
Abstract
Periodontitis and Alzheimer's disease (AD) are chronic disorders that share an underlying inflammatory component and emerging epigenetic mechanisms influencing disease progression. Alterations in DNA methylation are increasingly recognized as critical in modulating immune responses and neurodegenerative processes. This review examines the evidence linking periodontal inflammation to AD pathogenesis and evaluates the potential of blood-based epigenetic biomarkers for early diagnosis and risk stratification. A narrative review was conducted by integrating findings from both preclinical and clinical studies that investigated the relationship between periodontitis and AD. Emphasis was placed on research assessing DNA methylation profiles, gene expression alterations, and the impact of proinflammatory mediators on the central nervous system. Particular attention was given to studies examining the role of the immune and complement systems in mediating the effects of chronic oral inflammation. The evidence indicates that chronic periodontal inflammation can trigger systemic responses that compromise the integrity of the blood-brain barrier, thereby facilitating the accumulation of amyloid-β plaques and hyperphosphorylated tau proteins in the brain. Aberrant methylation patterns in genes related to immune regulation and protein processing suggest converging molecular pathways between periodontitis and AD. Moreover, emerging data reveal that epigenetic alterations detectable in peripheral blood closely mirror cerebral changes, opening new avenues for early detection. As a narrative review, our synthesis is hypothesis-generating and does not establish causality; the proposed epigenetic link between periodontitis and AD remains provisional pending longitudinal and interventional confirmation.
Introduction
Periodontitis and Alzheimer's disease (AD) share a chronic inflammatory background that may converge on systemic immune activation and tissue-specific responses, suggesting potential mechanistic intersections relevant to disease onset and progression.1–3 Chronic inflammation has been consistently associated with a wide spectrum of persistent clinical conditions, including periodontitis4,5 and AD.6,7 The use of epigenetic biomarkers in blood has grown significantly in recent years, and several studies propose that peripheral signatures could provide accessible readouts of disease risk, stage, and trajectory.8–12
Blood-based molecular measures are attractive because they enable minimally invasive collection, longitudinal monitoring, and integration with clinical phenotypes.13–15 Additionally, peripheral blood has been identified as a source of immune and inflammatory signals that reflect broader physiological states, including those implicated in neurodegeneration.16–19 Several investigations have shown that peripheral inflammatory mediators correlate with neuroinflammatory processes, and that leukocyte-derived molecular patterns can track pathological changes linked to AD.20–24 In other settings, epithelium-derived DNA methylation provides a minimally invasive readout, supporting its use as a source of epigenetic biomarkers. 25
Given these systemic inflammatory alterations, blood-based biomarkers may capture shared biological pathways connecting oral and neural tissues, helping to delineate risk factors and candidate mechanisms of disease crosstalk.26–29 DNA methylation provides a stable, integrative layer of regulation that responds to environmental exposures, chronic inflammation, and disease-related stressors.30–33
Epigenetic mechanisms regulate multiple aspects of cellular biology, including gene expression, immune signaling, and cell fate, and they can encode persistent molecular “memory” of prior inflammatory states.34–37 These properties position DNA methylation as both a historical record of exposure and a candidate biomarker for early cellular transformation.38–42 The relationship between chronic inflammatory processes, such as periodontitis, and neurodegeneration in AD has prompted growing interest in identifying convergent epigenetic signals that may serve as indicators of susceptibility, progression, or response to intervention.43,44 Emerging evidence suggests that epigenetic variation in accessible tissues can mirror disease-relevant changes, while also capturing systemic components of immune dysregulation that influence both oral and brain health.45–49 Related lines of research propose that periodontal disease represents a modifiable risk factor for broader inflammatory burden, which may, in turn, shape epigenetic landscapes that contribute to neurodegenerative trajectories.50,51
Periodontitis
Periodontitis is a chronic, multifactorial inflammatory disease linked to dysbiotic biofilms and progressive loss of supporting tissues, manifested as clinical attachment and alveolar bone loss. 26 An unregulated, host-dependent immune response drives irreversible tissue destruction through interactions among bacteria, host immunity, and tissue-homeostasis mechanisms. Accordingly, susceptibility reflects environmental and genetic determinants of phenotype and individual risk.52,53 However, it can lead to dysbiosis, which is associated with uncontrolled and persistent inflammation. This process is characterized by epithelial ulceration, as well as the destruction of periodontal ligament fibers and the alveolar bone.54,55 Periodontopathogens and their virulent metabolic factors induce proliferation and increased permeability of the junctional epithelium, as well as the production of tissue-destroying metalloproteinases, 56 which enable these factors to reach the underlying connective tissue. Consequently, periodontal tissues generate proinflammatory mediators that initiate a local inflammatory response.
Notably, microbial products attract proinflammatory cells, which migrate from the bloodstream into the gingival sulcus. As the inflammatory response progresses, it extends into the connective tissue and alveolar bone. However, if homeostasis is disrupted and proinflammatory mediators reach excessive levels, a loss of connective tissue and bone tissue occurs. If this inflammation persists, it progresses to periodontitis, which is characterized by an immunoinflammatory content in the deeper tissues of the periodontium. This infiltrate affects the periodontal ligament, alveolar bone, and cementum, resulting in the formation of deep periodontal pockets, clinical attachment loss, and radiographic signs of bone loss. In patients with compromised periodontal health, inflammatory mediators and periodontopathogenic microorganisms may enter the bloodstream, stimulating the production of acute-phase proteins such as C-reactive protein, fibrinogen, and haptoglobin, which are considered “biomarkers” of the systemic inflammatory response. 57 It has been reported that the systemic inflammatory response, triggered by local infectious processes and chronic inflammation, increases the risk of developing cardiovascular diseases, adverse pregnancy outcomes, diabetes complications, atherosclerosis, and rheumatoid arthritis. 53
Pathophysiology
Gingivitis results from a nonspecific inflammatory reaction in the gingival tissues due to biofilm accumulation, which alters the subgingival environment by increasing concentrations of proinflammatory mediators and connective tissue degradation products in the gingival fluid. This process promotes the excessive growth of “periodontal pathogens” within the subgingival biofilm. If the host's inflammatory response is sufficient, and there are favorable genetic and environmental factors, the lesion remains as gingivitis without progressing to periodontitis, thereby becoming a stable lesion. 57 However, if the host's immune and inflammatory responses fail to achieve such stability, and the individual is epigenetically susceptible and influenced by unfavorable environmental factors, the disease may progress and clinically manifest as periodontitis. Traditionally, periodontitis was viewed solely as an infection of microbial origin leading to periodontal destruction.
Periodontitis is a multifactorial condition in which bacteria are necessary, but not sufficient, for its development; the inflammatory infiltrate observed in gingivitis and periodontitis lesions reflects the host's response to the polymicrobial challenge presented by the biofilm.58,59 These cytokines play a role in the differentiation of B cells into antibody-secreting plasma cells, 60 which are predominant in periodontitis lesions. Recent studies have shown that these cells in diseased human periodontal tissue represent a significant source of the osteoclastogenic factor secreted by activated T cells, which are a cytokine that induces osteoclast differentiation. 53
Local inflammation in periodontitis
Periodontal inflammation serves as a physiological defense mechanism against microorganisms, which can become chronic due to the production of regulatory cytokines by epithelial cells and phagocytes during the initial phase and by lymphocytes in later stages. 61 Inflammation begins as a protective response to microbial challenge characterized by vascular dilation, increased capillary permeability, enhanced blood flow, and leukocyte recruitment. Polymorphonuclear neutrophils are the first leukocytes to respond and accumulate at the inflamed sites. These cells produce reactive oxygen species and enzymes, which contribute to local inflammation, 62 leading to the destruction of connective tissue, 63 when their recruitment is uncontrolled or when the microbial challenge is not adequately managed, as seen in chronic systemic disorders. 20 Furthermore, neutrophils act as the first line of defense in the innate immune system, performing phagocytic and microbicidal functions. 64 Monocytes also migrate to the site of inflammation, differentiate into macrophages, and produce cytokines and potentially destructive proinflammatory molecules which are associated with alveolar resorption. 64 These macrophages are activated by microbial agents to enhance their phagocytic ability and are induced by cytokines to support immunoregulation, cell proliferation,65,66 and tissue repair. 67 Recently, distinct macrophage phenotypes, such as M1 and M2 populations, have been identified. The polarization of these macrophages results in either a proinflammatory or proresolution response. Several factors amplify the inflammatory processes and increase the severity of periodontitis. An increase in cytokine levels leads to a hyperinflammatory response, resulting in tissue damage.
