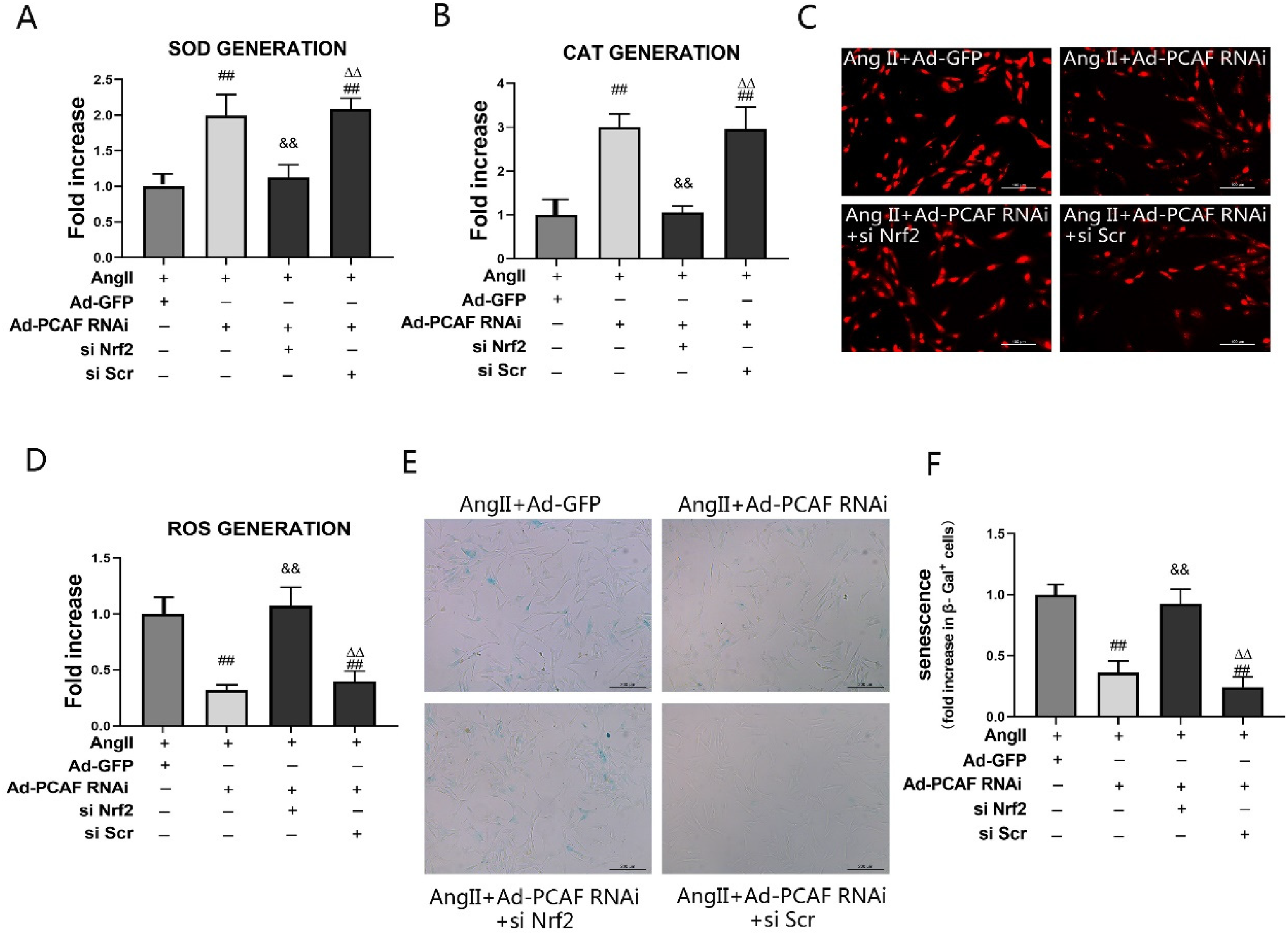
Nrf2 silencing reverses the effects of downregulation of PCAF on attenuating oxidative stress damage and senescence in VSMCs. (A) SOD activity in VSMCs (n=5). (B) CAT activity in VSMCs (n=5). (C-D) ROS levels in VSMCs (scale bar=100 μm, 400×; n=5). (E-F) β-gal activity in VSMCs (scale bar=200 μm, 200×; n=5). ##P<0.01 vs. AngII+Ad-GFP group; &&P<0.01 vs. AngII+Ad-PCAF RNAi group; △△P<0.01 vs. AngII+Ad-PCAF RNAi+si Nrf2 group.
