
Abstract
Background
The role of histone deacetylase 6 (HDAC6) in neurodegenerative diseases, particularly Alzheimer's disease (AD), has attracted significant research interest. Peroxiredoxin 2 (Prx2), a key antioxidant enzyme and HDAC6 substrate, plays a neuroprotective role against oxidative stress-mediated apoptosis.
Objective
This study systematically investigates the neuroprotective mechanism of the HDAC6-Prx2 axis in both cellular and transgenic AD models.
Methods
An AD model was established by bilateral hippocampal microinjection of Aβ1-42 oligomers in mice. The assessments of mice or their brain samples were included behavioral tests, immunofluorescence, western blot, NADP+/NADPH ratio, and oxidative stress assays. HDAC6-mediated acetylation of Prx2 was confirmed via co-immunoprecipitation, and the specific site was identified.
Results
The disruption of the HDAC6-Prx2 interaction can significantly alleviate the apoptosis of hippocampal neurons in AD mice and salvage learning/memory deficits. Inhibiting HDAC6 can increase the acetylation level of Prx2 K196, thereby enhancing its antioxidant activity. Acetylated Prx2 inhibits the excessive production of reactive oxygen species (ROS), which is mechanically linked to HDAC6-dependent neuronal apoptosis This pathway mechanistically links HDAC6 activity to oxidative stress-induced apoptosis. HDAC6-mediated deacetylation of Prx2 K196 was shown to exacerbate oxidative damage and cognitive decline.
Conclusions
The study identifies a novel pathway where HDAC6 inhibition elevates Prx2 K196 acetylation, breaking the vicious cycle of ROS and apoptosis. Dual targeting of HDAC6 activity and Prx2 acetylation status represents a promising therapeutic strategy for AD.
Keywords
Introduction
Dementia is a major cause of disability and death worldwide. It includes the types of dementia such as Alzheimer's disease (AD), vascular dementia, Lewy body dementia, and frontotemporal dementia, among which the most common type is AD.1,2 AD is a neurodegenerative disorder that is both progressive and irreversible, and has significant social consequences.3,4 It is defined by a gradual loss of cognitive function, including behavior and mental capacity, as well as a decrease in learning ability. The pathological characteristics of AD are believed to be the extracellular accumulation of amyloid-β (Aβ) in senile plaques and the aggregation and hyperphosphorylation of tau protein in neurofibrillary tangles inside neurons.5–7 These pathologies can result in apoptosis and neuronal loss, impaired neuronal function, and brain atrophy.8–10 Since the aging population is rising, the discovery of cellular and molecular mechanisms that precipitate the onset of AD is crucial. Consequently, developing effective treatment strategies remains a pressing challenge.
HDACs, a type of deacetylase enzymes, have garnered significant attention in degenerative diseases. There are 18 HDAC isoforms classified into four classes, based on their phylogenetic features: class I (HDAC1, HDAC2, HDAC3, and HDAC8), class IIa (HDAC4, HDAC5, HDAC7, and HDAC9), class IIb (HDAC6 and HDAC10), and class IV (HDAC11), with HDAC6 having unique features that distinguish it from other HDACs.11,12 Unlike other HDACs, HDAC6 is localized in the cytoplasm and uniquely contains two homologous catalytic domains.13,14 The expression of HDAC6 protein increases significantly in various neurodegenerative diseases, including AD.15–17
Previous studies have shown that administration of HDAC inhibitors can alleviate cognitive impairment in AD mice models by targeting histone acetylation.18–21 Moreover, recent research indicates that epigenetic factors regulating memory-related genes and long-term synaptic plasticity may be linked to the pathogenesis of AD. Histone acetylation in the hippocampus is regulated by Histone acetyltransferases (HATs) and HDACs, which play crucial roles in memory formation. Inhibition of HDAC6, which increases gene acetylation, promotes hippocampal-dependent memory formation and hence is a critical factor in regulating learning and memory function in AD.22,23
Oxidative stress-mediated neuronal death plays a part in the pathogenesis of various neurological diseases during their progression.24,25 Peroxiredoxins (Prxs) are antioxidant enzymes responsible for decomposing peroxides such as H2O2 and reactive oxygen species (ROS), regulating cell signal transduction by modulating redox signaling.2,26,27