Neutrophils, key cells of immune system, play a crucial role in maintaining periodontal tissue homeostasis. Beyond their role in acute inflammation, neutrophils are also implicated in chronic inflammatory disorders such as rheumatoid arthritis, psoriasis, and atherosclerosis, among others. Their ability to modulate antigen presentation in lymph nodes and to produce various types of chemokines and proinflammatory, anti-inflammatory, or immunoregulatory cytokines makes them key players in the regulation of adaptive immunity.20,68 On the other hand, macrophages, phagocytic cells of the immune system, also play a significant role in periodontitis. Although their presence is relatively low in healthy periodontal tissues, their numbers increase in inflamed gingival tissue, where they can secrete proinflammatory molecules that contribute to collagen degradation and the severity of periodontal disease. Moreover, the association of macrophages with periodontal inflammation and the colonization of pathogenic periodontal bacteria, such as Porphyromonas gingivalis (P. gingivalis), highlights their role in promoting detrimental inflammatory responses.69–71 T and B lymphocytes are also crucial immune cells in periodontitis. For instance, T helper 1 (Th1) cells secrete interferon-gamma, which is involved in cell-mediated immunity, while T helper 17 (Th17) cells mediate inflammatory responses and recruit neutrophils. Their involvement in antibody production can influence disease progression.72–75
Systemic inflammation in periodontitis
Systemic inflammation associated with periodontitis is linked to the hematogenous dissemination of periodontopathogenic bacteria or the release of inflammatory mediators from periodontal tissues into the bloodstream. The ulceration of the epithelium in periodontal pockets exposes a large surface area, allowing both bacteria and their virulence factors to enter the circulation and leading to documented bacteremia in patients with periodontitis. Additionally, extraoral inflammation can arise from the ingestion of periodontal bacteria, contributing to intestinal dysbiosis and gut-mediated systemic inflammation or pulmonary aspiration that potentially leads to pneumonia. Elevated systemic inflammation associated with periodontitis can have multiple systemic complications. 1 One of the key etiological factors of periodontitis is subgingival dysbiosis, characterized by a dramatic shift from a symbiotic to a dysbiotic microbial community, which is associated with systemic inflammation.
The systemic translocation of local immune responses, as well as certain pathobionts to distant organs, 34 along with shared genetic and epigenetic factors that predispose to a hyperinflammatory response, helps explain the complex associations between periodontitis and other comorbidities. 76 In periodontitis, locally produced proinflammatory cytokines enter the circulation system and induce an acute-phase response characterized by elevated levels of C-reactive protein, fibrinogen, and serum amyloid A, contributing to atherosclerosis. This immune response triggers the release of proinflammatory molecules that characterize low-grade inflammation, a chronic, subclinical process with systemic manifestations. This persistent inflammatory state represents a significant risk factor for the development of various chronic diseases, including neurodegenerative disorders. 77 In addition, the dentogingival epithelial area, particularly the periodontal pocket epithelium in contact with the subgingival biofilm, serves as a critical interface through which local inflammation can initiate systemic effects. 78 In this context, several studies have evaluated the impact of periodontal treatment on systemic inflammation, demonstrating beneficial effects. For instance, nonsurgical periodontal therapy has been shown to reduce metabolic markers in patients with chronic conditions such as type 2 diabetes and chronic kidney disease.79,80 Similarly, periodontal therapy has been associated with a decrease in HbA1c and C-reactive protein levels, suggesting a positive effect on metabolic control and systemic inflammation reduction. 81
Periodontitis and its association with systemic diseases
Numerous studies have demonstrated associations between periodontitis and various chronic inflammatory diseases, including rheumatoid arthritis, type 2 diabetes,82,83 and cardiovascular disease. 84 It is also linked to colorectal cancer, 85 obesity, 86 Parkinson's disease, 87 respiratory infections, 88 and adverse pregnancy outcomes.89,90 Bacteremia and circulating endotoxins from periodontal lesions drive persistent systemic inflammation and endothelial dysfunction,84,91,92 which in turn promotes insulin resistance.82,83 Additionally, co-infections (e.g., viruses) may further amplify these effects.91,92 Lymphocytes activated in periodontal tissues can migrate to distant sites and exacerbate systemic inflammation; 93 chronic periodontitis can also reprogram hematopoietic progenitors via “trained myelopoiesis”, generating a sustained surplus of proinflammatory myeloid cells.94,95
In addition to metabolic and cardiovascular conditions, periodontitis has been implicated in neurodegenerative disorders such as AD and Parkinson's disease.87,96 These conditions are characterized by chronic neuroinflammation driven by activated microglia releasing cytokines, chemokines, reactive oxygen species, and proteases. Although acute neuroinflammation can be protective, persistent activation exacerbates neuronal damage and accelerates neurodegeneration.97,98 Moreover, a bidirectional oral–gut–brain axis has been proposed: experimental periodontitis can induce both gut dysbiosis and neuroinflammatory changes, 99 suggesting that periodontitis-driven systemic inflammation may perpetuate a cycle of dysbiosis and neural injury. The periodontitis-AD link has gained significant attention in recent years due to overlapping inflammatory and immune pathways between these diseases.
Alzheimer's disease
AD is a severe and irreversible neurodegenerative disorder characterized by a slow and progressive cognitive decline that interferes with both essential and instrumental activities of daily living. It primarily manifests as a memory disorder, where recent information fails to be consolidated into long-term memory.100,101 This progressive condition affects key brain regions responsible for memory and cognitive functions, leading to their gradual degeneration. Consequently, patients experience impairments in learning, reasoning, communication, and the ability to perform daily activities, resulting in a significant decline in quality of life. Additionally, AD imposes a considerable socioeconomic burden on both individuals and public healthcare systems worldwide. 102 A key feature of AD is neurodegeneration, marked by reduced neuritic arborization and synaptic dysfunction, particularly in the hippocampus. These alterations contribute to cognitive deterioration and the loss of autonomy in affected individuals. 18
Etiology and pathophysiology
