
Abstract
Background
Lewy body disease (LBD) is a neurodegenerative disease characterized by Lewy bodies, and it clinically presents dementia with Lewy bodies (DLB) and Parkinson's disease (PD). Alzheimer's disease (AD) pathologies frequently coexist with LBD, complicating the clinical manifestation.
Objective
We evaluated the impact of AD pathologies, including amyloid-β and tau depositions, on cognitive dysfunction and glucose metabolism in LBD using multiple positron emission tomography scans.
Methods
Our study cohort consisted of 14 patients diagnosed with LBD, including five from the PD spectrum and nine from the DLB spectrum. In addition, 12 amyloid-negative cognitively healthy controls (HCs) and 13 amyloid-positive AD-spectrum patients were included. We subsequently explored the influence of amyloid and tau deposition on cognitive dysfunction and glucose metabolism among the LBD patients.
Results
In the LBD group, 44.4% of the DLB patients were amyloid-positive, and all PD patients were amyloid-negative. While tau accumulation was lower than in AD and similar to HCs at the group level, tau accumulation in the AD signature region was correlated with cognitive dysfunction. Among the changes in glucose metabolism, the cingulate island sign (CIS) index was elevated compared to AD. However, as cognitive impairment progressed, the CIS index decreased, reflecting reduced metabolism in the posterior cingulate gyrus, which was closely associated with tau accumulation in the same region.
Conclusions
Our findings indicate that AD pathologies, and particularly tau accumulation, significantly impact both cognitive dysfunction and glucose metabolism in LBD. This underscores the importance of addressing AD-related changes in the clinical management of LBD patients.
Keywords
Introduction
Lewy body disease (LBD) is a clinico-pathological spectrum characterized by the neuropathologic accumulation of alpha-synuclein in the central nervous system, and it is broadly classified into Parkinson's disease (PD), PD with dementia (PDD), and/or dementia with Lewy bodies (DLB), depending on their clinical course. In LBD, Lewy bodies are thought to be widely distributed not only in the central nervous system but also in the peripheral nervous system, exhibiting a vast clinical spectrum that encompasses motor, cognitive, and autonomic symptoms.1,2 In addition, Alzheimer's disease (AD) pathologies such as amyloid-β and tau depositions are often associated with LBD.3,4 Within LBD, the incidence of amyloid-β pathology is higher in DLB, although amyloid deposition is also seen in PD.5,6 It has been documented that amyloid-β accumulation in the cortex shortens the duration from the onset of motor symptoms to the development of dementia.7,8 Similar to synuclein accumulation in the cerebral cortex, tau pathology is believed to be linked to cognitive deficits in LBD.5,9 Thus, the clinical trajectory of LBD is frequently compounded by the effects of concurrent AD pathology, alongside the multifaceted nature of its pathogenic evolution. With the integration of disease-modifying therapies, such as anti-amyloid-β protein agents, into clinical practice, evaluation and comprehension of the AD pathology associated with LBD has become imperative. This understanding is crucial both for identifying indications for the use of anti-amyloid-β protein drugs in treating LBD and for the development of disease-modifying therapies aimed at targeting alpha-synuclein.
Recent developments in AD core biomarkers, including positron emission tomography (PET) biomarkers, have made it possible to classify an individual's brain pathology based on the amyloid-β deposition, pathologic tau, and the neurodegeneration (ATN) system published as the research framework in 2018. 10 On the other hand, the presence of AD pathology in LBD makes clinical differentiation difficult. Verification by amyloid PET imaging confirmed that patients with LBD exhibiting positive amyloid PET results demonstrated a pattern of cerebral atrophy akin to that observed in patients with AD. 11 Validation using tau PET has reported increased accumulation in LBD, which is useful for differentiating AD from LBD in the dementia phase. 12 However, its usefulness for differentiating AD from LBD in the early stages of the disease, including the mild cognitive impairment (MCI) phase, requires additional validation. Furthermore, 18F-fluorodeoxyglucose (FDG)-PET, acting as a surrogate marker of neurodegeneration, has been reported to show that the cingulate island sign (CIS) is useful for differentiating AD. CIS differentiates DLB from AD with 81% accuracy. 13 However, it is known that CIS is affected by AD pathology, and one report has shown that the degree of CIS is related to the Braak NFT stage, suggesting that CIS may be a biomarker that measures the relative contribution of the background pathology to dementia.14,15 Most of these reports, however, have been evaluated using a single modality, with few having used multiple modalities to examine how AD pathology in LBD is involved in cognitive dysfunction and neurodegeneration, as represented by glucose metabolism.
Therefore, the aim of this study was to clarify the impact of co-existing AD-pathologies on cognitive decline and glucose metabolism by performing multiple PET scans according to the ATN frameworks in cognitively healthy controls (HCs), AD patients as disease control, and LBD patients.
Methods
Participants
This study follows a prospective design. Fourteen patients with LBD, including five PD spectrum and nine DLB spectrum, participated in the present study. Clinical diagnoses were also based on the criteria of the Queen Square Brain Bank, Emre's criteria of possible/probable PD with dementia (PDD), and McKeith's criteria of possible/probable DLB.16–18 All participants underwent neuropsychological batteries, including Mini–Mental State Examination (MMSE), to assess their cognitive performance. Cases with MMSE scores of 23 or below were classified as demented, and those with scores of 24 or above as not demented. Among the not-demented cases, the presence or absence of mild cognitive impairment (MCI) was determined based on Petersen's diagnostic criteria. 19 Motor disability was evaluated using the Movement Disorder Society-Sponsored Revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS) Part III. Ultimately, 3 PD, 2 PDD, 5 MCI-Lewy bodies (LB), and 4 DLB composed the LBD group. Moreover, cases that met McKeith's criteria for possible or probable DLB also fulfilled the possible or probable DLB classifications according to the revised 2017 DLB diagnostic criteria.17,20 Incidentally, among the LBD patients, dopamine transporter imaging, 123Iodine-metaiodobenzylguanidine myocardial scintigraphy (MIBG), and polysomnography were not performed in all cases, but only in a subset of patients.