The Prxs family is composed of six isoforms (Prx1-Prx6), with Prx2 being an abundant neuron-specific Prx located in the cytoplasm of neurons in the brain.28,29 Prx2 not only regulates oxidative stress in neurodegenerative diseases but also plays a role in cancer, pulmonary hypertension, hematological, and other diseases.30,31 Numerous studies reveal that Prx2 overexpression occurs in AD and exerts a significant neuroprotective role in oxidative stress-induced cell death, delaying apoptosis.32–34 Notably, age-related cognitive function decline is accelerated with a reduction of Prx2. The acetylation of Prx2 plays a pivotal role in regulating nerve cell death associated with neurological diseases. The acetylation of Prx2 participates in the regulation of pathological processes in neurological diseases through multiple mechanisms, with its core roles primarily including anti-oxidative stress, inhibition of neuronal apoptosis, and maintenance of cellular functions.28,35 The involvement in this elevated neuronal antioxidant response under oxidative stress means that acetylation of Prx2 can enhance its reducing activity and its resistance to superoxidation and transition to high molecular-mass complexes. Oxidative stress can severely damage DNA, proteins, and lipids, initiating neuronal apoptosis due to ROS generation. 36 However, acetylated Prx2 can protect against neuronal apoptosis by reducing ROS production through anti-oxidant stress.35,37
HDAC6, as a deacetylase enzyme, plays a crucial part in redox regulation and cellular stress response. 38 In this study, we discovered that Prx2 is a novel substrate of HDAC6, and HDAC6 regulates Prx2 antioxidant capacity through modulating its acetylation at K196 site. HDAC6-Prx2 signal pathway has a close association, and HDAC6 deficiency leads to an increase in the acetylation level of Prx2. This study conducts a detailed study on whether the HDAC6-Prx2 signal pathway protects HT22 murine hippocampal neuronal cells against oxidative stress-mediated cell death and prevents hippocampal neuronal apoptosis for improved cognition by regulating ROS levels. Furthermore, the HDAC6-Prx2 signal pathway may offer a potential therapeutic strategy for AD.
Methods
Animals
Professor Yao from Duke University School of Medicine kindly provided adult male HDAC6 knockout (HDAC6−/−) mice, while adult male C57BL/6 (WT) mice were acquired from the animal model institution of Nanjing. 39 The mice were housed under controlled temperature, humidity and light conditions (Animals were kept under a 12 h light/dark cycle) with water and rodent chow diet freely available. All mouse used in the experiments were males aged 10 weeks old and weighed between 25–30 g. All mouse experiments were conducted in accordance with guidelines of Institutional Animal Care and Use Committee (IACUC) and under an approved IACUC protocol of Xuzhou Medical University (IACUC 20140404W009). The euthanasia of mice using carbon dioxide was performed in strict accordance with the guidelines established by IACUC.
Antibodies and plasmids
Commercially available primary antibodies used were as follows: anti-HDAC6 (cat# 7612, 1:1000, CST), anti-Bax (cat# 14796, 1:1000, CST), anti-Bcl-2 (cat# 3498, 1:1000, CST), anti-Cleaved Caspase 3 (cat# 9661, 1:1000, CST), anti-β-actin (cat# ab8227, 1:1000, Abcam), anti-mouse IgG (cat#115-035-003, 1:2000, Jackson lmmuno Research Laboratories), anti-Myc (cat# 2276, 5 μg/sample for IP, 1:1000 for WB, CST), anti-Flag (cat# F3165, 5 μg/sample for IP, 1:1000 for WB, Sigma-Aldrich), anti-Acetylated-Lysine (cat# 9441, 1:1000, CST), anti-Aβ1-42 (cat# ab201060, 1:200 for IF, Abcam), anti-NeuN ((cat# ab177487, 1:200, Abcam), and DAPI (cat# D9542, 1:10000 for nuclear staining, Sigma-Aldrich). All fluorescent-labeled secondary antibodies were purchased from Vector Laboratories, Inc. (United States). All the plasmids such as HDAC6, wild-type Prx2, acetylation-mimetic K196Q mutant, and acetylation-deficient K196R mutant were obtained from Shanghai Genechem Co., Ltd (China).
Drug administration
Aβ1-42 oligomers were prepared using the following described method. In brief, 1 mg of human beta amyloid (1-42) (Aβ1-42) (Millipore, USA) was dissolved in 1% NH4OH at a concentration of 1 mg/mL and sonicated for 30 s to 1 min until it was fully dissolved. Subsequently, 1.49 mL of phosphate-buffered saline (PBS) was added to 1 mg of solution Aβ1-42 to prepare a concentration of 0.67 mg/mL, and the mixture was incubated at 37°C for 4–5 days. Aliquots were then stored at −80°C until further use.