AD is classified by age at onset into early-onset (<65 years) and late-onset forms; both share core neuropathology, display heritability, and occur as sporadic or familial cases, with sporadic AD predominantly late-onset. 103 However, several hypotheses have been proposed to explain its underlying mechanisms: Protein Aggregation: One of the hallmark microscopic features of AD is the presence of extracellular plaques primarily composed of amyloid-β (Aβ), which is a byproduct of the sequential cleavage of amyloid-β protein precursor (AβPP) by β- and γ-secretases. Additionally, intracellular aggregates of hyperphosphorylated tau, known as neurofibrillary tangles, accumulate predominantly in neurons. Other pathological findings include dystrophic neurites (axon and dendrite segments containing aggregated hyperphosphorylated tau), reactive astrogliosis, and activated microglia, particularly around plaques.104,105 These molecular and cellular alterations drive neurodegeneration that is characterized by synaptic and neuronal loss. 106 Neurodegeneration: The progression of AD is mediated by disrupted synaptic plasticity and neuronal integrity. Aβ1–42, a proteolytic product of AβPP metabolism, accumulates in the neuronal endoplasmic reticulum and extracellularly. The most critical pathological event is synaptic loss and selective neuronal death. Furthermore, neuronal damage is hypothesized to result from the conversion of nontoxic Aβ monomers into toxic oligomers. The abnormal accumulation of Aβ oligomers in nerve terminals has been associated with synaptic damage and neurodegeneration, highlighting their neurotoxic role.107,108 Oxidative Stress: Given the essential role of oxygen in cellular and tissue homeostasis, systems for the production and clearance of reactive oxygen species (ROS) are crucial. While ROS exhibit important physiological functions, excessive levels lead to oxidative stress, which is implicated in various diseases. The brain is particularly susceptible to oxidative damage, which disrupts neuronal membrane integrity. This process includes the oxidation of lipids, proteins, and nucleic acids, as well as the impaired clearance of Aβ due to oxidation of the low-density lipoprotein receptor-related protein 1 (LRP1). 109 Oxidative stress is believed to occur early in AD and is closely linked to Aβ presence. Elevated levels of Aβ1–40 and Aβ1–42 correlate with increased oxidative damage markers in the hippocampus and cortex, whereas the cerebellum, which exhibits low Aβ levels, does not show significant oxidative stress. 110
Infectious-inflammatory hypothesis: Aβ has been proposed to function as an antimicrobial peptide (AMP) against various microorganisms, supporting the hypothesis of a connection between infectious processes and AD. 111 Another central pathogenic event is thought to involve glial dysfunction. 112 Scientific evidence suggests that chronic inflammatory processes contribute significantly to AD pathogenesis by promoting the generation and secretion of proinflammatory mediators that interact with neurodegeneration at multiple levels, ultimately leading to neuronal death.113,114 The association between neuroinflammation and AD is supported by both neuropathological and epidemiological studies, which indicate that inflammation exacerbates the defective processing of Aβ and AβPP, thereby promoting Aβ aggregation, a process further intensified under proinflammatory conditions. 115 Microglial receptors recognize the structural components of viruses, bacteria, fungi, abnormal endogenous proteins, as well as complement system proteins, antibodies, chemokines, and cytokines, triggering microglial activation. This leads to increased proliferation and morphological changes from a ramified state to an amoeboid phenotype. 116
Local inflammation in AD
Neuroinflammation is a brain response aimed at mitigating potential damage, mediated by microglia and astrocytes. The activation process of glial cells is characterized by the expression of molecules involved in the inflammatory response, including cytokines, components of the complement cascade, acute-phase reactants, proteases, protease inhibitors, and neurotoxic products. 117 It has been shown that such inflammatory mediators are produced locally in the brain and selectively increase in the regions affected by AD. 118
There is solid evidence that certain compounds, such as CatB, CatD, CXCL10, MMP-9, and Aβ, are directly neurotoxic and are expressed and released by microglia into the extracellular space within the brain. This suggests that distinct stimuli may trigger the release of specific subsets of neurotoxins or even lead to unique features of microglial toxins.119,120 The release of proinflammatory molecules can lead to synaptic dysfunction, neuronal death, and the inhibition of neurogenesis. The complement system may also become activated, promoting microglial phagocytosis and potentially leading to inappropriate synaptic pruning. During neuroinflammation, anti-inflammatory cytokines such as IL-1RA, IL-4, IL-10, and IL-11 are produced, which may be part of a mechanism to prevent excessive inflammation. However, in neurodegenerative diseases, neuroinflammation tends to be chronic and does not resolve spontaneously, representing a key factor in disease progression. 121
Systemic inflammation in AD
There is substantial evidence supporting bidirectional crosstalk between systemic inflammation and the central nervous system (CNS). Peripheral immune cues can influence the brain through converging routes: specialized interfaces with reduced barrier stringency (e.g., circumventricular regions) that permit cytokine access; endothelial activation and transporter-mediated signaling at the blood-brain barrier (BBB); neural sensing via afferent pathways; and, under inflammatory conditions, leukocyte trafficking across a compromised barrier.122–124 Early-life infectious or inflammatory exposures can modulate CNS immune set points and increase late-life vulnerability to AD.122,125 Longitudinal studies show that plasma inflammatory proteins are elevated years before the clinical diagnosis of dementia, aligning peripheral inflammation with prodromal neurodegeneration. 126 Moreover, aging and multimorbidity sustain low-grade systemic inflammation that may accelerate AD-relevant pathophysiology. 123
Taken together, these observations support the view that chronic peripheral immune activation can exacerbate central processes relevant to AD. In this context, peripheral inflammatory inputs promote microglial priming and astrocyte polarization toward a reactive A1 phenotype, driving complement-mediated synaptic loss, increasing tau pathology via kinase upregulation and hyperphosphorylation, and stimulating the release of proinflammatory cytokines that can induce neuronal and glial injury via apoptotic and inflammasome pathways. 123
Plausible pathways linking periodontitis and AD through inflammation
As previously mentioned, evidence suggests a plausible relationship between neurodegenerative processes and chronic peripheral immunoinflammatory conditions such as periodontitis. 127 The inflammatory hypothesis proposes that chronic and progressive brain inflammation may be a key factor in neurodegeneration. The proinflammatory cascade involves the production of cytokines such as TNF-α, IL-1β, IL-6, and C-reactive protein by glial cells. These molecules, in turn, stimulate the further production of Aβ1–42, P-Tau, and other proinflammatory mediators through paracrine and/or autocrine signaling pathways. 38 Periodontitis, particularly in its moderate to severe forms, plays a significant role in systemic inflammation and its potential involvement in the etiology and progression of AD (see Figure 1).
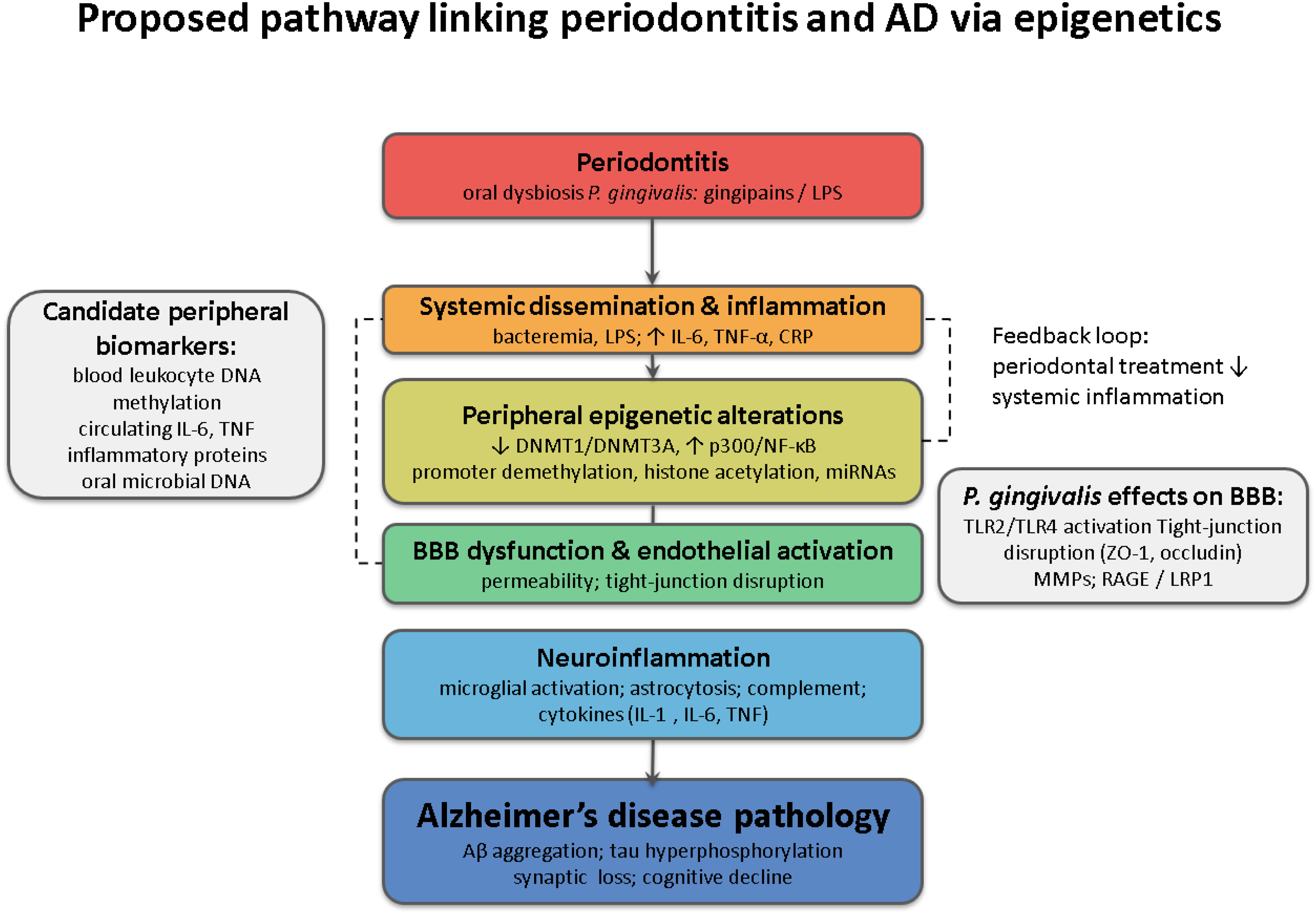
Conceptual model linking periodontitis to Alzheimer's disease through shared inflammatory and epigenetic pathways. Periodontal dysbiosis (P. gingivalis and other keystone pathogens) releases LPS/gingipains that modulate epigenetic enzymes (↓DNMT1/↓DNMT3A; ↑NF-κB/p300), promoting promoter hypomethylation and histone acetylation at inflammatory loci (e.g., IL6, TNF). Systemic spillover drives leukocyte trafficking, endothelial activation, BBB disruption (TLR2/4, tight-junction loss), neuroinflammation, and AD-relevant regulation captured by peripheral blood DNA methylation (DNAm) signatures and downstream Aβ/tau pathways.