From the same cohort, 13 AD-spectrum individuals and 12 HCs were included in the study. All participants underwent PET scans as outlined below, with amyloid-negative cognitively healthy individuals serving as HCs, and individuals with amyloid-positive preclinical AD, MCI, and AD dementia patients comprising the AD-spectrum group. The AD spectrum consisted of three preclinical AD, three subjects with MCI, and seven subjects with AD dementia. None of the HCs exhibited any neurological signs, and they had no history of any other neurological or psychiatric disorders. Clinical diagnoses of AD and MCI were made under the National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association and Petersen's criteria, respectively.19,21 The classification of amyloid-negative or amyloid-positive was based on the amyloid PET examination, as described below. The inclusion flow chart is described in Supplemental Figure 1.
All participants were recruited from affiliated hospitals between July 2012 and March 2018. Written informed consent was obtained from all subjects and from spouses or other close family members if the participants were cognitively impaired. This study was approved by the institutional review board of the National Institutes for Quantum Science and Technology (QST) in accordance with the ethical code of QST and the ethical guidelines for clinical studies of the Ministry of Health, Labor and Welfare in Japan, as well as the Declaration of Helsinki. The study was registered with UMIN Clinical Trials Registry (UMIN-CTR; number 000017978).
PET and magnetic resonance imaging (MRI) acquisition
We conducted triple PET scans on the same subjects using 11C-Pittsburgh Compound-B (PiB) for amyloid, 11C-pyridinyl-butadienyl-benzothiazole 3 (PBB3) for tau, and 18F-FDG-PET for glucose metabolism, aligning with the ATN biomarker framework. The scan protocols were described in detail in our previous reports. 11 Briefly, PET images were acquired using a Siemens ECAT EXACT HR1 scanner (CTI PET Systems, Inc., Knoxville, TN). Dynamic scans were conducted for 70 min with 11C-PiB, 11C-PBB3, and for 60 min with 18F-FDG. The doses administered for each tracer were as follows: 11C-PiB, (457 ± 91 MBq), 11C-PBB3 (430 ± 64 MBq) and 18F-FDG (184 ± 30 MBq). All PET images were reconstructed by the filtered back projection method (Hanning filter, cut-off frequency: 0.4 cycle/pixel). Attenuation and scatter corrections for each image were performed using a 10-min transmission scan with a 68Ge-68Ga line source and the single-scatter simulation method, respectively.
All MRI examinations were performed with a 3-Tesla magnetic resonance scanner (Signa HDx, GE Healthcare, WI, USA, or MAGNETOM Verio, Siemens Healthcare, Erlangen, Germany). Subjects were scanned with a 3D T1-weighted turbo-gradient echo sequence (repetition time/echo time range, 7/2.8 ms; field of view, 260 × 244 mm; matrix, 256 × 256; 170 contiguous axial slices of 1.0-mm thickness).
Data preprocessing
All images were preprocessed using PMOD software version 4.304 (PMOD Technologies Ltd, Zurich, Switzerland) and Statistical Parametric Mapping software (SPM12, Wellcome Department of Cognitive Neurology, London, UK), operating in the MATLAB software environment (version 9.12; MathWorks, Natick, MA, USA). At first, motion correction was applied to all PET images. Then, each PET image was coregistered to individual T1-weighted magnetic resonance images and anatomically normalized (MNI152 standard space, Montreal Neurological Institute, Montreal, QC, Canada) using Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra. 22 Parametric 11C-PiB, 11C-PBB3, and 18F-FDG PET images were generated by a voxel-based calculation of the standard uptake value ratio (SUVR) to the cerebellar cortex at 50–70 min, 30–50 min, and 30–60 min after the injections, respectively. 11
Subsequently, we established AD-signature regions of interests (ROIs) based on the Automated Anatomical Labeling template for each PET image as follows. 23 These ROIs included the frontal, temporal, and parietal cortices, precuneus, anterior striatum, and insular cortex for amyloid PET, and the entorhinal cortex, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal areas for tau PET.24,25 We set ROIs on the posterior cingulate gyrus (PCG) and precuneus plus cuneus (PpC) in the FDG-PET images to evaluate the CIS index. The CIS index was defined as the SUVR value of the PpC divided by the SUVR value of the PCG (Figure 1). CIS was visually evaluated by three independent observers. For the CIS index, we utilized the numerical values as continuous variables in our analysis without establishing a cut-off value. For amyloid PET, the centiloid scale was also evaluated, and a cut-off value of 15 points was used to determine whether amyloid is positive or negative.24,26
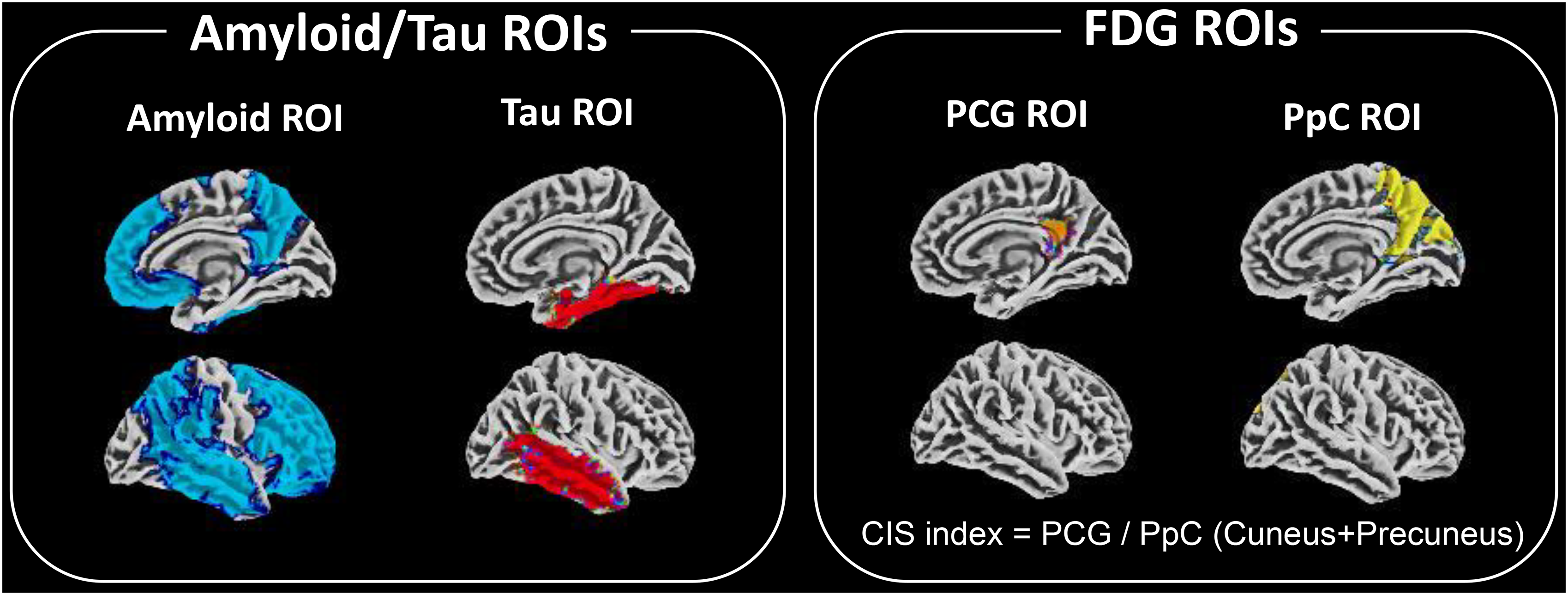
Regions of interest (ROIs) used in this image analysis.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 9.5.1 (GraphPad Software, Boston, Massachusetts, USA) and the SPM12 module. We conducted one-way analysis of variance to compare demographic data and SUVR values from ROI analyses among the three groups, and Bonferroni correction was used for multiple comparisons. To investigate the correlation between cognitive decline and SUVR parameters in the LBD group, Pearson's correlation analysis was conducted using MMSE, each SUVR value, and the CIS index.