The detailed protocol for Aβ1-42 administration in the dorsal hippocampus of the brain has been described previously.40,41 Adult male mice were anesthetized using 2% isoflurane and placed in a stereotaxic apparatus (Kopf Instruments, USA). The area of incision was disinfected with 75% ethanol, and a hole was drilled through the left part of the skull. Subsequently, the mice were bilaterally injected into the dorsal hippocampus (coordinates from Bregma: AP = –2.0 mm, ML = ±1.5 mm, and DV = −2.0 mm) using a 10 μl microsyringe with either 5 μl of aggregated Aβ1-42 (0.67 mg/ml) or 5 μl of 0.9% saline (control group) at a rate of 0.5 μl/min. 40 These injections were administered over six days at a specific time of day. Following a seven-day period post-injection, the mice were trained in the Morris water maze (MWM) over six days, and all group performances were recorded.
Morris water maze test
The MWM test is a well-established method of evaluating the spatial learning and memory abilities of mice.42,43 The test comprises of a five-day acquisition trial and a one-day probe test. The experiment takes place in a circular water pool, with water at a depth of 40 cm and a temperature of 22 ± 1°C. White ink is added to the water to render it opaque, and the maze is partitioned into four quadrants. A transparent platform (10 cm in diameter) is hidden 1 cm below the water surface and placed at the midpoint of one quadrant, which is removed on the last day for the probe test. During the acquisition trial, the mice are subjected to four contiguous trials per day for five consecutive days, with a hidden platform. The time limit is 90 s, and if the mouse reaches the platform within that time and stays there for 10 s, the clock stops automatically. If a mouse cannot find the platform in 90 s, it is manually moved to the platform and allowed to stay on it for 15 s. The escape latency of the mice is recorded, and the average escape latency is analyzed for each day (using ANY-maze). During the probe test on the sixth day, the platform is removed, and the mice are allowed 90 s to search for the absent platform. The mice's time spent in the target quadrant (where the platform was located during hidden platform training) and the distance traveled are recorded and analyzed.
Open field test
The open field test involved recording the spontaneous locomotor activity of mice in a square arena (50 cm wide × 50 cm deep × 40 cm high). Before the test, the mice were acclimatized to the testing room for 30 min. Once inside the open field, the mice had the freedom to explore for 3 min, and their spontaneous activity was monitored using a computational tracking system. Standard parameters used to assess locomotor activity (ambulatory distance) and exploratory behavior (time spent in the center and periphery) were recorded using ANY-maze. The room temperature was kept constant, and noise kept to a minimum. To avoid any scent cue, the apparatus was cleaned with 75% ethanol before and after each test.
Aβ immunofluorescent staining
Mice were anesthetized and perfused to harvest brains. The brain tissues were fixed with 4% paraformaldehyde for 4–6 h, then sectioned into 40 μm-thick slices using a vibratome. After air-drying the brain slices, antigen retrieval was performed. Sodium citrate antigen remediation solution (Proteintech, PR30001) was diluted with double-distilled water (ddH2O) at a ratio of 1:50. Immerse the brain slices in the diluted solution, heat at 100°C for 15 min, and then let them cool down to room temperature prior to subsequent steps. Following retrieval, the slices were removed and washed three times with PBS, 5 min each time. A 0.05% Triton X-100 solution was prepared for permeabilization at room temperature for 30 min. After permeabilization, 10% goat serum was prepared for blocking at room temperature for 1 h. Subsequently, the prepared primary antibody anti-Aβ1-42 dilution was added to the slices, which were then incubated overnight at 4°C. After washing with PBS, the slices were incubated with the corresponding fluorescent secondary antibody at room temperature for 1 h. After secondary antibody incubation, the slices were washed three times with PBS, followed by incubation with DAPI for 10 min. Finally, the slices were washed three times with PBS again and mounted with anti-fluorescence quenching mounting medium. Fluorescent images were captured using a confocal laser scanning microscope (LSM880, Zeiss, Germany).
DHE staining
To evaluate ROS generation in hippocampal neurons, dihydroethidium (DHE) fluorescence assay was performed on brain cryosections, as described previously.44,45 Briefly, coronal brain sections (15 μm thick) were prepared using a cryostat microtome. After fixation in 4% paraformaldehyde, sections were incubated with 10 μM DHE (Sigma-Aldrich) in light-protected humidified chambers at 37°C for 30 min. Following three washes with phosphate-buffered saline (PBS, pH 7.4), nuclei were counterstained with 1μg/mL DAPI (4’,6-diamidino-2-phenylindole) for 10 min at room temperature. Fluorescence imaging was performed using a fluorescence microscope equipped with TRITC (Ex/Em: 535/610 nm) and DAPI (Ex/Em: 358/461 nm) filter sets. Quantitative analysis of DHE fluorescence intensity, reflecting superoxide anion levels, was conducted using ImageJ software (NIH) with consistent threshold settings across all samples.
