
Abstract
The prevalence of Alzheimer's disease (AD) is on the rise due to the global aging population. AD is the most common cause of dementia, accounting for 60–80% of all cases, which makes it a significant health concern. In recent years, the failure to develop targeted pathological drugs has led researchers to shift their focus. The blood-brain barrier (BBB) is a specialized structure that separates the circulating blood from the brain tissue and tightly regulates the passage of molecules, ions, and cells between the blood and the brain. Through a comprehensive review of current literature, we highlight the multifaceted role of BBB dysfunction in the pathogenesis of AD and discuss the complex mechanisms involved. Changes in the structure and function of endothelial cells, pericytes, and astrocytes, along with elevated expression of the highest-risk gene APOE4, can all lead to BBB damage, thereby promoting the onset of AD. Furthermore, we explore potential therapeutic targets to preserve BBB integrity and function to mitigate AD progression. This review underscores the significance of ongoing research efforts in elucidating the intricate interplay between BBB integrity and AD pathology, offering valuable insights for future investigations and therapeutic strategies.
Introduction
More than a century ago, Dr Alois Alzheimer, a German doctor, described a “strange disease” that he later named (Alzheimer's disease, AD). Alzheimer's is defined as senile dementia with plaques and neurofibrillary tangles.1–4 AD is a progressive and untreatable neurological condition that affects cognitive functions, representing the most prevalent cause of dementia. It is estimated that around 5% of males and 6% of females aged 60 and above worldwide are affected by AD. 5 It can lead to progressive cognitive decline. 6 In 2019, more than 55 million individuals globally were living with this incapacitating illness, and there is a growing trend, with 82 million cases expected by 2030.7,8 The blood-brain barrier (BBB) is a specialized physical barrier, a dynamic multicellular interface that regulates the central nervous system's (CNS) homeostasis, preventing peripheral toxins and pathogens from infiltrating the brain. The BBB consists of endothelial cells (ECs), pericytes, the basement membrane (BM), and astrocyte endfeet. 9 Currently, the focus of research on the pathogenesis of AD still revolves around two major pathological changes. However, the successive failures in developing related drugs have led us to shift our focus from traditional pathological, hypotheses the “Amyloid hypothesis” to the BBB. This research focus shift may result from insufficient understanding of AD pathogenesis and drug-development strategies. In recent years, research on AD has increasingly focused on changes in the BBB. Dysfunction of the BBB, whether as a precursor or a result, facilitates the occurrence and advancement of AD. However, the mechanisms by which BBB damage leads to AD onset are still unclear. 10 Elucidating the multifaceted role of the BBB in AD pathogenesis has become a research imperative. This review systematically integrates current evidence on BBB-AD pathophysiological interactions, characterizes both structural and functional alterations of BBB cellular components in AD progression, and mechanistically links BBB dysfunction to impaired amyloid-β (Aβ) clearance dynamics. Furthermore, we critically evaluate innovative therapeutic strategies targeting BBB restoration, with particular emphasis on recent advances in pharmacological interventions and their translational potential for AD management.
Overview of Alzheimer's disease
AD is the most prevalent type of dementia worldwide. 11 The main clinical features of AD are progressive short-term memory loss and cognitive dysfunction. The prevalence rate, mortality rate, and prevalence rate of this disease are increasing year by year. Its pathological mechanism is complex: in the early stage, the toxic protein Aβ activates the inflammatory signaling pathway, and the Ca2+ signaling pathway induces neuronal apoptosis and synaptic loss. In the late-stage of the disease, Aβ accumulation is key in the pathological process. Aβ interacts with neurons, activating kinases like glycogen synthase kinase 3β (GSK-3β) and cyclin-dependent kinase 5 (Cdk5). These kinases phosphorylate tau protein. The hyperphosphorylated tau loses its binding affinity to tubulin, causing microtubule disassembly. Then, phosphorylated tau aggregates into paired helical filaments, which further form neurofibrillary tangles. These tangles disrupt axonal transport, synaptic function, and inter-neuronal communication, leading to severe neuronal damage and cognitive and memory disorders in patients.12,13 Neuroimmunity has emerged as a hallmark of AD and is recognized as a key pathogenic driver.14–16 Microglia, the resident immune cells of the central nervous system, exhibit a dual role in AD progression. 17 During early stages, they actively phagocytose Aβ plaques, pathological protein aggregates characteristic of AD brains.18–20 However, chronic microglial activation in later phases results in sustained release of pro-inflammatory cytokines, including interleukin-1β (IL-1β) and tumor necrosis factor-alpha (TNF-α). This neuroinflammatory cascade not only perpetuates CNS inflammation but also directly contributes to synaptic loss and neuronal death, ultimately exacerbating cognitive deterioration in AD. 21 Dementia is also associated with impaired synaptic plasticity, APOE4 gene expression in glial cells, 22 many other CNS diseases, and damage to the BBB.
Blood-brain barrier
AD drug development: past, present, and the pivotal role of BBB
From 1998 through 2024, the FDA approved only eight drugs for AD, of which only three targeted AD pathological processes: aducanumab, lecanemab, and donanemab.23–25 Notably, donanemab is the most recent anti-amyloid monoclonal antibody approved by the FDA. Importantly, it is the first and only limited course amyloid-targeted therapy that can be stopped upon achieving sufficient amyloid plaque clearance, thereby significantly reducing treatment burden. In contrast, the remaining five dementia medications (tacrine, donepezil, rivastigmine, galantamine, and memantine) only temporarily alleviate symptoms without modifying disease progression. 26 Specifically, the first four are cholinesterase inhibitors that boost neurotransmitter levels, while memantine uniquely blocks excessive glutamate activity via NMDA receptor antagonism. 6 This differential mechanism makes memantine complementary to existing therapies by targeting distinct neurodegenerative pathways. Given these therapeutic limitations, recent focus has shifted following repeated Aβ/tau targeting failures, with 70% of development now concentrated on non-Aβ/non-tau targets. One of the key pathophysiologies of AD is the BBB, which offers a fresh perspective for AD medication research and development. Subsequently, we will expound in greater detail on the composition and function of the BBB.
BBB overview and function
BBB is a functional unit that separates brain tissue from the peripheral environment.27–29 The basic framework is formed by the tight junctions (TJs) of vascular ECs through TJ proteins such as zonula occluden-1 (ZO-1), occludin, and claudin-5. Additionally, pericytes and astrocytes eventually cover ECs, thus limiting the passage of peripheral macromolecules, cytotoxic substances, and immune cells entering the CNS. This barrier helps maintain the stability of the internal environment and the normal function of brain cells. 30 It is the best microenvironment for maintaining the healthy functioning of the CNS. 31 Although the BBB typically prevents peripheral pathogen infiltration into the CNS, studies have demonstrated that herpes simplex virus type 1 (HSV-1) infection can disrupt BBB integrity. Research confirms that HSV-1 infection downregulates the expression of the Golgi structural protein GM130 in brain microvascular ECs, inducing Golgi fragmentation and activating the caspase-3-dependent apoptotic pathway. This cascade leads to a significant reduction in the protein levels of TJ proteins occludin and claudin-5, ultimately resulting in impaired BBB function, as evidenced by decreased transendothelial electrical resistance (TEER). 32 AD pathology may facilitate herpesvirus entry into the CNS by compromising BBB integrity. For instance, Liu et al. found an HSV-1 infection rate of 0.44% in AD patients, significantly higher than the 0.24% rate in the control group (p < 0.0001). 33 Furthermore, 5XFAD mice rapidly developed Aβ deposits following intracranial HSV-1 injection, with viral load positively correlating with plaque density, 34 indicating a potential interplay between pathogen infiltration and AD pathology. The BBB is a key component of the neurovascular unit (NVU). The NVU comprises vascular ECs, pericytes, astrocytes, and neurons, which collectively regulate BBB integrity and function (Figure 1). Through tight neurovascular coupling mechanisms, these cellular components maintain the selective permeability of the BBB, ensuring CNS homeostasis.35–37 Therefore, damage to components of the NVU, such as ECs, by HSV-1 may further exacerbate BBB dysfunction and accelerate neurodegenerative processes.