Direct role of periodontopathogens in AD
Several bacteria closely associated with periodontitis, such as Aggregatibacter actinomycetemcomitans, P. gingivalis, and Treponema denticola, invade periodontal tissues and evade the host immune system by releasing a wide range of virulence factors. Peripheral blood cytokines such as TNF-α, TGF-β, monocyte chemoattractant protein (MCP-1), IL-8, macrophage migration inhibitory factor (MIF), interferon-γ, and IL-6 have been implicated as potential biomarkers for AD in serum and plasma. 128 Among these, TNF-α plays a critical role in neurodegeneration, as it is associated with gliosis, demyelination, BBB disruption, and apoptosis. 129 Furthermore, herpes simplex virus type 1 (HSV-1) has been proposed as a factor linking periodontitis to the accumulation of Aβ deposits, which is a hallmark of AD. 130 Species of Treponema associated with periodontitis have been isolated from the brains, blood, and cerebrospinal fluid (CSF) of AD patients, with bacterial antigens detected in the trigeminal nerve, ganglia, brainstem, and hippocampus. 131 Additionally, studies have reported associations between P. gingivalis, Treponema denticola, Tannerella forsythia, and Fusobacterium nucleatum and brain tissue in AD cases.132,133 This microorganism can cause tissue damage and invading weakened epithelial layers, contributing to systemic diseases. 134 Notably, its virulence factors, such as gingipains and lipopolysaccharides, play a crucial role in complement system activation in the brain, where Aβ plaques serve as the activator. 130
Sensory information induced by P. gingivalis LPS via IL-1β stimulation activates microglial cells. This interaction helps orchestrate emotional and behavioral stress responses through reciprocal connections with other brain regions, including the central amygdala, which serves as the brain's alarm and emotion center, and the frontal cortex, which plays a critical role in regulating complex cognitive, emotional, and behavioral functions. 135 P. gingivalis plays a significant role in AD pathogenesis through the secretion of specific peptidases known as gingipains. Furthermore, it inhibits IL-8 biosynthesis, affecting immune cell chemotaxis. 132 The identification of gingipain antigens in the brains of individuals with AD, as well as in those without a dementia diagnosis, suggests that P. gingivalis brain infection could be an early event that explains the pathology observed in middle-aged individuals before cognitive decline. Additionally, gingipain immunoreactivity in the brains of AD patients has been found to be significantly higher compared to unaffected controls. 133 The detection of P. gingivalis DNA in brains of AD patients and in the CSF of living subjects diagnosed with probable AD suggests that this genetic material could serve as a potential differential diagnostic biomarker.133,136 P. gingivalis is a potent modulator of the immune system. Its LPSs and gingipains inhibit the deposition of opsonins such as IgG, C3b, and C5b-9 on the bacterial surface, effectively blocking C3, a key component of all complement pathways. These mechanisms are fundamental to the virulence of microbial communities where P. gingivalis is present. 113 Lysine-specific (Kgp) and arginine-specific (RgpA and RgpB) gingipains are proteases that degrade complement components C1 to C5, preventing C3b deposition on the bacterial surface and capturing the regulatory protein C4b-binding protein (C4 bp). Thus, gingipains not only degrade complement proteins through proteolysis but also inhibit their activation.142 In the context of AD, these gingipains may exacerbate the effects of complement gene mutations, promoting local infections. Tau protein, a key component of tangles in AD patients, has been reported as a substrate for gingipains.113,133 Outer membrane vesicles from P. gingivalis disrupt epithelial junctions and degrade tissue, affecting genes related to the complement system. These vesicles are responsible for transducing proinflammatory signaling cascades that drive cognitive lesion development. 121 Additionally, the presence of P. gingivalis fimbriae has been associated with the dysregulation of peripheral monocytes 126 and specific T-cell subtypes in extraoral lesions, promoting the establishment of a mixed microbial community in the brain.137,138 Furthermore, studies analyzing postmortem brain tissues from AD patients have demonstrated the presence of multiple bacterial species, including P. gingivalis and Treponema denticola.133,137
Role of common inflammatory mediators
Local periodontal treatment has been shown to reduce systemic inflammatory markers and improve the progression of associated diseases. Therefore, a bidirectional relationship exists between periodontitis and certain systemic conditions. 1 Chronic inflammation induced by periodontitis produces proinflammatory molecules that affect cranial nerve endings, continuously exposing the host to cytokines and other systemic inflammatory markers, such as C-reactive protein. 138 Studies have demonstrated a significant effect of systemic inflammatory markers (CRP and IL-6) in the relationship between periodontitis and Aβ peptides.
The indirect effect of periodontitis on Aβ peptides, mediated by CRP and IL-6, has been significant. 139 Among the virulence factors, LPSs from periodontal pathogens stimulate cytokine production. 140 In recent years, the indirect relationship between chronic inflammation induced by infections such as periodontitis and various systemic diseases has gained increasing attention.43,141 Sustained elevation of proinflammatory cytokines due to periodontal inflammation can cross the BBB and activate microglial cells, establishing a link between periodontitis and AD.
Moreover, studies have explored the association between serum cytokines and AD risk. 127 However, when these molecules accumulate, as observed in AD, they promote neurodegeneration.142,143 These inflammatory molecules can enter the brain via systemic circulation or neuronal transport, triggering microglial activation, which leads to neuronal damage, cerebral amyloid accumulation, and cognitive dysfunction.144,145 Peripheral proinflammatory cytokines can also activate the vagus nerve, transmitting signals to the CNS through neural pathways. Additionally, activated peripheral monocytes can be recruited to the brain, where they secrete cytokines such as TNF-α, further activating microglia and exacerbating neuroinflammation. 146
Shared role in cerebrovascular pathology
AD and cardiovascular diseases share cardiometabolic and lifestyle-related risk factors. These same factors are linked to indicators of abnormal brain aging, including reduced brain volume and increased white matter hyperintensities observed in brain imaging studies. 147 Risk models incorporating these shared factors have been developed to assess dementia risk, integrating cardiovascular risk factors, aging markers, and dementia predictors, such as blood pressure, cholesterol levels, and diabetes. 148 Atherogenesis has been linked to an increased risk of AD and has also been associated with periodontitis.
Periodontitis has been identified as a contributing factor due to the presence of high-risk periodontopathogens.149,150 Key inflammatory and immune response agents in periodontitis include LPSs from periodontopathogens, which induce inflammatory cell infiltration into blood vessels, vascular smooth muscle proliferation, vascular lipid degeneration, and intravascular coagulation, potentially triggering thromboembolic events. Beyond thrombus formation, platelets promote AβPP formation, which subsequently increases the production of neurotoxic Aβ in the brain. These findings confirm that cerebrovascular pathology induced by periodontitis may accelerate AD progression.127,151
Role of periodontitis-induced oxidative stress in AD
Aging is associated with increased oxidative stress in the brain, contributing to the onset and progression of AD. The brain is particularly vulnerable due to its high concentration of oxidation-prone lipids, elevated oxygen consumption, and limited antioxidant capacity. Chronic periodontitis, recognized as a significant source of systemic oxidative stress, may play a role in this process. It is plausible that free radical production induced by periodontitis surpasses the body's antioxidant defenses, negatively impacting brain tissue. 152 The pathophysiology of AD is linked to the neurotoxicity of Aβ plaques and P-Tau neurofibrillary tangles, both of which are closely associated with oxidative stress. Initially, these plaques activate microglial cells, which at low concentrations serve a neuroprotective function, maintaining brain homeostasis by acting as mononuclear phagocytes. However, with aging and genetic factors, Aβ and P-Tau accumulation in neurons triggers a chronic inflammatory state in the microglia, leading to the release of inflammatory mediators such as TNF-α, IL-1α, IL-1β, and complement C1q. These mediators not only directly damage neurons but also transform astrocytes into their neurotoxic A1 variant, promoting neuronal death and exacerbating synapse destruction. 153
Recent studies suggest that exacerbated oxidative stress, resulting from mitochondrial dysfunction or the accumulation of transition metals, promotes both Aβ production and aggregation as well as the phosphorylation and polymerization of P-Tau. This establishes a vicious cycle that amplifies oxidative stress and inflammation, accelerating neurodegeneration. 154 A comparative analysis of genome-wide association study (GWAS) datasets from human populations exposed to pathogens in early childhood and AD patients found that overexpressed genes in the hippocampal transcriptome of AD were related to immune and inflammatory processes that were upregulated during infection. Early-life resistance to infections led to an immune-mediated gain of function in APOE4, CR1, and TREM2, which was later linked to Aβ deposition and late-life AD. This suggests that chronic low-grade infections and antimicrobial immune responses in the brain may contribute to neurodegeneration.
Key aspects of the blood–brain barrier in the periodontitis-AD connection
Comprised of endothelial cells, astrocytes, and pericytes, its dysfunction is closely linked to AD and is associated with neuroinflammatory processes. The BBB regulates substance exchange between the blood and the brain, playing a fundamental role in brain health and responses to external factors. 155 In the context of periodontitis, a significant factor that may trigger neuroinflammation is P. gingivalis. Its LPSs and gingipains, detected in both animal models and humans with AD, can cross the BBB. This alteration may influence key proteins such as LRP-1, RAGE, tau, and ApoE, affecting the immune response. Although P. gingivalis is not the only microorganism capable of crossing the BBB, it stands out due to its ability to induce dysbiosis even at low concentrations. 50 Certain proinflammatory mediators can be actively transported across the BBB, allowing the brain to receive inflammatory signals and respond to systemic infections. This interaction is essential for coordinating the brain's response to infections and maintaining homeostasis. 156 Dysbiosis associated with periodontitis can also affect BBB integrity and functionality. Pathogens and their byproducts may alter barrier permeability, promoting neuroinflammation and contributing to the development of neurological and psychiatric disorders. It has been speculated that bacteria may reach the brain via the trigeminal and vagus nerves, suggesting that some forms of dementia could originate from dysbiotic processes in the mouth or gut.99,124 Since periodontitis and cognitive decline share risk factors such as age, smoking, and diet, it is essential to consider the influence of confounding factors in their potential association. Cardiovascular events and strokes, which are linked to periodontitis, may directly impact cognitive function. 157 Traditionally, the advanced destruction of periodontal structures in old age has been regarded as a consequence of lifelong cumulative damage rather than a contributing factor or a result of an increased rate of destruction. 158 Both conditions share environmental and lifestyle determinants, including immune response and genetic predisposition. 159 The proper control of confounding factors is crucial for analyzing the existing literature. Furthermore, the cumulative effects of confounding factors throughout the life course may significantly influence final estimates. 160
Epigenetics in the periodontitis and AD connection
Gene expression is regulated by multiple mechanisms, including chromatin structure, transcription factors, and post-transcriptional modifications such as splicing and mRNA polyadenylation, as well as regulatory molecules like microRNAs. 161 While genetics studies biological inheritance by analyzing DNA sequences, epigenetics focuses on changes in gene expression that are not encoded in the DNA sequence, such as chemical modifications of DNA and chromatin remodeling.162,163 Although the genetic sequence is stable, the epigenome is dynamic and responds to environmental and lifestyle factors, bridging genetics and the environment. 164 Epigenetic traits can modify gene expression without altering the DNA sequence and are influenced by environmental factors, affecting disease susceptibility and longevity,165,166 and epigenetic mechanisms involve heritable and reversible changes that regulate gene expression.167,168 DNA methylation is a key focus in these studies, requiring cellular deconvolution processes to analyze cellular diversity in epigenomic profiles. 169 Cellular diversity, determined by transcription factors, growth factors, and cytokines, is crucial for cellular function and presents a technical challenge in these studies. 170 Environmental signals modulate cellular differentiation, as seen in hematopoietic stem cells, which can switch lineages due to transcriptional and environmental influences. 171 Distinct epigenetic patterns in each cell type determine gene expression and response to external stimuli, impacting disease susceptibility. 172 Alterations in these mechanisms can lead to diseases, making epigenetics essential for diagnosis, prognosis, and treatment. 173 Epigenetic mechanisms include gene silencing mediated by microRNAs and post-translational histone modifications such as methylation, acetylation, and phosphorylation, which affect chromatin structure and DNA accessibility. 174 These modifications can interact to regulate gene expression. Gene silencing occurs through mRNA degradation by the RNA-induced silencing complex or chromatin remodeling. Additionally, histone modifications, carried out by methyltransferases and acetyltransferases, influence chromatin organization and transcriptional activity.173,175,176 In periodontitis, exposure to P. gingivalis lipopolysaccharide downregulates DNMT1 and DNMT3A and perturbs histone marks at pro-inflammatory loci, facilitating NF-κB-dependent transcriptional activation in epithelial and keratinocyte models, including IL6 and CCL20.177,178 These axes overlap with AD blood DNA methylation (DNAm) findings enriched for immune and inflammatory pathways, and with methylomic signatures that correlate with CSF biomarkers of neuroinflammation and neurodegeneration.179,180 At a gene-centric level, convergence spans complement and lipid-handling loci implicated by blood epigenome-wide association study (EWAS) in AD and by transcriptional or epigenetic alterations in periodontal tissues. 178 Together, these observations provide a coherent pathway-level bridge between periodontal epigenetic dysregulation and AD-relevant immune signaling, while remaining hypothesis-generating.