TUNEL staining
Apoptotic cell death was quantified using the TUNEL assay (Roche, Germany) according to manufacturer's protocol with modifications.46–48 Briefly, mouse brains were harvested and immediately frozen in optimal cutting temperature (OCT) compound. Coronal sections (15 μm thick) were obtained using a cryostat (Leica CM1950) at −20°C chamber temperature. Prior to staining, the sections were fixed with 4% paraformaldehyde for 15 min and washed twice with phosphate-buffered saline (PBS) before being pretreated in 0.5% Triton X-100 in PBS (pH 7.4) for 1 h and washed twice with PBS. The TUNEL reaction agent, protected from light on ice, was applied by incubating the cells for 1 h at 37°C in a moist chamber. Finally, apoptotic cells were observed as fluorescein-stained neurons using a fluorescence microscope (BX-43, Olympus, Tokyo, Japan).
Immunofluorescence staining
Immunofluorescence analysis was performed as previously described. 49 Initially, 15 μm frozen sections were incubated in a 37°C incubator for 30 min and subsequently washed with PBS. The slices were then fixed with 4% paraformaldehyde for 15 min. After thorough cleaning with PBS, they were blocked with 0.1 M PBS containing 10% donkey serum and 0.3% Triton-X for 1 h at 37°C. Brain slices were incubated with primary antibodies, Rabbit anti-NeuN, diluted with 0.3% Triton X-100 in PBS, overnight at 4°C. After washing in PBS, the secondary antibody was added and incubated at room temperature for 1.5 h and then washed in PBS. Finally, the sections were mounted with an anti-fluorescence quencher and observed and photographed under a fluorescence microscope.
Immunoblotting
Mice brain and cells tissues were harvested for western blot as described previously.47,48 Tissues were lysed/homogenized in RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100) and 1 mM phenylmethanesulfonylfluoride (PMSF), 1 mM EDTA, 5 mM sodium fluoride, 2 mM sodium orthovanadate and protease inhibitors, 15 min at 4°C and centrifuged at 12,000 × g. Tissues lysates were analyzed by western blotting with Rabbit anti-HDAC6, Rabbit anti-Bax, Rabbit anti-Bcl-2, Rabbit anti-cleaved caspase-3, Rabbit anti-β-actin. Primer antibody was incubated overnight at 4°C. The membranes were incubated with a horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody or anti-mouse IgG at RT for 2 h. All membranes were normalized to the endogenous control β-actin. Immunoreactive bands were visualized using enhanced chemiluminescence (Odyssey, LI-COR Company, USA). The images were obtained under identical settings and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Cells treatments
The murine hippocampal neuronal cell line (HT22) was obtained from ATCC cell lines and maintained in Dulbecco's modified Eagle's medium (DMEM, Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Sijiqing, China) and 0.1% penicillin/streptomycin (Vicmed, China) at 37°C with 5% CO2. Subculture was carried out based on cell density. The concentration and treatment duration of Tubastatin A (TubA) used in HT22 cells were determined based on previously well-established studies focusing on the inhibition of HDAC6 expression.38,50,51 Subsequently, the HT22 cells were randomly divided into four groups: 1) Control group: HT22 cells treated with an equal volume of DMSO without additional interventions; 2) Aβ1-42 + Prx2 group: HT22 cells transfected with the Prx2 plasmid, followed by treatment with 5 μmol/L Aβ1-42 for 24 h; 3) Aβ1-42 + Prx2 + TubA group: HT22 cells transfected with the Prx2 plasmid and treated with Aβ1-42, then cultured with 2.5 μmol/L TubA for 12 h; 4) TubA group: HT22 cells treated solely with 2.5 μmol/L TubA for 12 h.
Cell transfections
Cells cultured at approximately 70% confluence were transfected with plasmid DNA using Lipofectamine 2000 (cat. 11668, Invitrogen) following the manufacturer's instructions as previously described. 52 Meanwhile, before our studies with these plasmids, we consistently achieved a transfection efficiency of ∼60–70% (assessed via EGFP labeling and flow cytometry) when using the same transfection reagent and cell seeding density as in the current work. For plasmid transfections in HT22 cells, three Prx2 variants—wild-type Prx2, acetylation-mimetic K196Q mutant, and acetylation-deficient K196R mutant—were individually transfected. Additionally, co-transfection experiments were performed with Prx2 and HDAC6 plasmids. The transfection procedure was conducted as follows. Two 1.5 ml EP tubes, referred to as A and B, were prepared before beginning the experiment, and 50 μl of empty medium (a penicillin-streptomycin mixture (PS) and DMEM without FBS) was added to each. To tube A, 2 μg of plasmid was added. To tube B, an equal volume of transfection reagent was added and both were left to stand for 5 min. The transfection reagent was then mixed into the plasmids and left for 8 min. Finally, the medium in the culture dish was replaced with the empty medium and the mixture in tube A was evenly added to the culture dish. After 12 h, the culture medium in the dish was changed to complete medium (DMEM with FBS and PS).