Subsequently, we also conducted group comparisons and correlation analyses in a voxel-wise manner. Using the two-sample t-test model, we compared the AD and LBD groups with the HC group; the diseased groups were also stratified into dementia and non-dementia subgroups for separate analyses. Furthermore, to explore the impact of amyloid and tau, we used a correlation analysis model, treating the SUVR values of each component as covariates, to identify brain regions in amyloid/tau PET images that correlate with these SUVR values. Statistical comparisons were conducted at the voxel level (p < 0.05 (uncorrected), voxel > expected voxels per cluster).
Results
Demographic data
Table 1 shows the demographic data. The LBD group consisted of 14 patients with an average age of 76.9 ± 6.5 years and an MMSE score of 22.3 ± 5.0. This group consisted of five patients with PD spectrum and nine with DLB spectrum, with four being amyloid-positive and ten amyloid-negative. Notably, all four amyloid-positive cases were from the DLB spectrum, while no PD spectrum cases showed amyloid positivity. The AD group consisted of 13 patients aged 73.4 ± 7.2 years and an MMSE score of 22.1 ± 5.6. Within this group, six patients were categorized as non-dementia and seven as dementia. The HC group consisted of 12 individuals with an average age of 63.4 ± 13.5 years and an MMSE score of 28.9 ± 1.4. Significant age differences were found between the HC group and both the LBD and AD groups (p < 0.05). A significant difference was observed in sex between the AD and HC groups (p = 0.05). Additionally, there were notable differences in cognitive function, as measured by MMSE scores, between the HC group and both the LBD and AD groups (p < 0.05). However, there were no significant differences in age or MMSE scores in the LBD and AD groups.
Demographic features of patients with Lewy body disease, Alzheimer's disease, and healthy control group.
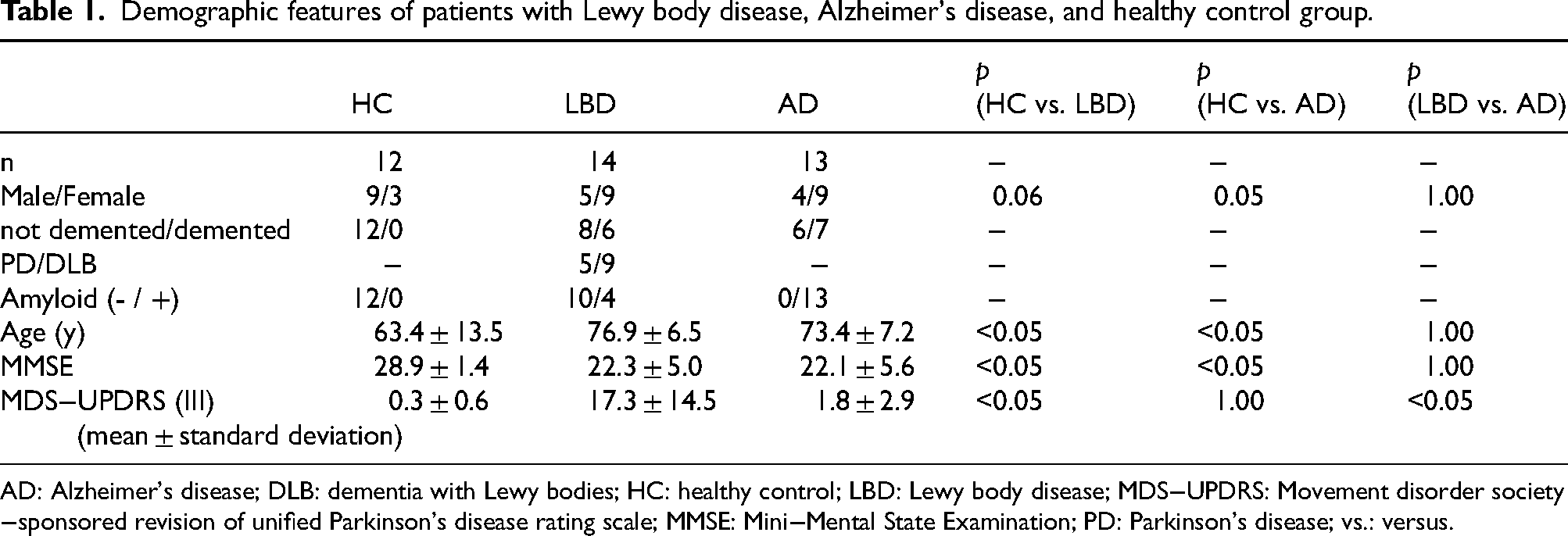
AD: Alzheimer's disease; DLB: dementia with Lewy bodies; HC: healthy control; LBD: Lewy body disease; MDS−UPDRS: Movement disorder society−sponsored revision of unified Parkinson's disease rating scale; MMSE: Mini−Mental State Examination; PD: Parkinson's disease; vs.: versus.