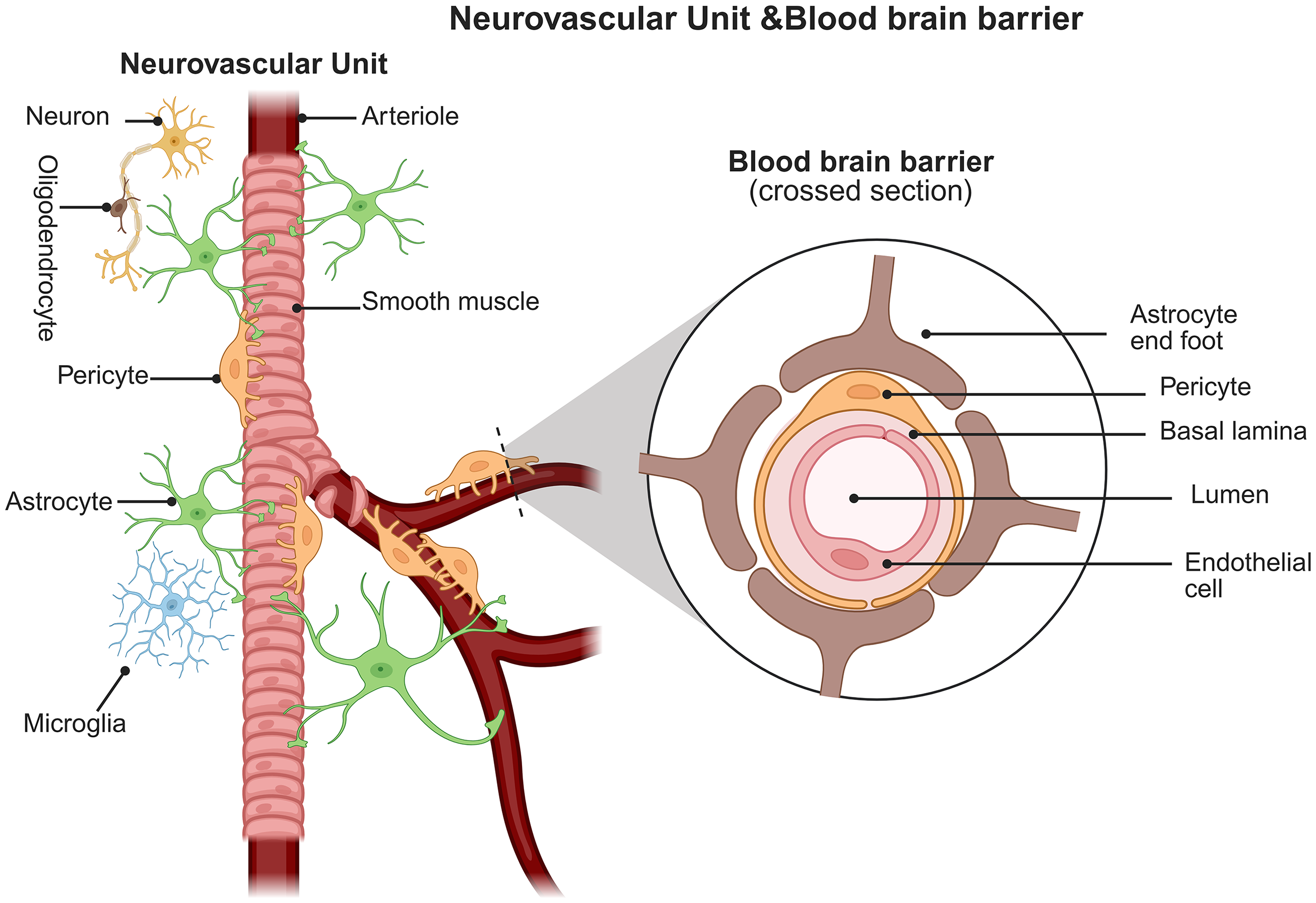
The neurovascular unit is a complex network of glial, neuronal, and endothelial cells, primarily responsible for responding to physiological or pathological stimuli within the central nervous system and regulating the metabolic needs of brain cells by modulating cerebral blood flow and solute traffic across the blood-brain barrier. Created in https://BioRender.com.
Endothelial cells
ECs are the main structure of the BBB, and their molecular properties are crucial for maintaining the function and integrity of BBB. In healthy conditions, BBB exhibits a high level of selectivity, which is determined by brain endothelial cells (BECs). 38 ECs in the CNS are special because of the TJ between ECs in the CNS, wherein dynamic opening and closing of adhesive junction (AJ) can regulate the permeability of BBB.39,40 The TJ complex is a distinctive molecular arrangement of the BBB, composed of adhesion proteins, occludins, ZO-1, ZO-2, ZO-3, and adhesion molecules. 41 ECs maintain the BBB's selective permeability, restricting the entry of pathogens while controlling the movement of vital nutrients and metabolites. 42 When ECs, especially BECs, are damaged, their ability to support the BBB's strength and performance is impaired. 43
Pericytes
Pericytes, located on the abluminal surface of ECs in the CNS's microvasculature, is crucial for maintaining the BBB. Structurally, they connect to ECs through a shared BM.44,45 Morphologically, they are spindle- or stellate-shaped with long, branched cytoplasmic processes for cell-cell interaction. 46
Pericytes maintain the functionality of the BBB by modulating the expression patterns of BBB-specific genes in ECs, such as glucose transporter 1 (Glut1) and the Transferrin receptor (CD71). Although most BBB markers do not exhibit significant changes at the mRNA level, CD71 shows a marked downregulation at the protein level. These findings suggest that pericytes play a crucial role in preserving the integrity and function of the BBB through the regulation of specific gene expression in ECs. 47
Pericytes are mural cells that ensheath the walls of microvessels. They modulate angiogenesis and support the development of the BBB. Furthermore, pericytes regulate blood flow by modulating the diameter of capillaries. 48 Defective pericellular coverage, impaired pericellular recruitment, and pericellular loss due to pericellular dysfunction all result in elevated endothelial cell endocytosis and permeability.47,49 ECs secrete platelet-derived growth factor B (PDGF-B), which is then located in the ECM and the surrounding microenvironment. 50 The platelet-derived growth factor receptor (PDGFR), expressed on the cell membrane surface of pericytes, plays a crucial role in cell-cell communication. 51 Research indicates that in AD, reduced production of PDGF-BB by cerebrovascular ECs leads to insufficient activation of the PDGFRβ receptor on pericytes, specifically inhibiting the extracellular signal-regulated kinase (ERK) signaling pathway. This ERK signaling deficiency diminishes pericyte proliferative capacity and increases their susceptibility to apoptosis, ultimately resulting in a reduction in pericyte number. The loss and functional impairment of pericytes are the direct cause of compromised BBB integrity (manifested as leakage and dysfunction), thereby exacerbating the pathological progression of AD (Figure 2). 52
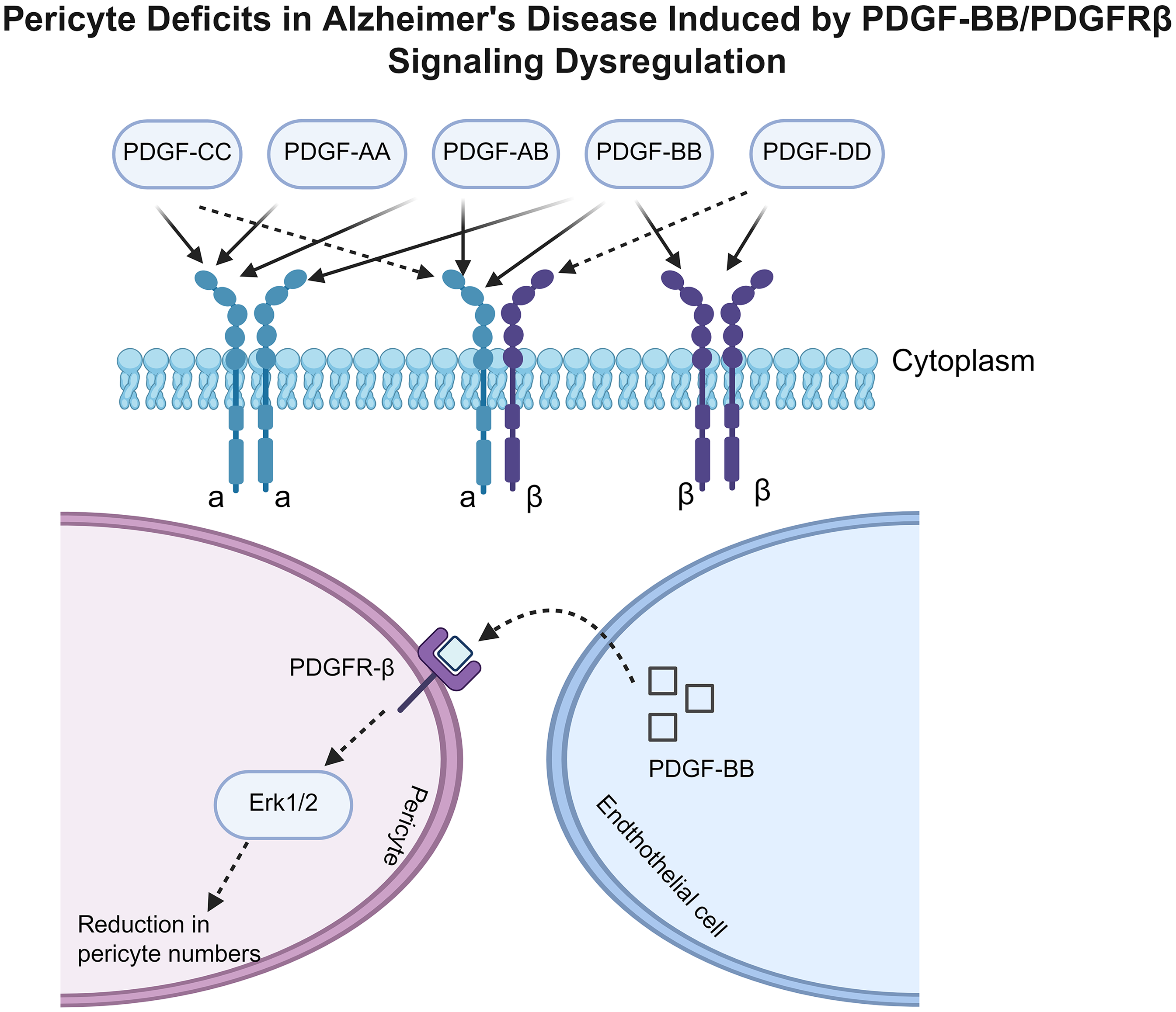
Pericyte deficits in Alzheimer's disease induced by PDGF-BB/PDGFRβ signaling dysregulation. The upper part of the figure illustrates different platelet-derived growth factor (PDGF) isoforms (PDGF-CC, AA, AB, BB, DD) binding to PDGFRα and PDGFRβ on the cell membrane. The lower section demonstrates that in Alzheimer's disease, reduced production of PDGF-BB by cerebrovascular endothelial cells leads to insufficient activation of the PDGFRβ receptor on pericytes, specifically inhibiting the extracellular signal-regulated kinase (ERK) signaling pathway. This ERK signaling deficiency diminishes pericyte proliferative capacity and increases their susceptibility to apoptosis, ultimately resulting in a reduction in pericyte number. Created in https://BioRender.com.
Astrocyte
Astrocytes form connections with blood vessels and are closely associated with the basal lamina, a structural element of the vessel wall. 53 They play a crucial role in the NVU, 54 integrating the functions of ECs, neurons, and pericytes, maintaining the microenvironment homeostasis within the NVU, and coordinating the bidirectional communication between the bloodstream and neural tissue. 55