Immunoprecipitation
The immunoprecipitation was performed as previously described. 52 Tissues and cells were lysed/homogenized in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, 1 mM PMSF, 1 mM EDTA, 5 mM sodium fluoride, 2 mM sodium orthovanadate, and protease inhibitors). Tissue protein extraction followed the same protocol as western blotting, while cells were centrifuged at 20000×g for 20 min at 4°C. For immunoprecipitation, 1 mg of protein lysate was incubated with anti-Myc antibody or anti-Flag antibody overnight at 4°C with gentle rotation. Next day, 50 μl protein G beads (11719416001, Merck) were added and incubated for 4 h at 4°C. Beads were washed thoroughly, centrifuged at 2600×g for 3 min at 4°C, resuspended in 1× loading buffer, and boiled at 100°C for 5 min. Proteins were separated by 8–15% SDS-PAGE, transferred to PVDF membranes, blocked with 5% milk, incubated overnight with primary antibodies, and then with corresponding anti-IgG secondary antibodies for 2 h at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence (Odyssey, LI-COR, USA) under identical settings, and images were analyzed using ImageJ software. Graphical display data were presented as mean ± standard error of the mean (SEM), Unpaired t-test was used for single two-group comparisons.
Detection of intracellular ROS
To measure the intracellular ROS level, an ROS assay kit (Kaiji, China) was utilized. 51 Cells were loaded with 50 μM DCFH-DA at 37°C for 30 min under a humidified atmosphere of 5% CO2, and then washed three times with PBS. Following treatment, cells were suspended in 200 μl PBS and intracellular ROS generation was assayed using a fluorescence microscope.
Measurement of intracellular NADP/NADPH ratio
Amplite™ Fluorimetric NADP/NADPH Ratio Assay Kit (AAT Bioquest, USA) was used.53,54 Intracellular NADP/NADPH was measured by enzymatic cycling methods as per instructions. The medium was removed from plate wells, and about 100 µL lysis buffer per 1–5 million cells (or 50–100 µL/well in a 96-well cell culture plate) was added, and kept at room temperature for 15 min. After adding the cell lysate, incubate at room temperature for 15 min, followed by centrifugation at 12,000×g for 5 min, and the supernatant was used for tests. The reaction was incubated at room temperature for 15 min to 2 h and protected from light. Fluorescence increase was monitored using a Fluorescence Microplate Reader (Synergy2, USA) at Ex/Em = 540/590 nm.
AAV injection
The generation and injection of Adeno-associated virus-9 (AAV9) viruses were carried out as previously described. 52 The expression of Prx2(K196Q) and Prx2(K196R) were driven by the CAG Promoter. The AAV viruses were packaged using the AAV9 serotype by Brain VTA (Wuhan, China). For intracranial injections, the mice were first anesthetized and then fixed a on stereotaxic plate (RWD Life Science, Shenzhen, China). The superior hippocampus was chosen for drilling with a hand drill (RWD). Subsequently, 1 μL of the transfected AAV virus was injected into the mouse hippocampus at the coordinates AP −2.0, ML +1.5, and DV −2.0 via stereotactic injection. The injection process lasted for 5 min. After the injection, the syringe was left in place for an additional 5 min before being pulled out. After that, the drilling holes were sealed, and the wound was sutured. Follow-up tests were conducted 4 weeks later.
Statistical analysis
Data were analyzed via SPSS 26. The Shapiro–Wilk method was used to analyze whether quantitative data followed a normal distribution. Data were presented as mean ± standard error of the mean (SEM) for graphical display, while mean values and standard deviation (SD) were reported in the text. Unpaired t-test was used for single two-group comparisons, and one-way ANOVA was applied for multiple-group comparisons. For the MWM, data from the place navigation test were analyzed using repeated measures ANOVA, while other data were examined by two-way ANOVA. After testing the homogeneity of inter-group variances, post-hoc multiple comparisons were performed: the LSD test was used if variances were homogeneous, and the Dunnett T3 method was applied if variances were heterogeneous. Statistical significance was defined as a p-value < 0.05, with significance levels marked as *p < 0.05, **p < 0.01, and ***p < 0.001.
Exclusion criteria: Samples with incomplete recording; Outliers identified using the Grubbs’ test (α = 0.05); Animals that failed to meet the minimum task requirements (Minimum task requirements for exclusion in the MWM test: 1) The average latency to find the hidden platform exceeded 60 s for three consecutive training sessions; 2) The average running time per trial was consistently longer than 45 s, with no obvious tendency of reduction; 3) The correct rate (number of correct trials/total training trials) was lower than 30% after the completion of all training sessions. Additionally, mice were excluded if they exhibited abnormal behaviors or physical discomfort, such as refusing to swim, being injured, or showing signs of illness. Minimum task requirements for exclusion in the open field test: 1) The total distance traveled in the 5-min test period was less than 30% of the average distance of the same batch of mice; 2) The duration of immobility exceeded 5 min, indicating excessive stress response; 3) Persistent abnormal behaviors such as curling up or trembling were observed throughout the test.).