Representative images from each of the three PET modalities in each group
Representative images of each group are shown in Figure 2. As mentioned above, amyloid PET images were negative in HC cases, PD cases and PDD cases. Conversely, DLB cases showed an increase in accumulation in amyloid PET images, although lower than the level observed in AD cases. Tau PET imaging demonstrated increased accumulation in patients with PDD, DLB cases and AD cases compared to HC cases and PD cases. FDG-PET imaging demonstrated hypometabolism in the occipital lobe in a large proportion of patients diagnosed with LBD, including PD cases, PDD cases and DLB case. Additionally, some cases with LBD displayed CIS, characterized by preserved metabolism in PCG as compared to the occipital lobe upon visual assessment. In particular, in cases with amyloid and tau accumulation in the region including PCG, glucose hypometabolism was also observed in PCG, leading to the absence of CIS. The mean images of the respective groups are shown in Supplemental Figure 1. The mean images of the subgroups in the LBD group are also shown in Supplemental Figure 3. In the mean image of the total LBD group, tau accumulation appears to be almost non-existent, but in the mean images of each subgroup in the LBD group, tau accumulation is observed, and particularly in the DLB group.
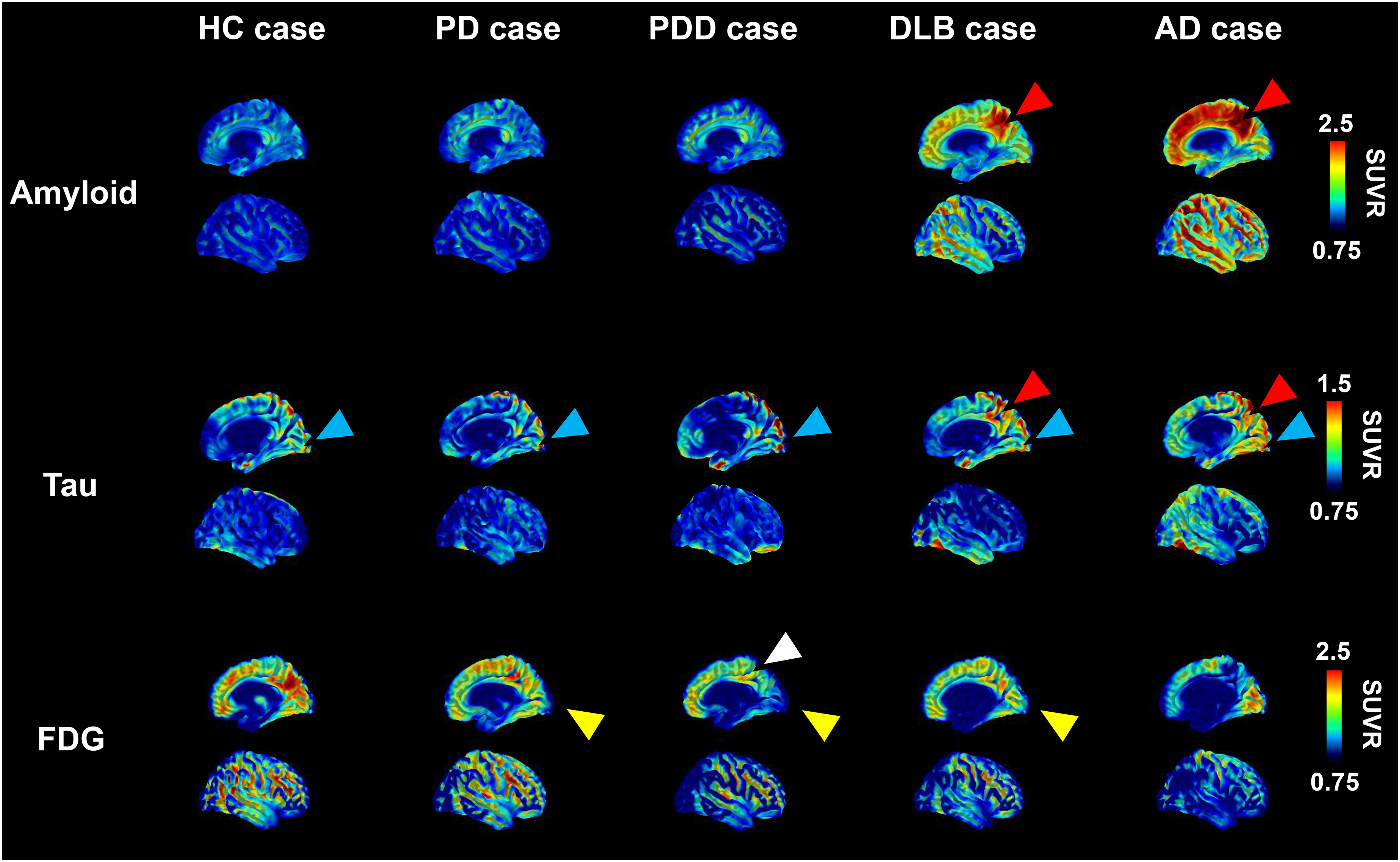
Representative images from each of the three positron emission tomography (PET) modalities in each group.
Each PET modality in AD and LBD with and without cognitive decline at the voxel level
Figure 3 shows the transitions of each PET modality in the AD and LBD groups, together with the severity of cognitive decline, as visualized by comparisons with the HC group. The non-demented LBD group includes PD and MCI-LB, and the non-demented AD group includes preclinical AD and MCI-AD. The demented LBD group includes PDD and DLB, and the demented AD group includes AD cases (Supplemental Figure 1). Analysis was performed by including variables that showed significant differences in the demographic data (age and sex) as covariates.