In states of normal health, astrocytes are consistently and vigorously engaged in the functioning of neural circuits. However, a range of CNS pathologies and injuries can shift their function to an “activated state” through the process of reactive astrogenesis. Astrocytes can nourish neurons by releasing vasodilators such as nitric oxide (NO) or arachidonic acid to regulate cerebral blood flow (CBF).56,57 In terms of maintaining the BBB function, they actively secrete a series of regulatory factors, which enhance the TJ proteins like occludin and claudins expression in ECs, thereby promoting the barrier properties of the BBB. 58 In addition, astrocytes establish close contact with cerebrovascular endothelial cells (CECs) via their endfeet and transfer mitochondria to ECs, thereby contributing to BBB integrity. 59 Studies demonstrate that Dmp1-expressing astrocytes regulate BBB function through mitochondrial transfer, while genetic deletion of Mitofusin 2 (Mfn2) in these astrocytes suppresses mitochondrial transfer and induces BBB leakage. Importantly, aging leads to reduced efficiency of this mitochondrial transfer mechanism, further compromising BBB integrity.
Nevertheless, in the context of neurodegenerative diseases such as AD, the role of astrocytes can take a pathological turn. Reactive astrocytes, as described, contribute to REDOX state imbalances by releasing cytokines, inflammatory factors, NO, and reactive oxygen species (ROS). This release leads to neuroinflammatory changes in AD, highlighting how the normal regulatory functions of astrocytes can be disrupted, and how these cells can instead contribute to the progression of the disease.60,61
Basement membrane
The cerebrovascular basement membrane (CVBM) is a special ECM composed of laminin, type IV collagen, proteogen, heparin sulfate, and proteoglycan. It surrounds ECs and pericytes, facilitating their interactions, and also engages with astrocytes through their distal processes. 62 The ECM is the acellular element of NVU that offers structural stability and impacts cellular activities, such as cellular adhesion, movement, specialization, and reproduction. 63 Previous studies have demonstrated that BM constituents are capable of controlling the cellular distribution of occludins in ECs, consequently impacting the stability of the barrier. 64 In AD, several changes in BM can be observed, mainly including the thickening of BM, 65 Deposition of Aβ 66 and BM protein composition changes.65,67,68 These may have greatly contributed to the development of the disease.
The mechanisms of BBB damage in AD
Causes of BBB injury
Simultaneously, the BBB oversees the passage of substances into and out of the CNS, rigorously maintaining the chemical makeup of the neuronal environment essential for proper neuronal function. 69 It was found that the BBB dysfunction caused by APOE4, a high-risk gene for AD, the aging of BBB structure, and the changes of BBB caused by related neurological diseases were the main causes of BBB injury. Next, focus on these aspects of the introduction.
BBB damage: overexpression of APOE4, the highest risk genetic factor for AD
APOE4 in AD: Genetic Basis and Multifactorial BBB dysfunction
There is a growing interest in the impact of the APOE4 allele on the BBB. Following numerous extensive genome-wide association studies and meta-analyses, the APOE4 continues to be the most significant genetic risk factor for sporadic AD.70–73 The APOE exists in three major isoforms (E2, E3, E4), with the E4 isoform exhibiting a cysteine-to-arginine substitution at position 112 (Cys112Arg) that alters protein conformation and reduces lipid-binding capacity. Compared to the neuroprotective E2 and neutral E3 isoforms, this structural change impairs both lipid-binding affinity and receptor interactions.22,74 Blumenfeld et al. 75 Review of the literature describes the various cell type-specific functions of APOE4 in AD and provides a deeper comprehension of the pathological mechanisms of APOE4, such as tau neurofibrillary tangle degeneration, astrocyte and microglia responses, and BBB disruption.
In astrocytes, APOE4 may disrupt end-foot interactions with cerebral vasculature (e.g., through impaired LRP1 receptor signaling), leading to end-foot retraction or deficient coverage that compromises BBB structural support. 76 Concurrently, attenuated binding of APOE4 to the TREM2 receptor on microglia diminishes phagocytic clearance of Aβ plaques. 77 Uncleared Aβ directly bind and activate DR4/DR5 death receptors, triggering synergistic activation of extrinsic apoptotic (caspase-8) and intrinsic mitochondrial (Bid-caspase-9) pathways in brain microvascular ECs, thereby increasing BBB permeability. 78 Furthermore, aberrant APOE4 expression induces hyperactivation of the RhoA (Ras homolog gene family member A) signaling pathway in pericytes, promoting pericyte migration and destabilizing BBB integrity. 79 These multicellular synergistic effects establish APOE4 as a central driver of BBB disruption.