DNA methylation
DNA methylation involves the addition of methyl groups (CH3) to cytosines, forming 5-methylcytosine (5mC), which is a modification primarily associated with gene silencing in regions such as transposons, imprinted genes, and the inactive X chromosome. 164 Although most CpG dinucleotides are methylated, CpG islands located in promoter regions generally remain unmethylated, allowing gene expression. 181 This process is catalyzed by DNA methyltransferases (DNMT3A and DNMT3B), which are responsible for de novo methylation, while DNMT1 maintains methylation patterns during replication. 182 Differential DNA methylation generates specific patterns that regulate gene expression across various tissues and developmental stages, influencing processes such as transcription, metabolism, and oncogenesis; the “methylome” refers to the genome-wide study of DNA methylation patterns, dynamically regulated by methyltransferase enzymes and dioxygenases such as the TET family and thymine DNA glycosylase, which participate in active demethylation. Azacitidine and decitabine, cytidine analogs, irreversibly inhibit DNMTs, leading to progressive hypomethylation, which is a process relevant to various physiological and pathological conditions. 175
Inflammation and DNA methylation
Chronic inflammation is a complex immune response central to the pathogenesis of chronic and age-related diseases. In contrast, DNA methylation signatures of the CRP could offer a more reliable assessment of chronic inflammation. As the DNA methylome reflects genome-wide variations, it serves as a powerful biomarker for low-grade chronic inflammation. Epigenetic determinants are increasingly recognized as a link between environmental factors and disease risk, with epigenetic modifications playing a crucial role in immune cell differentiation, which is a key component of the inflammatory process. 183 Moreover, low-grade systemic chronic inflammation has been recognized as a hallmark and a potential driver of individual differences in brain aging, which is consistently associated with dementia.
Moreover, low-grade systemic chronic inflammation has been recognized as a hallmark and a potential driver of individual differences in brain aging, which is consistently associated with dementia. 184 However, studies on peripheral inflammatory markers have yielded mixed results regarding cognitive performance. 185 Conole et al. identified DNA methylation of CRP as a more stable longitudinal marker, with stronger associations with cognitive performance than serum CRP levels. 186 Furthermore, systemic inflammation has been linked to cognitive decline, with evidence suggesting that this relationship is partially mediated by DNA methylation. 187
DNA methylation patterns in periodontitis
The field of epigenetics has recently been applied to dentistry, primarily focusing on inflammation, inflammatory markers, and the influence of environmental factors on oral health. 188 In the context of DNA methylation alterations, several oral diseases, including squamous cell carcinoma, tongue carcinoma, and odontogenic keratocysts, have been shown to exhibit epigenetic modifications.172,189,190 Epigenetic changes may play a crucial role in various aspects of periodontal disease progression, particularly in modulating genes involved in inflammation and immune responses. Moreover, this knowledge may help identify not only the factors that trigger the disease but also those that contribute to reduced therapeutic responses in certain patients. 163
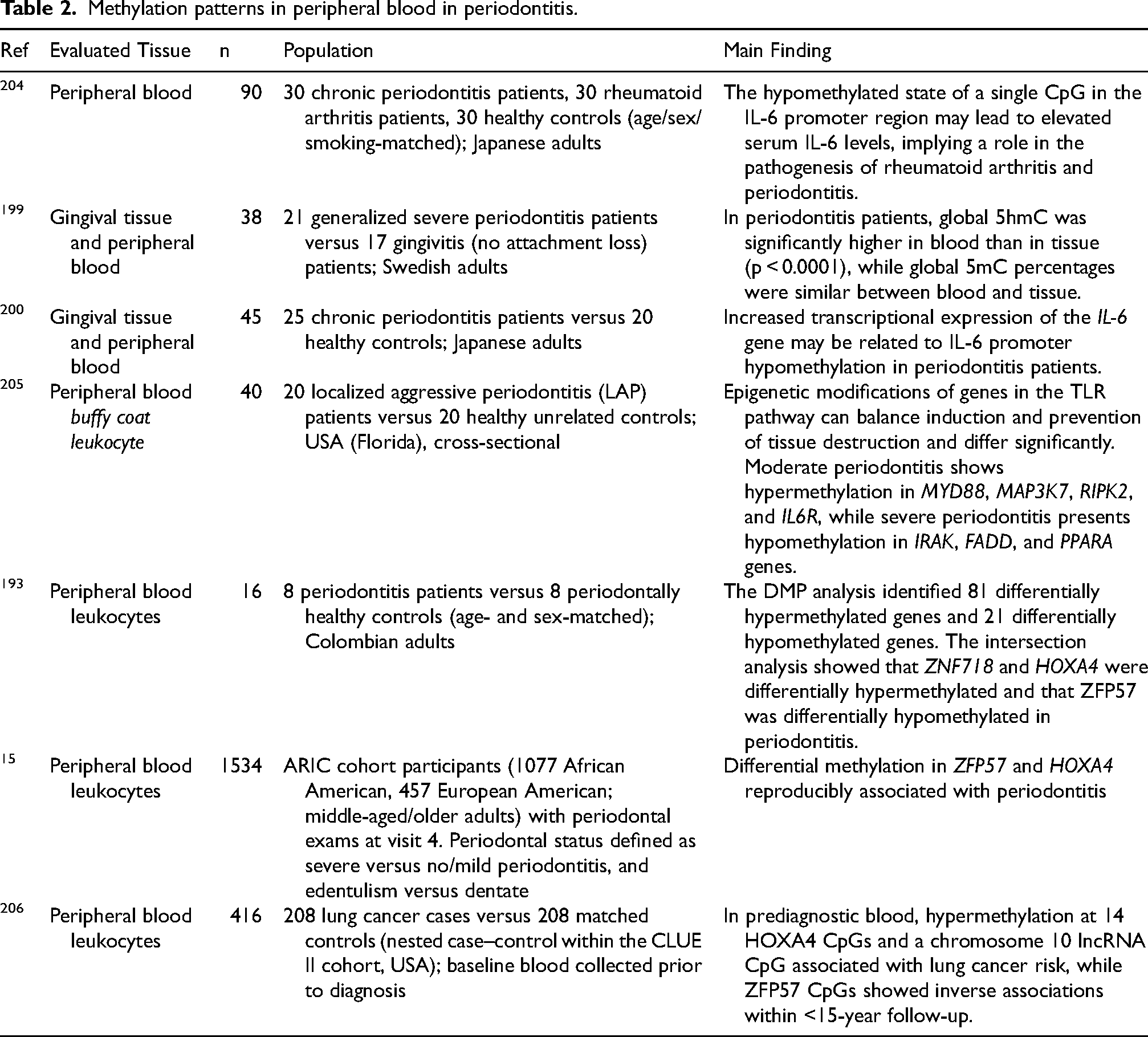
Epigenetic studies in periodontology have provided insights into how epigenetic variability accounts for differences in periodontitis manifestation among populations and individuals.2,191,192 Prior research has analyzed epigenetic alterations in gingival tissue biopsies from both healthy and inflamed sites, as well as in peripheral blood samples and experimental animal or cellular models. In patients with periodontitis, DNA methylation pattern alterations have been linked to characteristic inflammatory changes. These findings have been supported by studies conducted on both periodontal tissue and peripheral blood samples.83,193 Tables 1 and 2 summarize key findings from these studies.