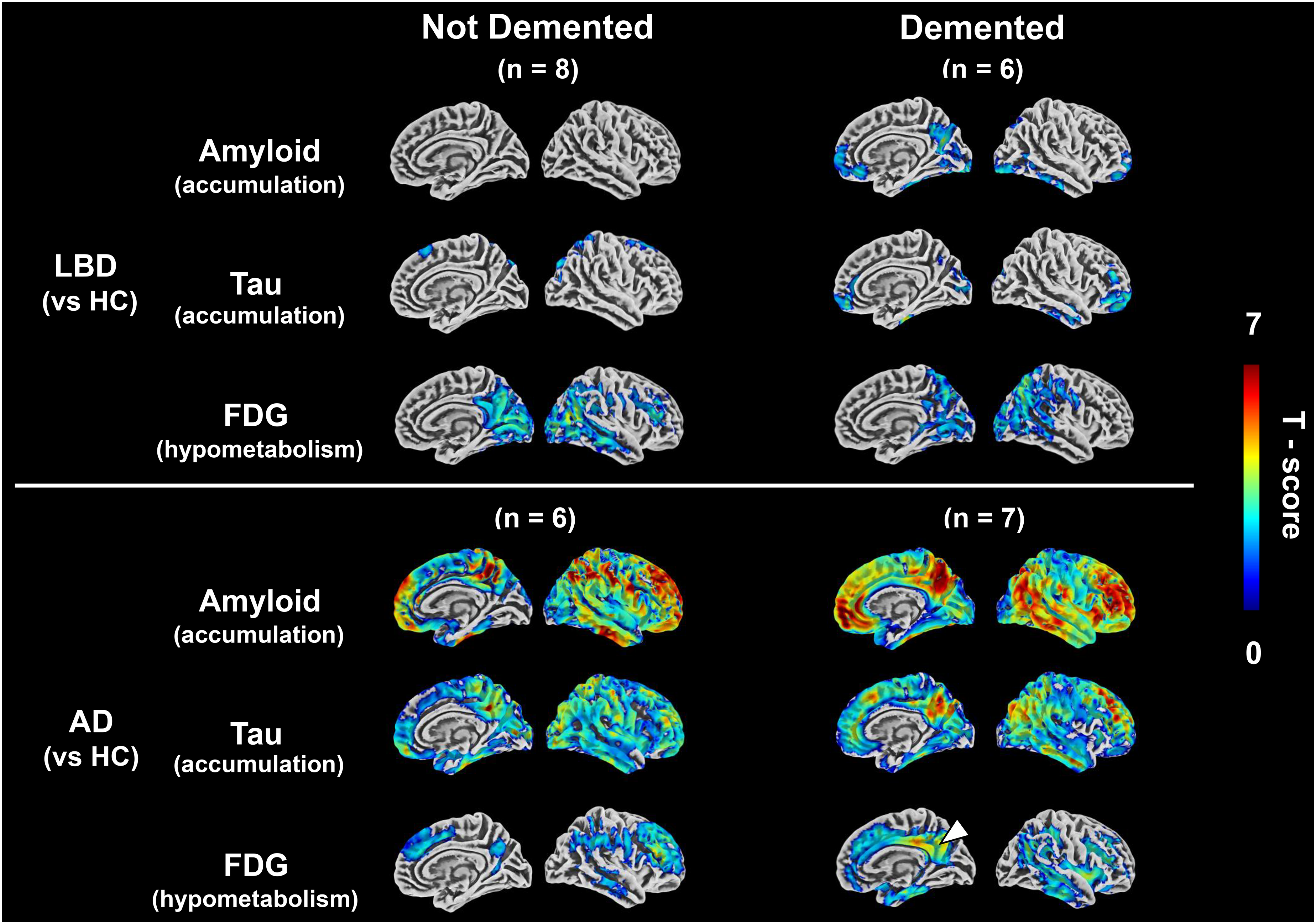
Changes of ATN positron emission tomography (PET) biomarkers along with cognitive decline.
Amyloid PET and tau PET highlight the areas of accumulation, while FDG-PET focuses on the areas of decreased glucose metabolism. In the AD group, high amyloid PET accumulation was consistently observed in the PCG and frontal lobes, as well as across the cerebral cortex of all disease stages. Tau PET accumulation, particularly in the dementia stage, increased predominantly in the regions extending from PCG to the lateral temporal-parietal and temporal lobes. FDG-PET revealed a pronounced decrease in PCG most notably in the dementia stage. In the LBD group, significant amyloid and tau PET accumulations were not evident in the non-dementia stage; however, in the dementia stage, accumulation began to manifest in similar regions as in the AD group, although to a lesser degree. Conversely, a hypometabolism in the occipital lobe was observed from the non-dementia stage, extending to the PCG and parietal lobes as cognitive dysfunction advanced. In this study, statistical analysis was performed at p < 0.05, but the results of analysis at p < 0.01 were also shown in Supplemental Figure 4. In addition, the results of a direct comparison between the AD and LBD groups are shown in Supplemental Figure 5.
ROI-based assessment
Using the ROIs shown in Figure 1, we quantified each PET image by calculating the corresponding SUVR values. In group comparisons (Figure 4, upper row), the AD group showed a significant increase in amyloid SUVR in the centiloid ROI and tau SUVR in the temporal meta-ROI compared to both the HC and LBD groups (p < 0.0001). The effect size, as measured by Eta squared, was 0.61 for amyloid SUVR in the centiloid ROI and 0.55 for tau SUVR in the temporal meta-ROI, respectively. The CIS index was significantly lower in the AD group compared with the LBD group (p < 0.001). However, it exhibited the smallest effect size among the PET modalities examined (Eta squared = 0.24). Furthermore, in a correlation analysis assessing cognitive impairment within the LBD group (Figure 4, lower row), no significant correlation was observed with amyloid SUVR in the centiloid ROI (p = 0.09, r = −0.47). Conversely, increased tau SUVR in the temporal meta-ROI and a reduced CIS index were significantly correlated with the degree of cognitive impairment (p < 0.005, r = −0.71, and p < 0.05, r = 0.61, respectively).