The typical postmortem neuropathological correlation is that carriers of the APOE4 exhibit a greater accumulation of Aβ plaque and more severe cerebral amyloid angiopathy.80–82
Mechanisms of APOE4-induced BBB damage
APOE4 has been shown to interfere with lipid metabolism and Aβ plaque formation and accumulation in the brain.83–86 In the human brain, APOE is mainly produced by astrocytes, and APOE4 has been shown to have several adverse effects on brain physiology.87–89 When the expression level of the APOE4 is significantly elevated specifically within astrocytes, it changes the morphology of lipoprotein particles and increases cholesterol in the brain, disrupting brain cholesterol homeostasis. Finally, hypercholesterolemia is induced, which in turn induces oxidative stress in the brain, activates the C/EBPβ/δ secretase pathway, promotes neuroinflammation, and damages BBB function. 90 Some studies have also shown that ApoE4 can reduce the transport and clearance efficiency of Aβ by affecting the transport of lysosomes. 91 At the same time, it can also stimulate the activator protein-1 (AP-1) transcription factor and increase the amyloid precursor protein (APP), leading to A large amount of Aβ deposition. 92
APOE4 is involved in peripheral inflammation and affects brain inflammation. Studies have proved that chronic low-grade inflammation in the periphery of APOE4 carriers can increase the risk of early onset of AD. 93 APOE4 mice immunized with peripheral injection of lipopolysaccharide (LPS) produced higher levels of proinflammatory cytokines, such as TNF-α, IL-6, and NO, which has an injury-inducing effect.94,95 Notably, emerging evidence highlights the role of APOE4 in modulating matrix metalloproteinases (MMPs), key mediators of extracellular matrix degradation and BBB disruption. A clinical study by Stomrud et al. 96 demonstrated that cognitively healthy APOE4 carriers exhibit elevated cerebrospinal fluid (CSF) levels of MMP-3 and MMP-9 compared to non-carriers, suggesting a potential link between APOE4-driven neuroinflammation and MMP-mediated vascular injury. These MMPs may further exacerbate BBB permeability by degrading TJ proteins and BM components, thereby accelerating neurovascular dysfunction in preclinical AD (Figure 3). These peripheral inflammatory changes may affect the inflammatory state in the brain through the BBB or interact with immune cells in the brain, and then promote the occurrence and development of neuroinflammation, indicating that there may be a potential link between systemic inflammation and brain inflammation, and its overexpression may indirectly enhance the inflammatory response in the brain through systemic inflammatory pathways
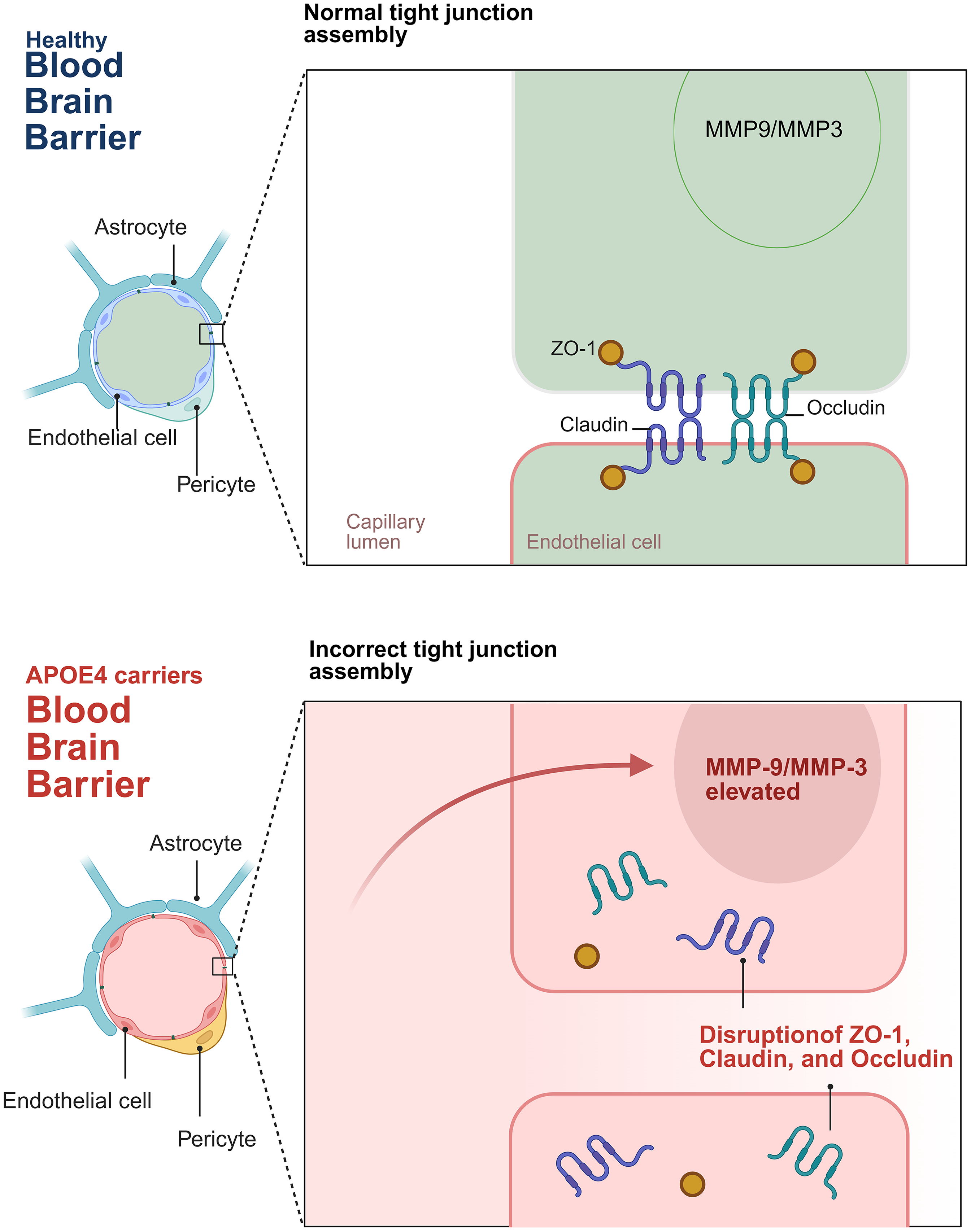
The figure depicts the distinctions in the BBB between healthy individuals and carriers of APOE4. In a healthy blood-brain barrier (BBB), the tight junctions are properly assembled, and their structural integrity is upheld by proteins including ZO-1, Claudin, and Occludin. The levels of MMP9 and MMP3 remain within the normal physiological range. Conversely, in the BBB of APOE4 carriers, APOE4 elicits an upsurge in the levels of MMP-9 and MMP-3. These elevated levels of MMP-9 and MMP-3 degrade the TJ proteins, thereby disrupting the assembly of TJ and augmenting the permeability of the BBB. Such alterations may potentially precipitate neurovascular dysfunction. Created in https://BioRender.com.