Discussion
In recent years, cardiovascular and cerebrovascular diseases have become the leading cause of human death.23,24 With the deepening of research on cardiovascular and cerebrovascular diseases, vascular aging has been recognized as one of the important risk factors for adverse cardiovascular and cerebrovascular events. 25 Vascular aging manifests as structural and functional alterations driven primarily by vascular cell senescence.26,27 In this study, we demonstrated that the important role of PCAF in the senescence of VSMCs for the first time. Our results showed that PCAF expression was significantly increased in AngII-induced senescent VSMCs, while downregulation of PCAF could effectively inhibit AngII-induced senescence in VSMCs. Further molecular studies demonstrated that PCAF downregulation counteracts VSMCs senescence, an effect mediated by the attenuation of oxidative stress damage via activation of the Nrf2-ARE signaling pathway. However, the above conclusion was based on VSMCs of male SD rats, and whether this conclusion was applicable to the female rats or human cell systems remains to be further elucidated, which is also a direction for our future research.
Vascular aging was a major risk factor for cardiovascular disease. Enhanced oxidative stress and aggravated inflammatory response were two important mechanisms of vascular aging. The latest study showed 28 that garcinol, a natural inhibitor of PCAF, could reduce the production of ROS in human VSMCs induced by high glucose. It is well known that the production of a large amount of ROS was an important inducer of senescence in VSMCs. In addition, we reported that PCAF plays an important role in regulating the lipopolysaccharide-induced inflammatory response of VSMCs. 15 Downregulation of PCAF in VSMCs could significantly reduce the lipopolysaccharide-induced inflammatory response and inhibit the proliferation and migration of VSMCs. Further studies have shown that this is related to the decreased activity of NF-κB signaling pathway in VSMCs after downregulation of PCAF. Although the previous studies suggested that PCAF might have some kind of regulatory effect on VSMCs senescence, this study was the first to directly demonstrate that downregulation of PCAF could alleviate the senescence of VSMCs in vitro.
Appropriate concentration of AngII is very necessary to maintain the normal physiological function of the cardiovascular system, but the accumulation of too much AngII in the body could promote the occurrence and progression of various cardiovascular diseases. In recent years, a large number of studies have shown that AngII could induce senescence of various types of vascular cells, and more and more researchers used AngII as an inducer of vascular cell senescence.29–31 The senescent cells present a series of specific manifestations, among which the enhanced β-galactosidase activity is one of the most prominent features.32,33 Therefore, the detection of β-galactosidase activity has become one of the most commonly used methods to identify cellular senescence. 34 In addition, senescent cells also have an active secretory function, a phenomenon known as SASP. 35 The main components of SASP include inflammatory cytokines, chemokines, adhesion molecules, proteases, and so on, but it is worth noting that different types of cells often have different main components of SASP.36,37 In this study, our detection of SASP after VSMCs aging mainly includes adhesion molecules VCAM-1 and ICAM-1. In addition, studies have shown that the expression of related proteins (p53, p21, p16) involved in the regulation of senescent cell cycle arrest could also effectively reflect the status of cell senescence.38–40 Similar to previous studies, our description of vascular cells senescence also mainly includes β-galactosidase activity, SASP and cell cycle arrest regulation-related proteins. Our results showed that the expression of PCAF was significantly increased after VSMCs senescence induced by AngII. At the same time, the activity of β-galactosidase and the expression of adhesion molecules VCAM-1 and ICAM-1, as well as the cell cycle arrest regulation related proteins p53, p21 and p16 were also all significantly increased. However, after downregulation of PCAF, the expression of the above mentioned senescence related molecules in VSMCs were all weakened. These results confirmed that PCAF is involved in the aging process of VSMCs, and downregulation of PCAF has the effect of inhibiting the aging of VSMCs.