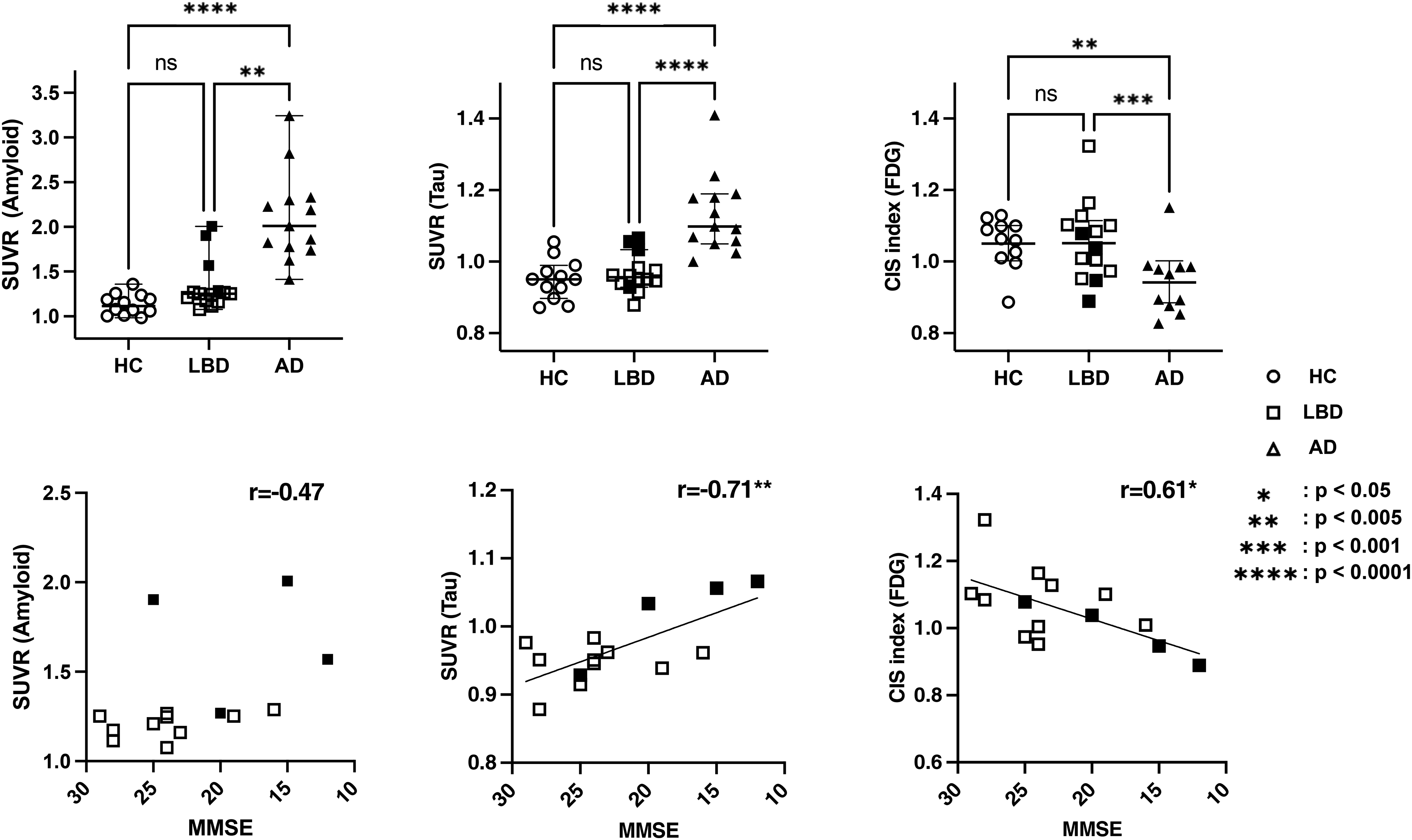
Region of interest (ROI)-based assessment of the ATN framework.
In Figure 4, the LBD group is indicated by white squares, while cases judged to be amyloid-positive based on the centiloid scale are shown as black squares. The centiloid scale was calculated using amyloid SUVR in the centiloid ROI used in this study, and the respective SUVR values, centiloid scales, and CIS indices for the LBD group are shown in Supplemental Table 1.
Multimodal assessments of CIS in LBD
In regions associated with CIS on FDG-PET imaging, SUVR in PCG was significantly correlated with cognitive impairment (p < 0.05, r = 0.57) (Figure 5A). Additionally, in whole brain analyses, the SUVR values of FDG-PET within PCG were found to correlate with tau PET accumulation in PCG (Figure 5B). Subsequent ROI analysis revealed that a reduction in FDG-PET accumulation was associated with an increase in tau PET accumulation within PCG (p < 0.05, r = −0.76) (Figure 5C). The accumulation of tau in PCG also demonstrated a correlation with cognitive impairment (p < 0.05, r = −0.62), with elevated SUVR observable from the early stages of cognitive decline (Figure 5D).
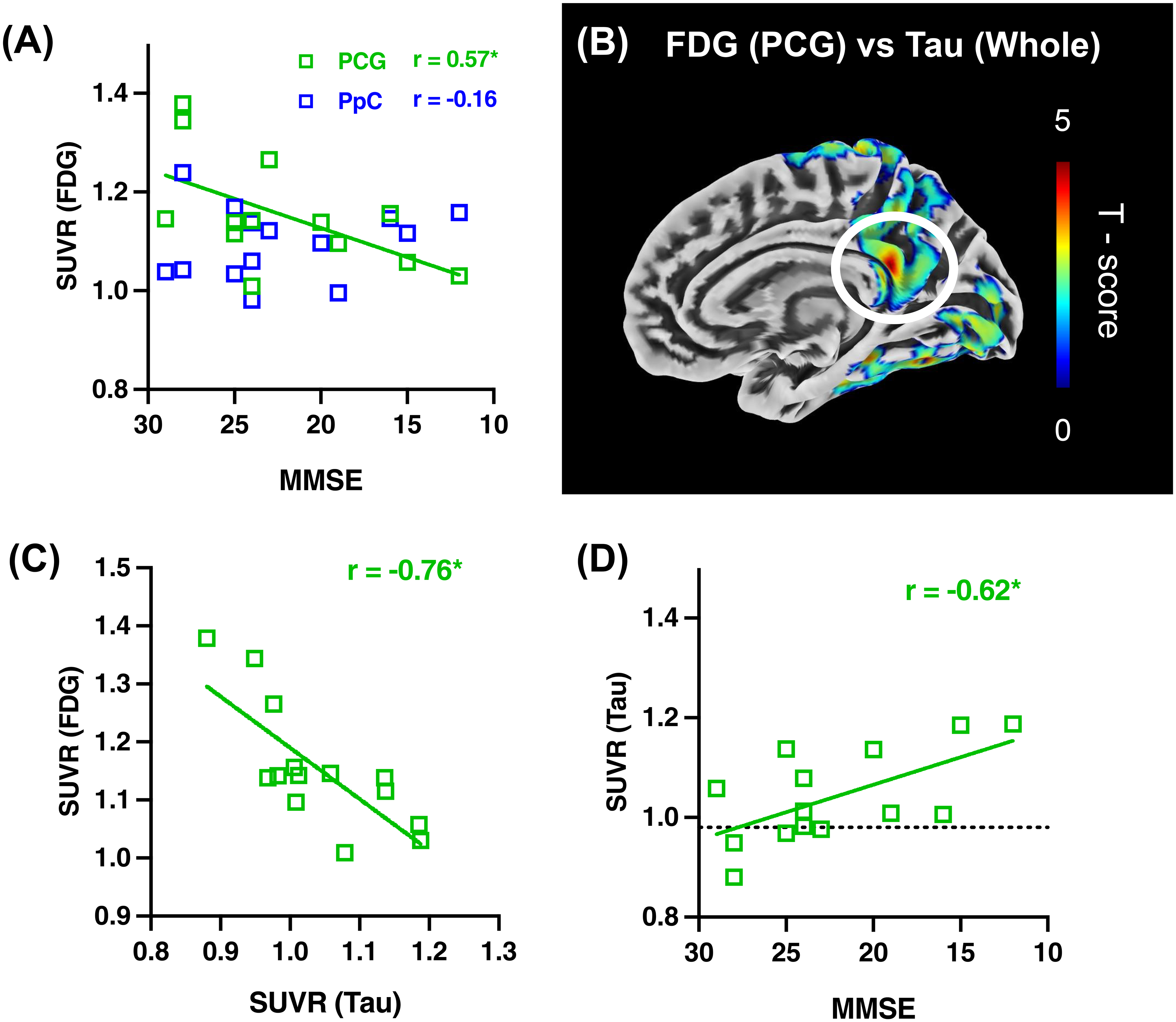
Multiple analyses of cingulate island sign (CIS) in Lewy body disease (LBD) group.