The cell structure of endothelial cells, pericytes, and astrocytes was changed
Endothelial cells: structural damage and BBB disruption
During AD, ECs experience alterations in their structure that interfere with their interconnections, consequently restricting their associations and communications with other components of the NVU (Figure 4). 97 In AD pathogenesis, MMP expression is elevated. 98 MMPs are a type of zinc-dependent proteolytic enzyme that can degrade the myelin layer in the deep white matter and cause vasogenic edema when they are active. 99 Furthermore, it has been demonstrated that MMPs further break down the proteins occludin, claudin, and ZO, which results in TJ disruption and an increase in BBB permeability. 100 Yoonjin et al. used microfluidic technology to 3D in vitro culture ReN cells and found that the level of Aβ increased, leading to a decrease in the activity of vascular ECs. The levels of claudin-1 and claudin-5 proteins on the cell membrane were decreased, and further detection showed that the levels of MMP-2 and ROS were increased. Activated MMP-2 loosens molecules and TJ proteins in the basal lamina surrounding ECs. 101 Multiple experiments have shown that the abnormal structure and function of ECs are closely related to BBB injury. At present, the regulatory mechanism of MMP in AD is still not fully understood, which provides an important direction for future research. Further exploration of the triggering factors of increased MMP expression and its upstream regulatory signaling pathways may provide new targets for the development of intervention measures.
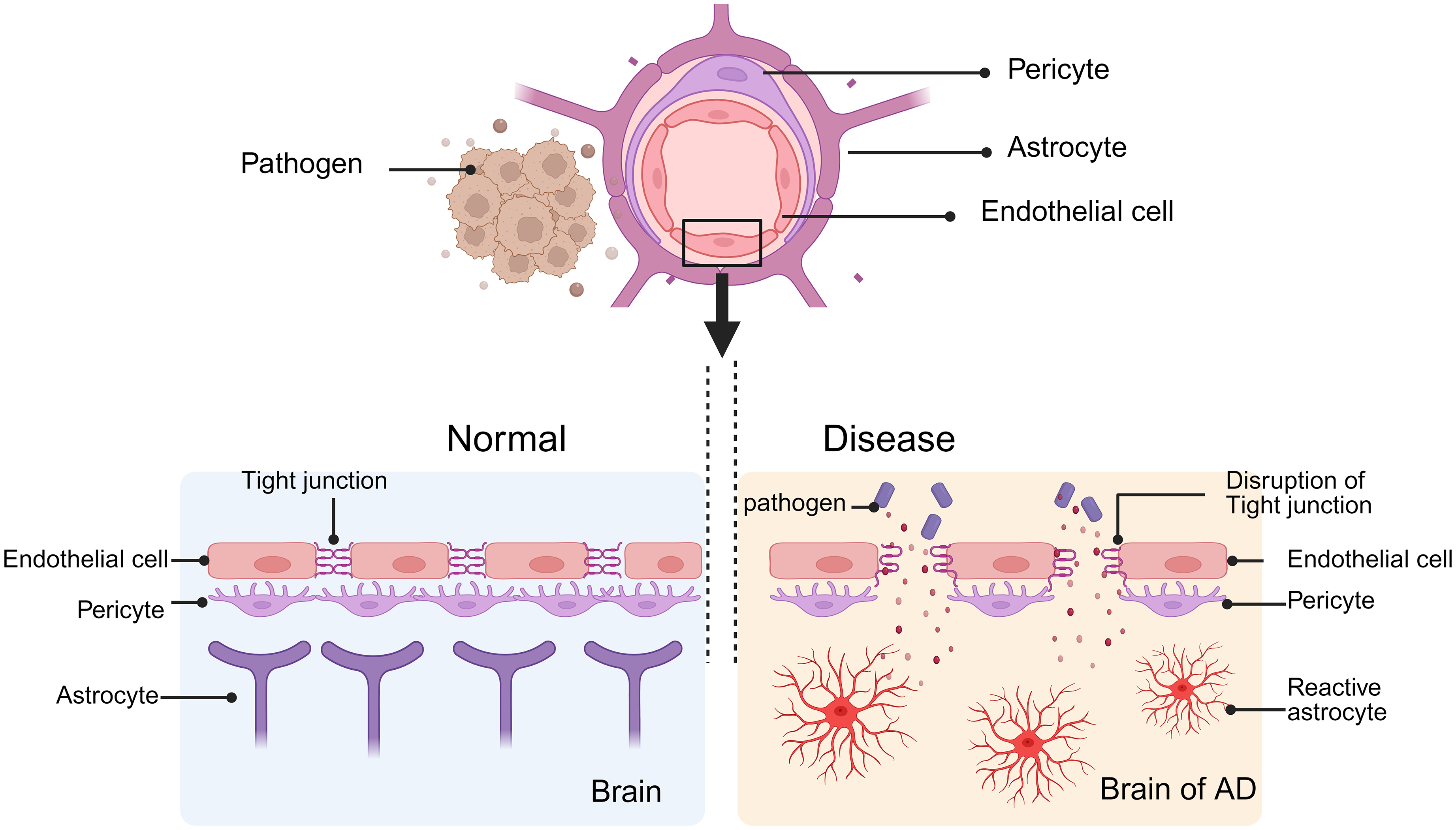
The figure illustrates the blood-brain barrier (BBB) under normal conditions and during pathological states. On the left, the BBB is intact, maintaining the stability of the brain and regulating its physiological functions. On the right, the breakdown of tight junctions allows pathogens to enter the brain through extracellular pathways, resulting in damage to the BBB. Created in https://BioRender.com.
Furthermore, integrated multi-omics analysis revealed significant downregulation of LEF1—a core transcription factor of the Wnt/β-catenin signaling pathway—in BECs of an AD model (5xFAD mice), leading to compromised BBB integrity. Experimental validation demonstrated a 50%-70% reduction in LEF1 protein levels in cortical and hippocampal vasculature of AD mice. 102 This LEF1 deficiency suppresses Wnt signaling, resulting in decreased expression of BBB transporters (e.g., GLUT-1, TfR) and subsequent disruption of glucose and iron homeostasis in the brain. 103 Further investigation identified a marked increase in senescent cell proportions within BECs of insulin-like growth factor receptor (IGF1R)-deficient mice, particularly pronounced in aged subjects. This cellular senescence significantly elevated BBB permeability, evidenced by increased leakage of fluorescent tracers across molecular weights (0.3 kDa to 40 kDa). 104 This study elucidates, for the first time, the multi-layered mechanisms of BBB impairment in AD from the perspective of endothelial-specific molecular networks, providing novel directions for precision therapeutics targeting endothelial repair.