Methylation patterns in periodontal tissues.
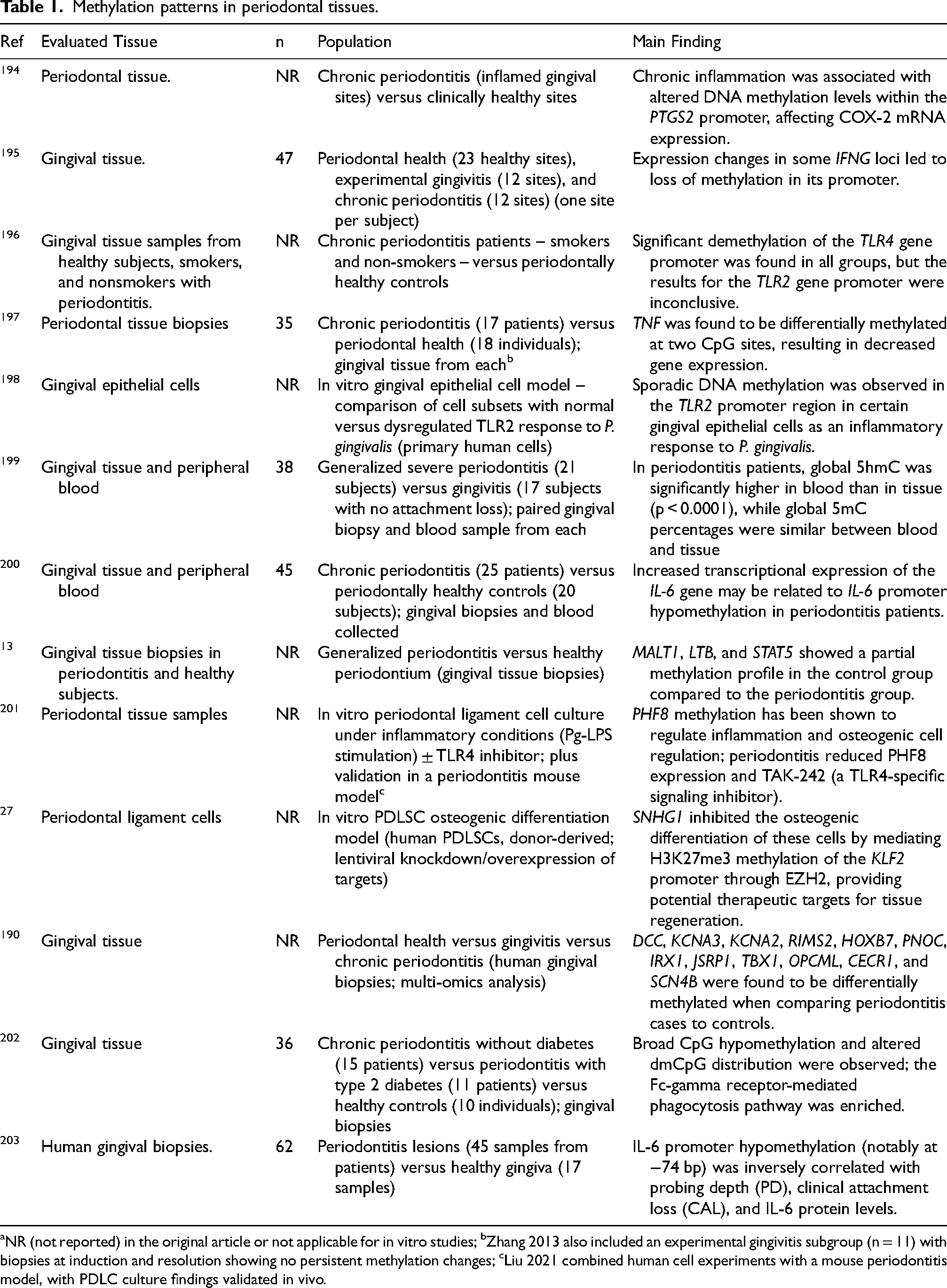
NR (not reported) in the original article or not applicable for in vitro studies; bZhang 2013 also included an experimental gingivitis subgroup (n = 11) with biopsies at induction and resolution showing no persistent methylation changes; cLiu 2021 combined human cell experiments with a mouse periodontitis model, with PDLC culture findings validated in vivo.
Methylation patterns in peripheral blood in periodontitis.
Methylation patterns associated with AD
Nongenetic factors contribute substantially to the etiology of AD; education level, hypertension, hyperhomocysteinemia, diabetes, obesity, depression, and stress have all been linked to epigenetic mechanisms. 207 Epigenetic regulation helps explain how these exposures influence AD pathogenesis. Epigenetic modifications, including DNA methylation/hydroxymethylation, histone modifications, and noncoding RNA regulation, modulate gene silencing, transcription, and post-transcriptional RNA processing, processes implicated in the onset and progression of AD.108,208 DNA methylation is highly tissue specific; however, the limited accessibility of human brain samples often necessitates the use of surrogate tissues such as blood to investigate associations between DNA methylation, brain function, and neurological health. 209 Postmortem brain analyses have focused on regions such as the entorhinal cortex, superior temporal gyrus, prefrontal cortex, and cerebellum. Tables 3 and 4 summarize findings from previous studies on this process in brain and in peripheral blood, respectively.
Methylation patterns in the brain in AD.
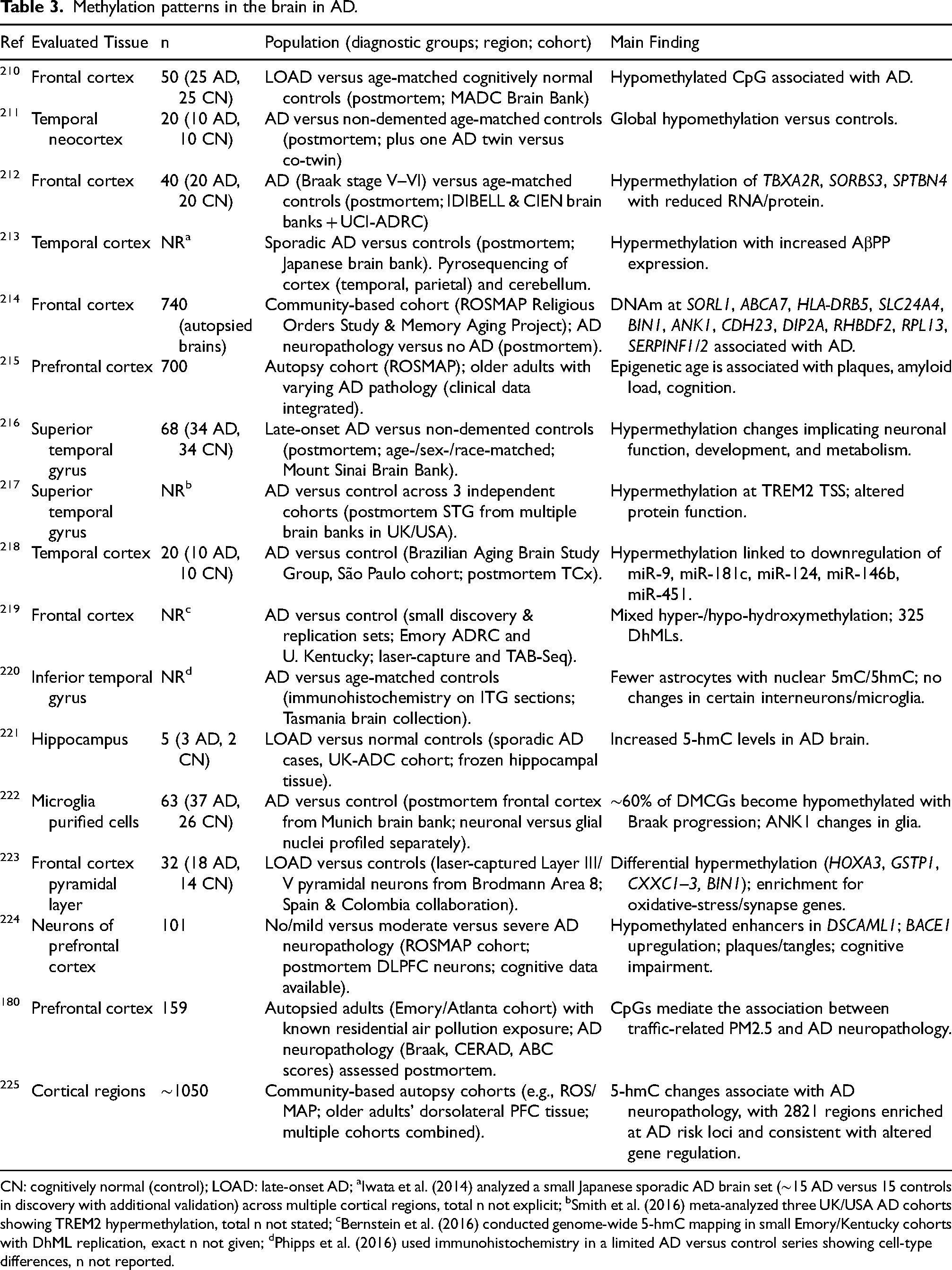
CN: cognitively normal (control); LOAD: late-onset AD; aIwata et al. (2014) analyzed a small Japanese sporadic AD brain set (∼15 AD versus 15 controls in discovery with additional validation) across multiple cortical regions, total n not explicit; bSmith et al. (2016) meta-analyzed three UK/USA AD cohorts showing TREM2 hypermethylation, total n not stated; cBernstein et al. (2016) conducted genome-wide 5-hmC mapping in small Emory/Kentucky cohorts with DhML replication, exact n not given; dPhipps et al. (2016) used immunohistochemistry in a limited AD versus control series showing cell-type differences, n not reported.