Discussion
We used multiple PET scans to evaluate the effects of amyloid and tau pathologies on cognitive impairment and glucose metabolism in LBD. Specifically, the tau accumulation associated with LBD impacts cognitive function and glucose metabolism, indicating that CIS, a pattern of altered glucose metabolism useful for differentiating between DLB and AD, is also influenced by the tau pathology.
The present study found no correlation between amyloid PET tracer accumulation and cognitive impairment in the LBD group, while DLB patients tended to show a higher rate of positive-amyloid PET scans compared to the PD/PDD patients. Pathological studies have demonstrated a higher rate of amyloid positivity in DLB compared to PDD. 27 Furthermore, previous PET imaging studies have shown that amyloid PET positivity is higher in DLB compared to PD/PDD, and is correlated with cognitive impairment.28–30 Another study demonstrated that PD patients without dementia show even lower amyloid PET positivity than HCs. 31 The role of amyloid and tau proteins in neurodegeneration among AD is posited to be that amyloid-β plaque accumulation promotes tau protein phosphorylation (amyloid cascade hypothesis), and that both contribute synergistically to synaptic dysfunction and neuronal cell death (dual-pathway hypothesis).32,33 Similarly, in LBD, amyloid lesions are believed to promote abnormal phosphorylation and accumulation of tau in neuronal cells. 34 It has also been demonstrated that amyloid lesions may promote the accumulation of synuclein pathology and neuronal damage, and their presence may exacerbate the development of tau-synuclein lesions and subsequently hasten the onset of cognitive impairment.7,8,35,36 Even in the absence of AD pathology, extensive neurodegeneration (white matter damage) due to Lewy body pathology is observed, although concomitant amyloid pathology contributes indirectly to some neurodegeneration via tau pathology. 37 Although there was no simple correlation between amyloid pathology and cognitive dysfunction, these results are consistent with previous studies and suggest that, in addition to Lewy body pathology alone causing neuropathy and consequent cognitive dysfunction, amyloid pathology may also be indirectly related to some of the neuropathies underlying cognitive dysfunction via tau pathology. This observation was further supported by additional cases scanned with a different PET scanner (Supplemental Figure 7), which showed increased tau PET accumulation in the PCG region among DLB cases, and particularly in those with amyloid positivity. Although these cases could not be included in the quantitative analysis due to different scanner specifications, they qualitatively reinforced our findings regarding the relationship between amyloid pathology and tau accumulation in DLB. However, as the small number of participants in our main analysis may have affected the results, we will need to be careful in interpreting the results of this study.
Indeed, the present study revealed that tau PET accumulation was also elevated in LBD and was correlated with cognitive impairment. A report from a meta-analysis on tau pathology in Lewy body dementia, including tau PET, cerebrospinal fluid levels, and pathological studies, demonstrated that cognitive function deteriorated with increasing tau burden. 38 Tau deposition, in conjunction with alpha-synuclein toxicity, induces cognitive dysfunction by the localized loss of synapses and neuronal cells.39–41 Similar to amyloid, the coexistence of tau and synuclein in vitro has been demonstrated to promote the polymerization of both proteins, indicating a synergistic interaction. 42 However, the fact that the present results showed no correlation between cognitive impairment and amyloid, but did show a correlation with tau, may suggest that tau plays a more significant role than amyloid-β in cognitive impairment in LBD with concomitant AD pathologies. Regarding the site of tau accumulation, some studies of tau PET using 18F-AV-1451 (also known as 18F-flortaucipir) have reported increased uptake in the inferolateral temporal and parietal/precuneus regions, similar to AD, while others have found higher accumulation in the temporoparietal and occipital cortices.12,41,42 In the present whole-brain analysis, tau accumulation was observed in the temporoparietal and occipital cortices, albeit to a lesser extent than in AD, suggesting that both the accumulation sites and its extent are consistent with those previously reported. In AD, regions with early tau accumulation are associated with neurological vulnerability. 43 While neuropathological vulnerability may influence the pattern of tau PET accumulation in LBD, the presence of α-synuclein pathology could also contribute to the differential accumulation observed between LBD and AD. The tau PET ligand 11C-PBB3 used in this study may bind to α-synuclein pathology, including Lewy bodies, at high tracer concentrations. However, it has been shown that at the low tracer concentrations of 11C-PBB3 used in human PET scans, while it may bind to α-synuclein pathology in some cases of multiple system atrophy (MSA) with high-density glial cytoplasmic inclusions, it does not detect α-synuclein in DLB. 44 In the present study, since the administered dose of 11C-PBB3 was low and no cases considered to be MSA were included, we believe that ligand binding to α-synuclein in tau PET did not occur.