Results
HDAC6 deletion can ameliorate learning and memory ability and improve environmental adaptability in AD mice model
The loss of HDAC6 is known to have positive effects on cognitive function, including improving learning and memory abilities and environmental adaptability in AD mice models. To further explore this topic, a study was conducted to evaluate whether the loss of HDAC6 in the hippocampus could prevent impairments in spatial learning and memory that are caused by i.c.v. injection of soluble Aβ1–42 in WT mice and HDAC6−/− mice. 55 The MWM place navigation task was used to test the mice's ability to find a hidden platform during 5 consecutive days (Figure 1A, B). Swim speed was comparable in all groups, indicating no impairments in motor performance. The results showed that while there was no initial distinction, the escape latency (the time taken to find the platform) of WT injected with Aβ1–42 (Aβ1–42 group) was significantly prolonged compared to the WT injected with normal saline (control group) and HDAC6−/− injected with normal saline (HDAC6−/− group) groups (Figure 1C). However, the HDAC6−/− injected with Aβ1–42 group (Aβ1–42 HDAC6−/− group) displayed a significant increase in target preference as compared to the Aβ1–42 group (Figure 1C). Therefore, the results suggest that the loss of HDAC6 can significantly improve spatial learning impairment in a mouse model for AD.
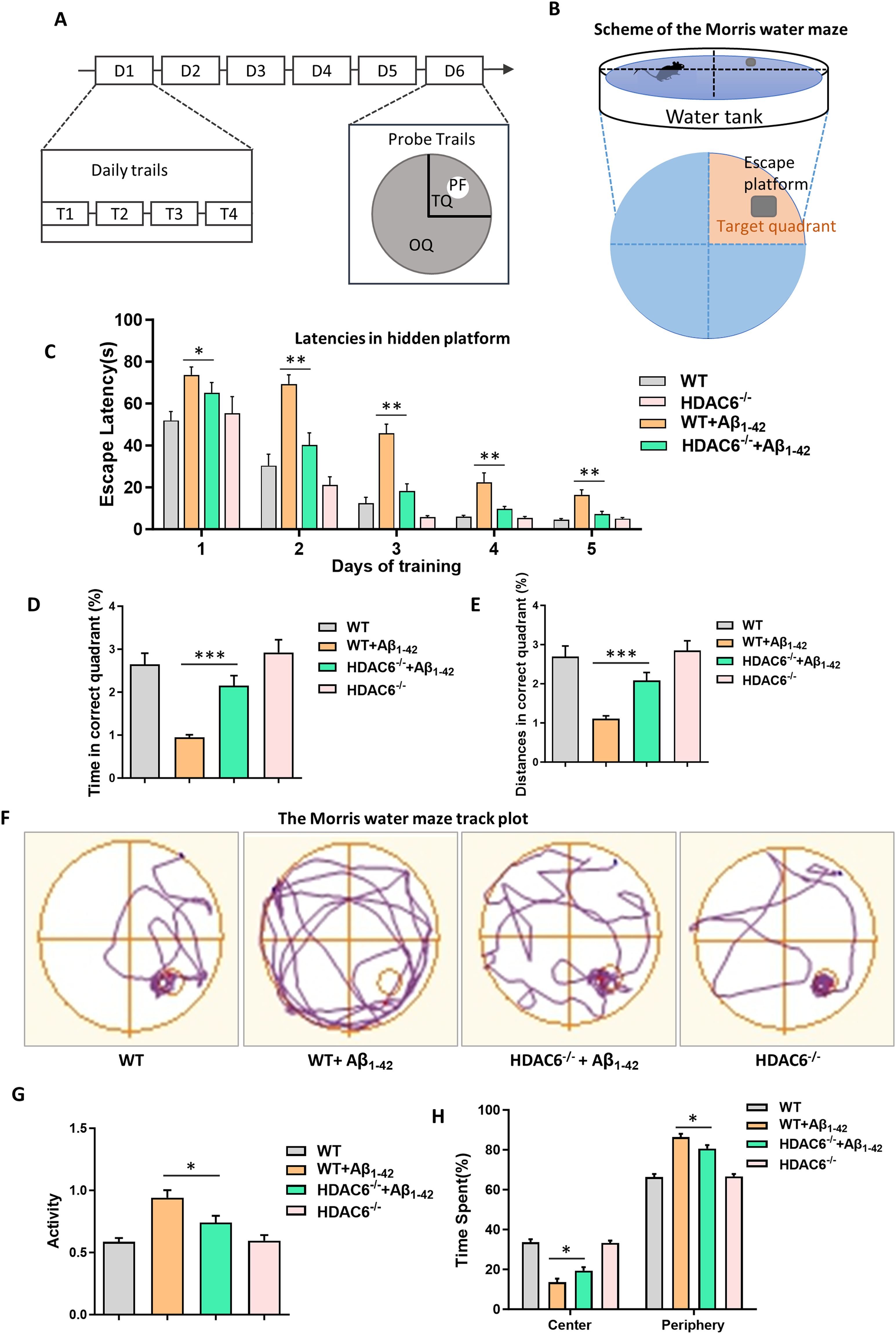
HDAC6 deletion can ameliorate learning and memory ability and improve environmental adaptability in AD mice model. (A, B) Training phase consisted of daily sessions (four trials per session: T1, T2, T3, and T4) during five consecutive days (D1–D5). The last trial the platform was removed, and memory retention was assessed during the probe trial on the sixth day(D6). PF: platform (white); TQ: target quadrant (small segment); OQ: other quadrants (large segment). Water maze illustration. (C) Average escape latency to find the hidden platform during 5 consecutive training days for four groups. (D) Swimming time in correct quadrant during the probe test for four groups. (E) Distances in correct quadrant during the probe test for four groups. (F) The upper panel indicates representative swim paths during the probe test. (G) Total activity of all groups in the open field. (H) Time spent in the center and periphery in the open field for all groups. The data are expressed as mean ± SEM. (n = 8) *p < 0.05, **p < 0.01, ***p < 0.001.