Methylation patterns in peripheral blood in AD.

AD: Alzheimer's disease; MCI: mild cognitive impairment; HC: healthy control; PBMC: peripheral blood mononuclear cell; DMC: differentially methylated CpG; DMR: differentially methylated region; EWAS: epigenome-wide association study; WGBS/WGMS: whole-genome (bisulfite) methylation sequencing; aGuan et al. did not report the control count in the abstract (study included 23 female AD and age-matched female controls); bKoetsier et al. trained on 623 EMIF-AD samples and validated in ADNI (n = 223), PPMI (n = 129), and BASE-II (n = 1017); cZhang 2025 meta-analyzed Framingham and ADNI (>1000 initially cognitively normal individuals); dChacón & Hernández 2025 reanalyzed GEO GSE59685 and GSE53740 datasets identifying differential methylation signals.
Genes associated with periodontitis and the pathophysiology of AD
Traditionally, literature has identified three genes strongly associated with the development of AD: APP, PSEN1, and PSEN2. The infection of gingival epithelial cells, gingival fibroblasts, and periodontal ligament cells with P. gingivalis and Treponema denticola induces modifications in the expression and activity of chromatin-modifying enzymes, along with both global and site-specific changes in DNA methylation and histone acetylation. 248 These genes have been shown to interfere with the physiological processing of the Aβ peptide and, due to their role in disease pathogenesis, have been included as causal genes in the new diagnostic guidelines for AD.103,249 Similarly, APOE has been extensively studied, with evidence suggesting that it influences tau-mediated neurodegeneration and microglial responses to AD-related pathologies. 250 Integrating AD GWAS with human brain proteomes identified genes whose cis-regulated brain protein abundance is consistent with a causal role in AD, including ACE, SNX32, DOC2A, and LACTB. 251
Recently, multiple studies have focused on identifying novel genes that may help elucidate the molecular pathways involved in AD. Some contribute to the increased production and aggregation of Aβ or tau (e.g., ADAM10, ABCA7, BIN1, CD2AP, CLU, FERMT2, CASSA, SLC10A2, SLC24A4/RIN3, DSG2, PLD3, BCHE, CTSD, MEF2C, APH1B, PICALM, SORL1, CR1, CD33, CST3, ABI3, PLCG2, and SHARPIN). Other genes, such as ACE, IGF1, and LKB1, are involved in altered clearance mechanisms of Aβ or tau, while UNC5C, PTK2B, and INSR have been linked to Aβ- or tau-mediated neuronal damage. SLC24A4/RIN3, BCHE, ACE, IGF1, and LKB1 have been implicated in tau phosphorylation.252,253 Given their role in AD pathogenesis, these genes represent promising candidates for future research aimed at developing novel diagnostic and therapeutic strategies.
In line with epigenetic evidence that DNA methylation proxies of chronic low-grade inflammation outperform serum CRP for brain and cognitive aging outcomes, the inflammatory gene programs below likely intersect with methylation-regulated immune pathways. 185 Functional enrichment analysis showed links to leukocyte cell-cell adhesion, phagocytosis, and transendothelial migration. 254 Previous GWAS and bioinformatics work has evaluated genetic markers for periodontitis: SNPs in NPY, IL37, and NCR2 associate with susceptibility to moderate or severe periodontitis, whereas a TLR9 marker associates with lower probability of severe disease; effects vary by sex and smoking. 255 Regarding periodontitis–AD connections, multiple genes have been implicated in onset and progression; CTSS, PLEK, IRF8, PTGS2, and FOSB emerge as candidates for periodontitis development and progression. 256
Identified genes within these pathways include IL6, IL10, IL1B, TNF, IFNG, CXCL8, CCL2, MMP9, and TLR4. 257 Regarding epigenetic relationship, as previously discussed, periodontitis and AD share multiple risk factors, with particular emphasis on the role of P. gingivalis and its impact on epigenetic modifications that may mediate this association. Defects in genes such as C1, CR1, C9, and CLU have been implicated in AD progression. Dysfunction in these genes impairs the ability of microglia to effectively remove waste proteins, including Aβ aggregates and extracellular neurofibrillary tangles. The APP gene encodes a peptide with antimicrobial properties, suggesting that Aβ accumulation may represent a response to previous infections. This supports the hypothesis that Aβ pathophysiology is linked to dysregulated innate immune responses and the microbial infection-amyloid cascade hypothesis.113,258 Lipopolysaccharides and gingipains from P. gingivalis have been shown to suppress C3b deposition, a critical convergence point of all complement pathways, thereby facilitating persistent infection. 259 Furthermore, the exposure of periodontal ligament cells to P. gingivalis LPSs induces epigenetic modifications, leading to reduced DNMT1 expression and increased NF-κB and p300 levels. 260 Similarly, the LPS has been found to downregulate DNMT3A and DNMT1 expression in keratinocytes.177,261 Taken together, P. gingivalis LPS and gingipains downregulate DNMT1 and DNMT3A while activating NF-κB/p300, promoting promoter hypomethylation and histone acetylation at inflammatory loci such as IL6 and TNF, thereby increasing transcription; this periodontal epigenetic shift is consistent with peripheral blood DNAm signatures in AD that relate to altered expression of genes such as APP and PIN1, supporting a plausible mechanistic continuum from periodontal infection to AD-relevant gene regulation.177,234,260,261 Aberrant promoter methylation profiles in genes involved in inflammatory activation, such as TLR2, PTGS2, IFNG, IL6, IL8, and TNF, have been identified in the gingival tissue, peripheral blood, and buccal mucosa of patients with periodontitis. These epigenetic alterations are associated with changes in gene expression and disease severity. Oxidative stress-related genes implicated in periodontitis reflect redox disequilibrium and inflammatory amplification relevant to neurodegenerative mechanisms.