The mechanism of aging is relatively complex and has not been fully clarified so far, but there are many theories about aging, such as neuroendocrine theory, genetic control theory, somatic mutation theory, immune theory, and oxidative stress theory.41,42 Among them, the oxidative stress theory is the one most recognized. 43 The theory of oxidative stress believes that there are oxidative and antioxidant systems in the body. Under normal circumstances, the oxidative and antioxidant systems are in a dynamic balance; but when aging occurs, the body's antioxidant capacity is significantly weakened. This dynamic balance is broken and biased to oxidation. At the same time, with the occurrence of aging, the production of ROS and other free radicals increases, and due to the weakened antioxidant capacity of the body itself, the scavenging speed of ROS and other free radicals is slowed down, resulting in the accumulation of ROS and other free radicals in the body, causing oxidative stress damage, and finally, the continuing oxidative stress damage will promote the occurrence and progression of aging.44–46 Therefore, attenuating oxidative stress damage in vascular cells plays a major role in improving vascular aging. Here we found that the oxidative stress related markers were regulated by PCAF, and downregulation of PCAF could significantly alleviate oxidative stress damage in VSMCs.
The Nrf2-ARE signaling pathway constitutes the core antioxidant defense mechanism mitigating oxidative stress damage.47,48 As the central transcription factor within this system, Nrf2 governs the expression of key antioxidant enzymes like HO-1 and NQO1. Under basal conditions, Keap1 typically sequesters Nrf2 in the cytoplasm; however, oxidative stress triggers Nrf2 dissociation, nuclear translocation, binding to antioxidant response elements (ARE), and subsequent upregulation of protective genes (e.g., SOD, CAT). This cascade reduces oxidative damage and delays cellular senescence.49,50 Crucially, our study identifies PCAF as a novel interactor with Nrf2. We demonstrate that PCAF downregulation directly activates Nrf2-ARE-driven antioxidant defenses. In senescent VSMCs, elevated PCAF expression correlated with diminished total and nuclear Nrf2 levels, heightened ROS, and decreased SOD/CAT activity. Strikingly, PCAF knockdown reversed these phenotypes: enhancing Nrf2 expression and nuclear accumulation while reducing ROS and elevating SOD/CAT levels. Furthermore, the functional dependence on Nrf2 was unequivocally established: Nrf2 siRNA transfection abolished both the anti-senescence and antioxidant benefits conferred by PCAF downregulation. Collectively, these findings indicate that PCAF regulates the Nrf2 pathway, and its downregulation attenuates VSMCs senescence primarily by activating Nrf2-ARE signaling to combat oxidative stress.
Conclusion
In conclusion, our results showed that PCAF expression is enhanced in senescent VSMCs induced by Ang II, and downregulation of PCAF could alleviate oxidative stress injury by activating the Nrf2-ARE signaling pathway, thereby ultimately inhibit the senescence of VSMCs. Therefore, PCAF may be considered as a promising therapeutic target for vascular aging.
Supplemental Material
sj-docx-1-chm-10.1177_13860291261456146 - Supplemental material for Downregulation of PCAF inhibits vascular smooth muscle cells senescence by reducing oxidative stress injury via activating the Nrf2/ARE pathway
Supplemental material, sj-docx-1-chm-10.1177_13860291261456146 for Downregulation of PCAF inhibits vascular smooth muscle cells senescence by reducing oxidative stress injury via activating the Nrf2/ARE pathway by Liqiang Qiu, Wenjing Li, Xiaoxiong Liu, Zhigang Che, Yi Liu, Changjiang Zhang and Changwu Xu in Clinical Hemorheology and Microcirculation
Footnotes
Acknowledgments
This work was supported by Grant from Natural Science Foundation of Hubei Province (2022CFB151) and the Fundamental Research Funds for Central Universities of the Wuhan University (2042023kf0029).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Fundamental Research Funds for Central Universities of the Wuhan University, Natural Science Foundation of Hubei Province, (grant number 2042023kf0029, 2022CFB151).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.