After acquisition trial, spatial memory ability of mice was tested by the probe test in the 6th day. The platform was removed, and the mice's trajectory was tracked via the video tracking system to record the time and distances to the original platform location. We observed that the Aβ1–42 group spent less time and swam less distances in the target quadrant compared with control group and HDAC6−/− group. However, the Aβ1–42 HDAC6−/− group spent more time and swam more distances in the target quadrant compared with the Aβ1–42 group, but spent less time and swam less distances compared with control group and HDAC6−/− group (Figure 1D, E). Through the trajectory, we found that Aβ1–42 group swam randomly with little or no preference for the platform location compared with the control group and HDAC6−/− group. But Aβ1–42 HDAC6−/− group swam more than Aβ1–42 group in the platform location (Figure 1F). These findings indicate that deficiency of HDAC6 can prevent the AD-induced spatial memory impairment.
Next, we tested exploratory behavior to observe anxiety state in the open field paradigm. When compared to the control group and HDAC6−/− group, Aβ1–42 group appeared abnormal autonomic behavior, which manifested as hyperactivity enhancement. However, the direct comparison of Aβ1–42 group to the Aβ1–42 HDAC6−/− group revealed that loss of HDAC6 rescued a mild hyperactivity phenotype observed in Aβ1–42 group (Figure 1G). In addition, we also analyzed the activity in the center and the periphery of the open field. The results showed compared with the Aβ1–42 group, Aβ1–42 HDAC6−/− group spent more time in the central area and less time in the peripheral area, indicating that basal anxiety levels were markedly altered (Figure 1H). This outcome reveals that the lack of HDAC6 is capable of relieving anxiety-related phenotypes in AD mice, accompanied by an increased duration of their activities in the central region of the test arena. In conclusion, HDAC6 deficiency can ameliorate autonomic behavior compared with AD mice.
HDAC6 suppression can attenuate neuronal apoptosis and reduce ROS and Aβ1–42 expression
The pathogenesis of AD involves a cascade of events initiated by the accumulation of Aβ. This leads to tau protein pathology, subsequent neuronal death, and ultimately, neurodegeneration. 56 In our study, to determine the protective effects of HDAC6 knockout against Aβ pathology, we performed immunofluorescence for Aβ1–42 expression in the hippocampal CA1 regions of each group mice. The results revealed that the increased levels of Aβ expression in the Aβ1–42 group and Aβ1–42 HDAC6−/− group compared with control group and HDAC6−/− group; meanwhile HDAC6 knockout mice performed a noticeable reduction in the Aβ expression within the Aβ1–42 group (Figure 2A, B).