Common genetic polymorphisms
Several studies have investigated susceptibility genes for AD through GWAS, identifying key candidates such as APOE, CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, CD2AP, and EPHA1. 262 Among these, the APOE ε4 allele is a well-established genetic risk factor for AD. Its involvement suggests that cholesterol metabolism and trafficking play a crucial role in AD pathogenesis, potentially reducing Aβ clearance either independently or through interactive mechanisms.179,263,264 Genetic polymorphisms in cytokines such as IL-1, TNF-α, and RAGE are shared risk factors for both periodontitis and AD. Variants of IL-1A-889 and IL-1B (−511 and +3953) influence AD susceptibility, with the T/T genotypes linked to increased IL-1β levels and a higher risk of both early- and late-onset AD. TNF-α polymorphisms also contribute to AD, with a haplotype on chromosome 6 showing significant association. Additionally, RAGE regulates plasma Aβ transport into the CNS, disrupting the BBB, promoting neuroinflammation, and increasing AD susceptibility. It also plays a role in periodontal tissue destruction through interactions with AGEs, leading to microvascular damage and excessive NF-κB activation.127,265
DNA methylation concordance between systemic diseases
Comparing the epigenome across different tissues is complex, yet studies have primarily focused on the correspondence of DNA methylation patterns (“methylome”) between them. Strong evidence supports the interaction between epigenetic dysregulation and inflammatory diseases.266–270 Loos and Van Dyke identified four shared genetic loci (CDKN2B-AS1, CAMTA1, VAMP3, and PLG) between atherosclerotic cardiovascular disease and periodontitis, along with a haplotype block in VAMP8, concluding that both conditions are linked to similar aberrant inflammatory pathways. 271 Inflammatory mechanisms have also been implicated in the relationship between AD and periodontitis, with DNA methylation markers emerging as potential tools for the risk prediction, screening, diagnosis, and prognosis of these conditions, which are areas with limited literature.272,273 Peripheral blood DNA methylation has been explored as a diagnostic biomarker for both AD and periodontitis.248,274 Studies assessing epigenetic markers in peripheral blood have revealed differential DNA methylation patterns in AD patients compared to healthy individuals, suggesting that a DNA methylation signature could serve as a diagnostic criterion for AD. 275 According to Cardona et al., given that total blood presents a gene expression profile like that of CNS tissues, while being more accessible, transcriptomic studies in blood could help identify pathways contributing to early disease diagnosis. 276 For instance, Jin et al. identified 48 key genes, including three transcription factors (FOS, MEF2C, and USF2) and several regulatory pathways (JAK-STAT, MAPK, NF-κB, and natural killer cell-mediated cytotoxicity), which influence intersection genes (C4A, C4B, CXCL12, FCGR3A, IL1B, and MMP3) as the most relevant candidates linking periodontitis and AD. 273 Additionally, Kubota et al. reported differential gene expression in gingival tissue affected by periodontitis, highlighting increased transcription of AβPP, IL-1β, and C1QA, as well as macrophages expressing AβPP in periodontitis-affected gingival tissues. 277 Other important genes in the progression of both AD and periodontitis include CD4, KDR, CXCR4, CXCL12, JAK2, and PTPN11. 278
Biomarkers in periodontitis and AD
As previously mentioned, numerous biomarkers have been reported as potential tools for early diagnosis and therapeutic monitoring in both periodontitis and AD. In line with recent large-scale cohorts, peripheral blood DNA methylation profiles correlate with core CSF biomarkers of amyloid, tau, neuroinflammation, and neurodegeneration, underscoring biological relevance beyond statistical association. 179 Moreover, multivariate blood-based methylation risk scores derived from EPIC-scale datasets support risk stratification for future cognitive impairment at the population level. 243 Additionally, whole-blood methylome profiling distinguishes cognitive status across mild cognitive impairment (MCI) and AD and predicts incident dementia in longitudinal cohorts, reinforcing the feasibility of minimally invasive monitoring across disease stages.245,246 In general, biomarkers offer advantages over clinical endpoints by being simpler, less costly, and amenable to repeated assessment over shorter intervals. 279 Recent literature also highlights the need for reliable biomarkers for diagnosis, prognosis, and treatment of neurodegenerative diseases.280,281 To enhance diagnostic accuracy, priority is placed on sensitivity, specificity, ease of measurement, and reproducibility. These attributes have driven the evaluation of CSF and blood biomarkers as potential predictors of neurodegenerative disease.282–285 Importantly, lack of standardization and external validation remains a key limitation in epigenetic research; thus, a multifaceted approach integrating epigenetics with other blood-based methods is advocated. 286
In dementia, decreases in epigenetic and related blood-based markers (e.g., DNA methylation levels, sirtuin activity, and BDNF expression) have been reported, suggesting that simultaneous evaluation may improve diagnostic precision; given the reversibility of epigenetic modifications, their measurement may also inform therapeutic response. 287 In the context of periodontitis, biomarkers have been investigated in both tissue and blood. A systematic review identified extracellular RNA as a promising diagnostic class, with four markers (SPRR1A, lnc-TET3-2:1, FAM25A, and CRCT1) showing over 71% sensitivity and 100% specificity in gingivitis samples, supporting point-of-care applications. 288 Additionally, microRNAs such as miR-146a, miR-155, miR-200b, miR-223, and miR-203 are strongly implicated in inflammatory and metabolic pathways, positioning them as diagnostic and prognostic candidates. 289 Epigenetic biomarkers thus open opportunities for diagnosis and prognosis in gingivitis and periodontitis, while transcriptomic profiling may enable personalized treatment and metagenomics may refine microbiome-informed strategies. 290 Collectively, these lines of evidence warrant robust, standardized designs to identify susceptibility and progression biomarkers and to establish prognostic value.
DNA methylation peripheral blood analysis provides a noninvasive and accessible method for diagnosing and monitoring AD. At the gene-centric level, peripheral blood studies have identified regulatory methylation shifts at key AD risk loci, for example APOE 5′UTR hypomethylation in blood with concordant patterns in brain tissue among AD cases, which supports the biological interpretability of blood-derived DNA methylation signals. 244 Complementarily, population-based analyses indicate that peripheral blood DNA methylation at selected loci associates with incident AD risk and with longitudinal progression metrics, supporting preclinical risk stratification potential. 180 In parallel, emerging clinical studies indicate that periodontal therapy is feasible in people with mild dementia and may yield short-term cognitive gains, though high-quality randomized evidence remains limited; registered trials in AD are underway.291–293 To operationalize this link between periodontal interventions and AD-relevant biology, forthcoming randomized trials of periodontal therapy should prospectively include peripheral blood DNA methylation endpoints anchored to AD pathophysiology, for example CSF-correlated methylomic signatures, APOE 5′UTR methylation, or multivariate methylation risk scores, enabling mediation analyses and early biomarker readouts aligned with cognitive outcomes.179,243,244
Large-scale studies have indicated that blood may serve as a surrogate for brain gene expression, especially for genes that are co-expressed between the two tissues and exhibit heritable variability in expression.288,289 Overall, these findings support the use of peripheral blood as a proxy for brain tissue and underscore the role of DNA methylation in dysregulated gene expression associated with the pathogenesis of cognitive disorders. 294 Epigenetic studies on peripheral blood have revealed differential DNA methylation patterns in patients with mild cognitive impairment or AD compared to healthy individuals. This suggests that a DNA methylation-based signature could serve as a potential diagnostic biomarker for AD. 274 Differential methylation in peripheral blood has already been used to predict disease onset and progression in autoimmune disorders, cancers, and cardiovascular diseases. Additionally, DNA methylation changes linked to both normal brain aging, and cognitive decline have been documented, with longitudinal measurements appearing to predict cognitive impairment more accurately. Blood DNA studies have identified biomarkers that may help diagnose AD and differentiate it from MCI. Associations between DNA methylation and the APOE genotype, the primary genetic risk factor for AD, have also been investigated.231,295,296 Silva et al. conducted a meta-analysis of genome-wide DNA methylation studies in blood, identifying five CpG sites associated with SPIDR, CDH6, and intergenic regions as potential diagnostic biomarkers for AD. 237 Other studies analyzing premortem blood samples have detected transcriptionally altered genes, even in the early stages of MCI.240,297,298 Additionally, efforts have focused on identifying protein changes in blood that reflect those found in CSF, aiming to develop more accessible biomarkers. Studies have demonstrated correlations between blood DNA methylation and CSF biomarkers, indicating that pathological changes in CSF are also reflected in the blood epigenome. These findings suggest that HOXA5 methylation may serve as a promising biomarker for AD.179,299 Peripheral blood analysis has also become a key tool in clinical trials to assess the efficacy of new AD therapies.
Studies on peripheral blood or peripheral blood mononuclear cells primarily provide insight into DNA methylation changes associated with disease status. In general, the blood DNA methylation profile differs between AD patients and cognitively normal individuals, highlighting the importance of analyzing omics data in both cognitively healthy subjects and those with preclinical AD. Blood biomarkers have been shown to measure drug effects on amyloid plaques and reflect changes in brain pathology and treatment response. Continuous monitoring of these biomarkers enables the evaluation of therapeutic efficacy over time.
Conclusions
Current scientific evidence suggests that periodontitis and AD share common pathophysiological mechanisms that are primarily mediated by chronic systemic inflammation and epigenetic dysregulation. Inflammation induced by periodontitis may contribute to neurodegeneration through multiple pathways, including the dissemination of inflammatory mediators, activation of the peripheral immune system, and disruption of the BBB, facilitating the entry of pathogens and proinflammatory cytokines into the CNS. Studies on epigenetic biomarkers in peripheral blood have demonstrated that DNA methylation and other epigenetic modifications can reflect gene expression alterations associated with both periodontitis and AD. These findings support the utility of epigenetic changes as diagnostic and prognostic tools, enabling the early identification of at-risk individuals and contributing to the design of personalized therapeutic strategies. Furthermore, the evidence suggests that both inflammatory and epigenetic mechanisms not only influence the progression of both diseases but may also modulate treatment response. Specifically, DNA methylation has been identified as a potential mediator of genetic susceptibility and inflammatory response modulation, highlighting its potential application in patient stratification and the monitoring of therapeutic interventions.
The growing recognition of the role of systemic inflammation in AD underscores the importance of adopting preventive approaches for chronic inflammatory diseases such as periodontitis. The identification of common epigenetic biomarkers could facilitate the development of early intervention strategies, as well as the optimization of treatments aimed at reducing systemic inflammatory burden. The association between periodontitis and the development of AD has been explored from multiple perspectives; however, a comprehensive analysis is still needed for a complete understanding. While the literature describes several potential mechanisms of interaction, no definitive pathogenic model has yet been established to explain this relationship. In this context, it is essential to continue exploring the underlying epigenetic mechanisms of both diseases. This will not only enhance the understanding of their relationship but also create new opportunities for the early diagnosis and prevention of AD through the control of periodontal inflammation.
In summary, converging evidence supports a plausible, epigenetically mediated connection between periodontitis and AD; however, current data are insufficient to infer causality. Future work should prioritize prospective cohorts with harmonized periodontal phenotyping, blood–brain multi-tissue methylation profiling, and interventional trials testing whether periodontal treatment modifies validated epigenetic biomarkers and cognitive trajectories.
Footnotes
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Universidad Santo Tomás, Colombia (project code MC-2023-DT007). The funder had no role in the study design; in the collection, analysis, or interpretation of data; in the writing of the manuscript; or in the decision to submit it for publication.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.