Pericytes: a multifaceted variation and functional relevance puzzle
In AD, the changes in pericytes are mainly reflected in three aspects: morphology, coverage, and marker expression.105–107 These changes are closely related to the progression of the disease, and there are many problems worth exploring in the current research. Initially, about pericyte markers. Pericytes are important sources of BBB BM proteins, including laminin 108 and vitreous adhesin, 109 and support the formation and stability of the inner tube, 45 a mouse model of pericellular deficiency caused by PDGF-BB/PDGFR-signaling abnormalities demonstrated impaired BBB formation and maintenance. Although PDGFRβ was initially considered a promising diagnostic biomarker for AD due to its role in the PDGF-BB/PDGFR-signaling pathway associated with the BBB integrity, recent investigations, as reported in reference, 110 have revealed that the concentration of PDGFRβ in the CSF of AD patients shows no significant difference compared to that of the control group. Consequently, PDGFRβ lacks the discriminatory capacity required for its use as a reliable diagnostic tool for AD. It is suggested that we may need to find more specific and sensitive markers, or further explore the dynamic change patterns of these markers in different stages of AD to accurately assess the functional status of pericytes and their relationship with the pathological process of AD. Secondly, in terms of pericyte morphology, progressive degeneration of pericytes in the brain or retina has been observed in AD patients and mouse models,105,111 but some studies have found no significant changes in pericytes in the frontal cortex of clinically diagnosed AD patients, which may be related to the different pericyte subtypes examined. 112 Future studies are needed to further clarify the characteristics of different subtypes of pericytes in morphological changes and how these changes affect their functions, which in turn affect the progression of AD. Finally, in terms of pericyte coverage, vascular amyloid fiber deposits lead to pericyte atrophy in AD, which is manifested as a large number of intracellular inclusions, pinocytosis vesicles, large lipid granules, and mitochondrial abnormalities, thereby causing pericyte coverage reduction. 113 The study by Vazquez-Liebanas et al. indicates that only <50% longitudinal pericellular coverage in the adult brain results in severe leakage problems, suggesting that pericellular coverage may also play a role in determining the threshold for BBB abnormalities. 114
Astrocytes: protein expression changes and functional duality
When Aβ builds up in AD, amyloid protein aggregates may form between the astrocyte endfeet and the vessel wall, giving the endfeet a bloated appearance and decreasing the blood artery's covering area. 115 This damage may lead to nutrient deficiencies in neurons and BBB dysfunction, thereby worsening the pathological condition of AD. 69 Protein expression in astrocytes varies in AD. Research has demonstrated that whereas AQP4 polarization is decreased in endfeet in AD patient brains and mice models of amyloidosis, AQP4 expression is often increased in cortical areas.116,117 This implies that as AD progresses, alterations in astrocyte protein expression exhibit complexity. Astrocytes have two sides, on the one hand, serving a protective role on the BBB in the physiological state, and on the other hand, they are destructive when in a pathological situation. For example, reactive astrocytes release factors that increase vascular permeability, such as vascular endothelial growth factor (VEGF), MMPs, NO, nitric oxide synthase (NOS), hypoxia-inducible factor-1 alpha (HIF-1α), and APOE4, which exacerbate the breakdown of the BBB. Conversely, astrocytes secrete protective factors, including Shh, Ang-1, retinoic acid (RA), and insulin-like growth factor-1 (IGF-1), which mitigate the destruction of the BBB (Figure 5). 10 Astrocytes are part of the integrity of the BBB, can nourish neurons, and have stabilizing significance for the CNS. Examination of cerebral cortical biopsies in individuals with AD has uncovered microscopic alterations in astrocytes proximal to accumulations in the brain tissue and blood vessels. These changes include constriction and expansion, as well as diminished levels of Glut1 and lactate transporters. This implies that impaired astrocyte function could contribute to the onset of early behavioral and cognitive deficits in AD mice. 118
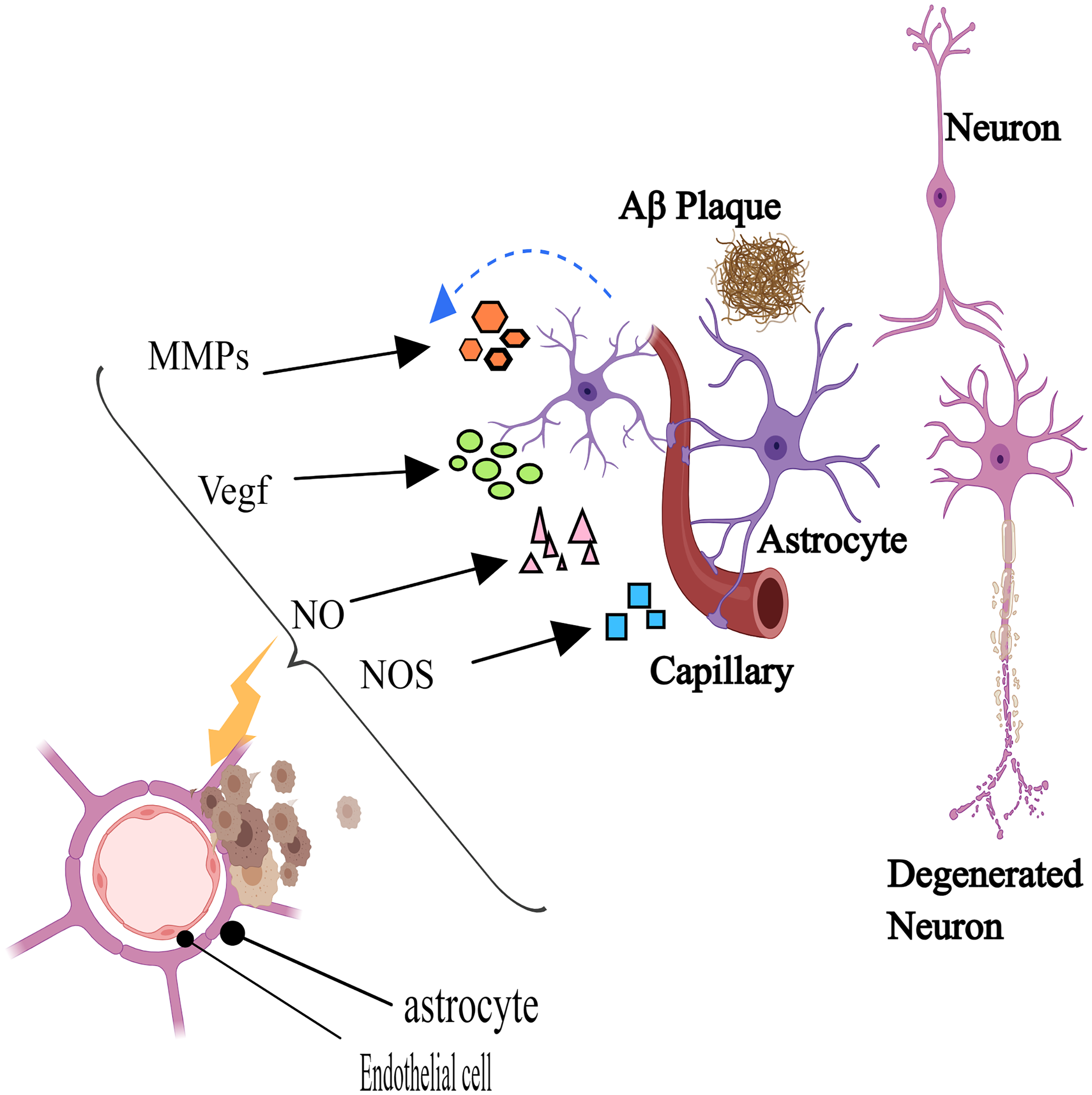
Reactive astrocyte secretes vascular permeability factors, including vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs), nitric oxide (NO), nitric oxide synthase (NOS), hypoxia-inducible factor-1 alpha (HIF-1α), and APOE4, which aggravate the destruction of the blood-brain barrier. Created with MedPeer (http://www.medpeer.cn).
Related diseases cause BBB dysfunction
BBB dysfunction is strongly linked to a spectrum of metabolic and vascular disorders, including atherosclerosis, type 2 diabetes mellitus (T2DM), hypertension, obesity, sleep disturbances, and hypercholesterolemia.119–121 These conditions exacerbate cerebral microenvironmental imbalances by disrupting BBB TJ integrity and compromising its selective permeability. Notably, aberrant BBB permeability plays a central role in the pathogenesis of neurodegenerative diseases such as AD. Specifically, BBB leakage permits the infiltration of peripheral neurotoxic proteins (e.g., fibrinogen, coagulation factors) into the brain parenchyma, which activates microglia and drives chronic neuroinflammation, thereby accelerating AD pathology.122,123 Moreover, advanced glycation end products (AGEs) induce phosphorylation of moesin—a cytoskeletal linker protein belonging to the ERM (ezrin-radixin-moesin) protein family—through activation of the RhoA/ROCK and p38 signaling pathways. Phosphorylated moesin triggers reorganization of the endothelial cytoskeletal protein F-actin, directly compromising the structural integrity of cerebral microvascular endothelium, thereby impairing barrier function and inducing pathological leakage. 124 Concurrently, significant dysregulation occurs in mucin-type O-glycosylation within the glycocalyx layer on BECs. Downregulation of key enzymes mediating mucin-type O-glycosylation (e.g., C1GALT1 and B3GNT3) results in aberrant glycan structures on mucin-domain glycoproteins (such as PODXL and CD34). This disrupts the physical and electrostatic barrier functions of the glycocalyx, increasing BBB permeability and facilitating the infiltration of blood-derived neurotoxic substances (e.g., albumin, IgG) into the brain parenchyma. 125
Moreover, these comorbidities often coexist and synergistically exacerbate BBB damage through shared pathological mechanisms, such as oxidative stress and inflammatory cytokine overproduction. 126 For example, in patients with concurrent T2DM and hypertension, hyperglycemia-mediated activation of the receptor for advanced glycation end products (RAGE) enhances Aβ influx, while hypertension-induced mechanical stress disrupts TJ proteins (e.g., occludin, ZO-1). These distinct mechanisms converge via oxidative stress and proinflammatory signaling, collectively aggravating BBB leakage. 126 Ultimately, such interactions establish a self-perpetuating vicious cycle of “BBB disruption–neuroinflammation–neuronal death”, markedly hastening AD progression.