In the LBD group, the CIS index correlated with cognitive dysfunction and decreased with progressive cognitive impairment. Alterations in the CIS index correlated with cognitive decline were predominantly influenced by the PCG metabolism, implying that tau deposition at the same locus may be involved. In autopsy pathology reports on CIS in DLB, it has been documented that longitudinal alterations in CIS were not associated with amyloid deposition but rather with lower neurofibrillary tangle Braak stages. 15 However, subsequent studies found no correlation with either amyloid or tau, leaving the issue unresolved. 45 It is well-established that PCG metabolism is diminished in AD from the early stages, which is hypothesized as resulting from tau accumulation in the parahippocampal region indirectly impacting PCG metabolism via the Papez circuit. 46 Conversely, the current study on LBDs indicates that tau deposition in PCG may directly influence alterations in CIS, while tau deposition around the parahippocampal region did not correlate with glucose hypometabolism in PCG. The mechanisms underlying PCG hypometabolism may differ between AD and LBD, with the presence of synuclein potentially accounting for these mechanistic discrepancies. As FDG-PET findings evolve with the progression of dementia, the timing for the assumption of the underlying pathology is also crucial. Previous reports on the pathology underlying CIS have primarily relied on post-mortem studies, with few investigations utilizing biomarkers.15,45 We posit that the application of amyloid and tau PET in this study enabled a temporally consistent examination of the underlying pathology of CIS.
Regarding MRI findings, the amyloid-positive LBD group showed a tendency for medial temporal lobe atrophy, as demonstrated in Supplemental Figure 6. This is considered to reflect the influence of AD pathology, as previously reported. 11 Characteristic MRI features of DLB, such as changes in the caudate nucleus volume, atrophy of the substantia innominata, relative preservation of the hippocampus and lateral temporal-parietal cortex compared to AD, and uniform brainstem atrophy distinguishing it from progressive supranuclear palsy and PD, have been reported in the literature.47–51 However, due to the limited number of DLB cases in our study, a detailed analysis was ruled out.
There are several limitations to this study. First, the HC group was younger than the LBD and AD groups. Additionally, a significant difference in sex distribution was observed between the AD and HC groups. However, in the whole-brain analysis using SPM, comparing results with and without the inclusion of age and sex as covariates revealed no substantial differences. Therefore, we consider the influence of these factors to be minimal in the context of this study. Second, the total number of participants was limited, resulting in an insufficient overall caseload. In particular, there were fewer amyloid-positive cases in the LBD group. In addition, although an analysis of the LBD group divided into subgroups was conducted, the small number of participants may have affected the results, so we need to be careful in interpreting the results of this study. Conversely, the significant impact of tau pathology on cognitive function and glucose metabolism, despite the low number of AD comorbidities, suggests that conditions like primary age-related tauopathy, which accumulate independently of amyloid, may also be involved in the range of the phenomena observed in LBD. 52 The limited sample size could certainly have influenced the results, and further validation with a larger cohort is essential in future studies. In addition, the synuclein biomarker was not measured in the present study. With the latest scientific and technological advances, the 2018 framework was updated to a new one in 2024. 53 In this latest recommendation, the biomarkers of alpha-synuclein pathology (S), as well as inflammation (I) and vascular brain injury (V), are now described in addition to the traditional ATN. Alpha-synuclein seed amplification assays (αSyn-SAA) in cerebrospinal fluid have been reported as the S marker. 54 αSyn-SAA in blood and αSyn-PET for LBD have also been reported. 55 Recently, there have been reports that suggested that the coexistence of synuclein pathology may influence clinical symptoms.56,57 Future multimodal imaging including synuclein PET is expected to provide us with a deeper understanding of the relationship among AD-related pathology, synuclein pathology and neurodegeneration. 58
Conclusion
The findings of this study imply that the presence of AD pathology, and particularly tau lesions, in LBD correlates with the advancement of cognitive dysfunction and further influences the pattern of glucose hypometabolism. However, as the number of cases was small, it will be necessary to conduct further studies with larger numbers of participants in the future. The implications of this research are that valuable insights will be gained toward the diagnosis and treatment strategies of LBD, and that they will also contribute to the development of disease-modifying therapies.
Supplemental Material
sj-docx-1-alz-10.1177_13872877251351220 - Supplemental material for Concurrent Alzheimer's disease pathologies in Lewy body diseases affect cognition and glucose metabolism in the posterior cingulate gyrus: A multimodal PET study
Supplemental material, sj-docx-1-alz-10.1177_13872877251351220 for Concurrent Alzheimer's disease pathologies in Lewy body diseases affect cognition and glucose metabolism in the posterior cingulate gyrus: A multimodal PET study by Kosei Nakamura, Kenji Tagai, Hitoshi Shinotoh, Shigeki Hirano, Soichiro Kitamura, Hironobu Endo, Keisuke Takahata, Yuhei Takado, Ming-Rong Zhang, Kazunori Kawamura, Osamu Onodera, Makoto Higuchi and Hitoshi Shimada in Journal of Alzheimer's Disease
Footnotes
Acknowledgments
The authors thank all patients, their caregivers, and volunteers for their participation in the present study, and clinical research coordinators, PET and MRI operators, radiochemists, and research ethics advisers at QST for their assistance with the current project. The authors also acknowledge the support of S. Moriguchi, T. Sasaki, H. Fujiwara, F. Kodaka, S. Furukawa, Y. Nakano, Y. Eguchi, K. Yamaoka, A. Isato, M. Maruyama, I. Kaneko, K. Suzuki, J. Ichikawa, S. Kawakami, Y. Toyota, M. Kurokawa, A. Kurose, Y. Iwasawa (no particular order). The authors acknowledge support for the recruitment of patients with cognitive decline by Y. Yoshiyama at the Inage Neurology and Memory Clinic, and by K. Kashiwado at Kashiwado Hospital.
ORCID iDs
Ethical considerations
This study was approved by the institutional review board of the National Institutes for Quantum Science and Technology (QST) in accordance with the ethical code of QST and the ethical guidelines for clinical studies of the Ministry of Health, Labor and Welfare in Japan, as well as the Declaration of Helsinki. The study was registered with UMIN Clinical Trials Registry (UMIN-CTR; number 000017978).
Consent to participate
Written informed consent was obtained from all subjects and from spouses or other close family members if the participants were cognitively impaired.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by AMED under Grant Nos. JP18dm0207018, JP19dm0207072, JP18dk0207026, JP19dk0207049 to Makoto Higuchi, and by JSPS KAKENHI Grant Nos. 26713031, 18K07543 and 23H02825 to Shigeki Hirano and Hitoshi Shimada, and by JST FOREST Program (Grant No. JPMJFR220R, Japan) to Hitoshi Shimada. The agencies had no further role in the study design, collection, analysis or interpretation of the data, writing of the report, or in the decision to submit the paper for publication.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.-R.Z., M.H., and H.S. hold a patent on compounds related to the present report (JP 5422782/EP 12 884 742.3).
Data availability statement
Anonymized raw data supporting the findings of the present study may be shared by the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.