T2DM is primarily characterized by insulin resistance, which manifests as excessive thirst, increased appetite, and frequent urination. The main hallmark of T2DM is hyperglycemia, which can result in EC dysfunction and inflammation. According to research, high blood sugar has been linked to thromboxane A2 receptor activation and BBB integrity and function impairment via the ROCK-PTEN-Akt-eNOS pathway. 127 When there is a lack of oxygen, the release of cytokines leads to an elevation in MMPs. These MMPs are destructive in nature and can cause further injury to the tissues and have significant implications for cerebrovascular integrity, potentially triggering a cascade of pathophysiological events that may exacerbate the existing condition. 128 Coucha et al. 129 mentioned that a faster increase in activated MMP-9 after stroke in diabetic mice ultimately disrupts the integrity of the BBB, thereby contributing to the development of cognitive impairment. These changes may facilitate the infiltration of neurotoxic substances, contributing to the pathological alterations seen in AD.130,131
Hypertension is the primary indication of increased blood pressure, which can result in numerous complications, and may even initiate vascular restructuring and neuroinflammation. Subsequently, it can impair the function of the BBB and enhance microvascular damage in AD patients. 132 The presence of sleep disorders is increasingly recognized as a contributing factor in impeding the removal of neurotoxic substances in individuals with AD, thus facilitating the progression of the disease. 133 Obesity is a metabolic disorder marked by inflammation and dysfunction of the ECs. Likewise, hypercholesterolemia can result in elevated generation of Aβ and compromised clearance mechanisms, leading to the accumulation of Aβ. 134 In the Pdgfbret/ret mouse model, due to the pericyte deficiency, the permeability of the BBB to water and a series of low- and high-molecular-weight tracers is significantly increased. In the experiment, after the mice were treated with a diet labeled with deuterated cholesterol, compared with the control mice, the enrichment of deuterated cholesterol in the brains of Pdgfbret/ret mice was significantly higher. This finding further confirms the importance of the integrity of the BBB in the brain. Disrupting the BBB allows cholesterol to flow from the circulating blood into the brain parenchyma, thereby affecting the cholesterol homeostasis in the brain. 135 BBB disruption reduced CBF and clearance of Aβ, contributing to neuronal dysfunction due to small blood vessel dysfunction. 126 However, the Baltimore Longitudinal Study of Aging (BLSA) cohort, which examined the relationship between AD and atherosclerosis, found no association between the degree of pathology of AD and atherosclerosis in the aorta, heart, and intracranial vessels, although only intracranial atherosclerosis was associated with dementia. 136 Therefore, related diseases promote BBB damage, providing favorable evidence for the pathogenesis of AD.
Furthermore, Vascular Cognitive Impairment and Dementia (VCID) is a degenerative disorder of the CNS characterized by progressive cognitive decline induced by chronic cerebral ischemia, whose pathological evolution is closely associated with BBB impairment. 137 Studies demonstrate that the APOE4 significantly exacerbates disease progression by disrupting lipid metabolism: this genotype promotes abnormal cholesterol ester accumulation in macrophages and smooth muscle cells, driving foam cell formation and accelerating atherosclerotic plaque development, thereby increasing the incidence of intracranial arterial stenosis. 138 Such vascular stenosis induces brain injury through dual pathways—large vessel disease causes significant regional blood flow reduction, while small vessel disease (including arteriolosclerosis and cerebral amyloid angiopathy) disrupts microcirculatory architecture. Research reveals that sustained ischemia activates HIF-1α, stimulating VEGF overexpression in pericytes and astrocytes. 137 VEGF treatment markedly enhances phosphorylation of occludin at Ser-490, triggering cytoplasmic translocation of occludin and claudin-5 from the plasma membrane and disrupting their polarized distribution. 139 Consequently, TJ pore expansion permits leakage of small molecules (e.g., ions, metabolites), directly compromising BBB integrity. 137 This provides a critical theoretical foundation for BBB-targeted intervention strategies.
Neuroimaging evidence of BBB disruption in AD
BBB dysfunction in AD has been extensively documented using neuroimaging modalities such as magnetic resonance imaging (MRI) and positron emission tomography (PET). 10
Recent advances in PET targeting cerebral glucose metabolism have revealed significant reductions in glucose utilization in AD patients compared to cognitively healthy individuals. These metabolic deficits predominantly emerge in the temporoparietal regions, including the precuneus and posterior cingulate cortex, during the early stages of the disease. 140 The observed decline in glucose uptake is hypothesized to reflect BBB dysfunction, as Glut1, the primary mediator of cerebral glucose transport, is exclusively expressed in BBB ECs rather than neurons. 141 Notably, dynamic contrast-enhanced PET studies using the radiolabeled glucose analog 18F-fluorodeoxyglucose (FDG) have demonstrated region-specific glucose hypometabolism in AD progression. Patients with mild cognitive impairment (MCI) exhibit reduced FDG uptake in the precuneus, cingulate cortex, and temporal lobes, whereas advanced AD patients show further metabolic decline in the hippocampus, parietal cortex, cingulate gyrus, and temporal cortices. 10
MRI plays a pivotal role in the clinical characterization of MCI and AD, serving as a non-invasive diagnostic tool encompassing both structural MRI (sMRI) and functional MRI (fMRI). In the early stages of AD, neuropathological changes predominantly manifest as cerebral atrophy and functional dysregulation. MRI enables precise detection of structural alterations, including tissue-specific atrophy patterns characteristic of AD. Notably, cerebral microbleeds (CMBs), indicative of vascular injury and associated with significant BBB disruption, have been reported in 10–30% of AD cases. 10 sMRI studies consistently reveal atrophy in the medial temporal lobe (MTL), entorhinal cortex (ERC), and hippocampus. Quantitative analyses demonstrate a 26–27% reduction in hippocampal volume and a 38–40% decrease in ERC volume in AD patients compared to age-matched controls. 142 These structural biomarkers not only correlate with cognitive decline but also provide critical insights into the spatiotemporal progression of neurodegeneration in AD.
The role and significance of BBB in the therapy of Alzheimer's disease
Optimization of drug delivery systems
Nanoscale drug carriers
To develop nanoscale drug carriers to increase the affinity of drugs to the BBB by surface modification and other means,143–146 a multi-faceted approach is being explored. Surface modification serves as a pivotal strategy. By attaching specific molecules like small molecules, antibodies, homing peptides, polymers, or targeting ligands to the nanocarriers, their surface properties are altered. This not only enhances the initial interaction between the nanocarriers and the BBB, but also enables them to overcome the physical and biological barriers presented by the BBB. Once the affinity is enhanced through surface modification, 147 these nanocellulators can carry drugs across the BBB and deliver drugs precisely to lesions in the brain, improving therapeutic efficacy and reducing peripheral side effects. Studies have shown that to improve the transport and cellular uptake of nanoparticles (NPs) in the BBB, molecules such as small molecules, antibodies, or homing peptides, polymers, or targeting ligands are often used to modify NPs.148,149 For example, NPs modified with transferrin, apolipoprotein, insulin-like growth factor I or II penetrate the BBB more efficiently than non-functionalized NPs. 150
Targeted delivery of drugs based on BBB-specific receptors
Using specific receptors expressed on the BBB, such as the Transferrin receptor (TfR), low-density lipoprotein receptor (LDLR), and Choline transporter (SLC5A7), the drug is linked to the receptor-ligand and transported into the brain by receptor-mediated endocytosis. This strategy can achieve targeted drug delivery and improve drug uptake and distribution in the brain.151,152 RVG (rabies virus glycoprotein) can target the brain by binding to nicotinic acetylcholine receptors (nAchRs) on the surface of neuronal cells. 153 Rvg-modified drug delivery systems have been used in a variety of studies. For example, RVG-peptide-linked trimethylated chitosan is used to deliver siRNA to the brain to treat AD. 154 Yizhou Dong's team 155 has developed a γ-secretase-mediated cross-BBB coupling (BCC) system, which uses γ-secretase-mediated transcytophoretic absorption to safely and effectively deliver biological macromolecules to the CNS by intravenous injection. It is referred to the use of BCC10 and targeted therapy for AD.
Drug development to regulate BBB function
Drugs regulating TJ proteins
Search for drugs that can modulate the expression and function of TJ proteins in BBB ECs to reinforce BBB integrity. A potentially viable approach is the activation of intracellular signaling pathways associated with TJ protein synthesis and assembly. For instance, certain natural compounds or small molecule drugs may alleviate neuroinflammation and Aβ accumulation by activating intracellular signaling pathways that facilitate the synthesis and assembly of TJ proteins and reduce BBB leakage. Bradykinin (BK) B2 receptors are constitutively expressed on BBB ECs, as has long been demonstrated. It can quickly and temporarily break up tight connections and increase BBB permeability after stimulation. 156 Guilherme Juvena et al. 157 also investigated the mechanism of action of BK in AD in their most recent paper. This study found that while HOE-140, a B2 receptor antagonist, decreased the expression of several genes, including C3, CCL12, and CCL5, BK treatment of transgenic neurospheres boosted the expression of immune-related genes, including Toll-like receptor 2 (TLR2) and CCL12. This suggests that BK plays a role in controlling the expression of genes linked to neuroinflammation and offers a possible method of controlling TJ protein production and function in BBB ECs by interfering with the BK signaling pathway. In addition, Qi Wang et al. 158 showed that activation of the Wnt/ β-catenin pathway by blue light stimulation using A newly developed optogenetic tool OptoLRP6 could rescue the damage of BECs induced by Aβ25–35. This restores the expression of Claudin-5, Zo-1, and Glut-1, which are essential for maintaining the TJ structure and function of the BBB. With the restoration of these proteins, BBB permeability improves. This provides new ideas and directions for solving problems related to BBB dysfunction in AD.
Anti-inflammatory drugs in AD: Effects on the BBB and new perspectives
In the study of neuroinflammation, the development of anti-inflammatory drugs targeting the destruction of BBB by inflammatory factors is of great significance for the treatment of AD. First of all, Xuebin Zhou et al. 159 studied the zebrafish brain injury model and cell co-culture model, and found that 20 μM andrographolide could significantly reduce the expression of CypA protein and block the activation of NF-κB/ MMP-9 protein. In addition, the drug also inhibits the secretion of adhesion molecules, such as VCAM-1 and ICAM-1, which play a key role in the recruitment, adhesion, and migration of inflammatory cells, and their reduction in secretion can alleviate the damage of the inflammatory response to the BBB. This will create favorable conditions for AD therapy. Secondly, two plant compounds, gingerol, and cucurbitacin B, have demonstrated potential therapeutic value by reducing neuroinflammation by inhibiting inflammatory factors and inducing autophagy. Polymeric NPs, which inherently do not contain transferrin or its receptor, can be loaded with these plant compounds. Then, they are labeled with ligands that specifically target transferrin receptors (TfR). As a result, the modified NPs can interact with TfR on the BBB, promoting the drug's passage across the BBB and improving the pathological process of AD. 160 Furthermore, Mia R. Burke et al. 161 pointed out that although glucocorticoids are generally considered to have anti-inflammatory effects, However, in AD, there is increasing evidence that GCs sensitizing brain cells to AD-related damage-associated molecular patterns by up-regulating TLR2,162,163 NLRP3 inflammasome,164–166 and P2Y2R, 167 among others. Thus amplifying AD-related neuroinflammation and affecting cognitive functions such as learning and memory. This suggests that we need to view the inflammatory effects of glucocorticoids on AD from multiple angles.
GLP-1 and VEGF: effects on ad treatment and BBB function
The application of growth factors has a significant protective effect in neurodegenerative diseases, especially in AD.168,169 Recent studies have shown that GLP-1, a growth factor, has significant neuroprotective effects in the treatment of AD. For example, Liraglutide in a phase II clinical trial of AD patients significantly slowed the aggravation of cognitive impairment, reduced the atrophy of brain temporal and parietal lobe volume, and reduced the atrophy of total gray matter cortical volume, indicating its protective effect on neurons and reduced neuronal loss.170,171 Traditional growth factors are difficult to cross the BBB, but GLP-1 drugs can easily penetrate the BBB to reach the lesion site, which makes us see the dawn of the treatment of AD. However, it has also been reported that VEGF is involved in the pathological process of AD, 172 and blocking VEGF function may delay AD pathology. Min Zhang et al. 173 blocked VEGF function by intraperitoneal injection of bevacizumab in 5×FAD mice that developed hippocampal amyloid deposition and memory deficits at 4 months of age. After one month, the long-term memory of the mice was significantly improved, and the anxiolytic effect was evident in female mice, indicating that early VEGF blockade had a protective effect on cognitive function in 5×FAD mice. By using orbital Evans blue injection to track cerebral vascular leakage in 5-month-old male and female 5×FAD mice, the researchers discovered that bevacizumab treatment decreased Evans blue leakage in the mice's cortex and hippocampus and successfully restored BBB integrity. This study reveals that VEGF is related to BBB function, which may affect BBB integrity and impair cognitive function, providing an important basis for further exploration of AD treatment strategies.
Conclusion
In conclusion, this review emphasizes the pivotal role of the BBB in the pathogenesis of AD. Most mechanistic insights have been derived from animal studies, which have revealed that BBB dysfunction often precedes typical pathological changes and drives the progression of AD. The high-risk gene APOE4 disrupts lipid and Aβ metabolism, triggering oxidative stress and inflammation, thereby impairing BBB function. Furthermore, structural and functional alterations in ECs, pericytes, and astrocytes contribute to BBB damage, while related diseases such as atherosclerosis and diabetes exacerbate this dysfunction, accelerating AD progression.
Regarding therapeutic strategies, several promising approaches have emerged. Firstly, nanoscale drug carriers and targeted delivery systems based on BBB-specific receptors have provided new avenues for drug delivery across the BBB. Secondly, drugs that regulate BBB function, such as those modulating TJ proteins and anti-inflammatory drugs, have shown potential in restoring BBB integrity. Lastly, growth factors like GLP-1 have demonstrated neuroprotective effects by penetrating the BBB, while research on VEGF blockade has offered new insights into AD treatment.
While animal studies have provided crucial insights, their findings need to be validated in humans. Future research should focus on further exploring the feasibility of the BBB as a therapeutic target and conducting more clinical studies to confirm the mechanisms identified in animal research. This will help to better understand the role of the BBB in AD and develop effective therapies.
Footnotes
Acknowledgments
First of all, I would like to sincerely thank everyone who helped me with this paper. I would like to begin by thanking my supervisor, Mr Meng Qiang, whose advice and encouragement enabled me to gain more insights into these translation studies. It was a great privilege and pleasure for me to study under his guidance and supervision. In addition, I am honored to benefit from his personality and diligence, which will be a lifelong asset to me. I can't thank him enough. I am also very grateful to all my friends and classmates who gave me help and companionship during the preparation of the paper. In addition, many thanks to my family for their love and unwavering support all the time. Finally, I am very grateful to those who took a lot of time to read this paper and gave me a lot of advice, which is very helpful for my future study.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Department of Science and Technology Program of Yunnan Province (Grant number: 202301AT070039, 202301AY070001-233), and The Yunnan Municipal Health Commission and First People's Hospital of Yunnan Province (Grant number: KUST-KH2022012Y, 2022-KHRCBZ-B04, KHBS-2022-017). National Natural Science Foundation of China (no. 81960228). Platform Open project of Yunnan Spinal and Spinal Cord Disease Clinical Medical Center, the First People's Hospital of Yunnan Province (Project number :2024JSKFKT-23). Yunnan Provincial Key Clinical Specialty in Neurology
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.