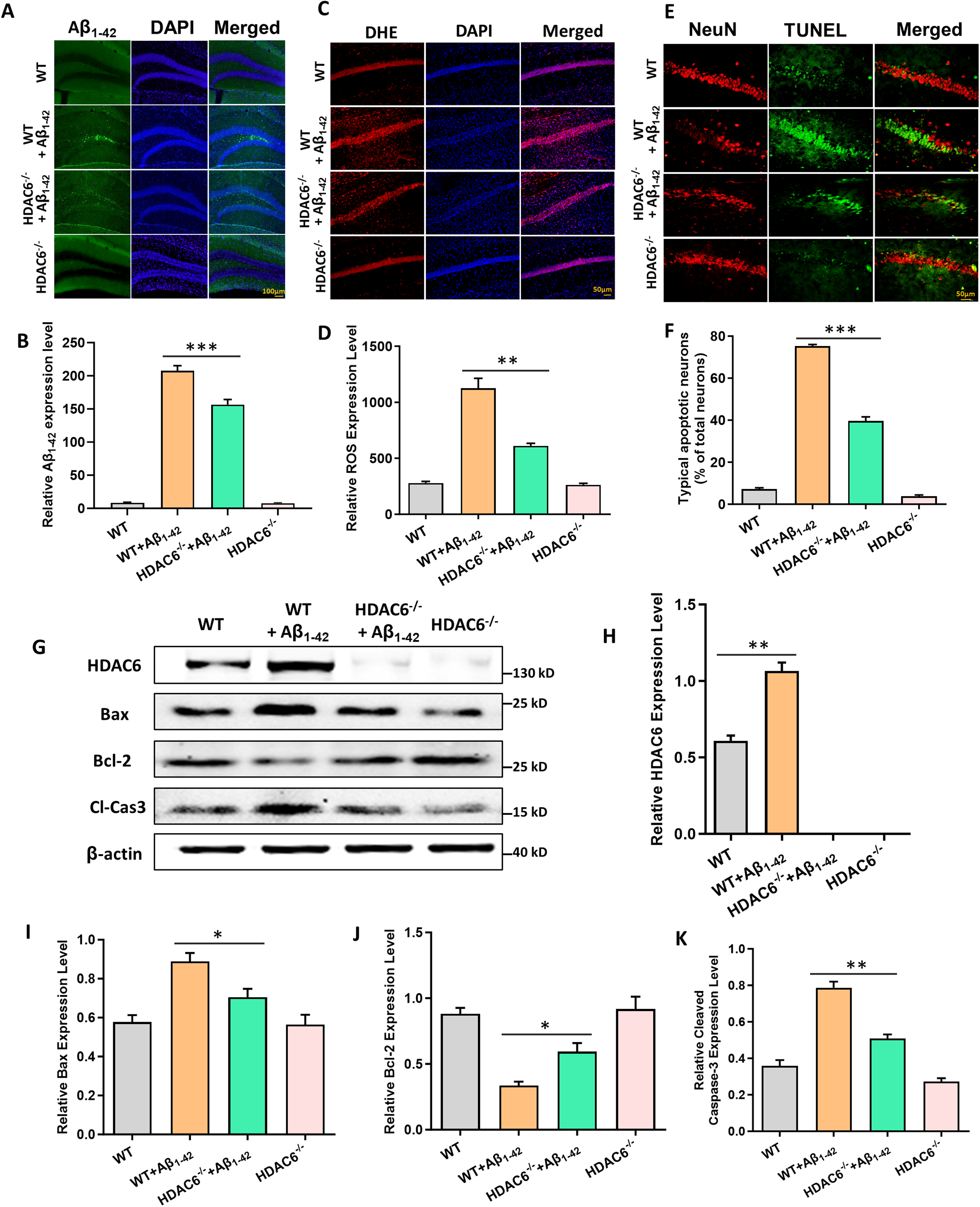
HDAC6 suppression can attenuate neuronal apoptosis and reduce ROS and Aβ1–42 expression. (A) The immunofluorescence for Aβ1–42 expression in the hippocampal CA1 regions of each group mice. (B) Quantification of Aβ1–42 expression of all groups. (C) ROS formation was visualized by DHE staining. (D) Quantification of ROS expression of all groups. (E) Neuronal apoptosis was marked by TUNEL and NeuN double-staining. (F) Quantification of neuronal apoptosis of all groups. (G) The expression of HDAC6, Bax, Bcl-2 and cleaved caspase-3 proteins by western blot in hippocampus of all groups. (H-K) Quantified results of HDAC6, Bax, Bcl-2 and cleaved caspase-3 proteins by western blot in hippocampus of all groups. The data are expressed as mean ± SEM. (n = 3) *p < 0.05, **p < 0.01, ***p < 0.001.
ROS generation in response to the level of oxidative stress was assessed by DHE staining. The results showed that ROS generation of Aβ1–42 group exhibited significantly elevated compared with control group and HDAC6−/− group; however, Aβ1–42 HDAC6−/− group reduced the elevation of ROS production significantly compared to Aβ1–42 group (Figure 2C, D).
To confirm the role of ROS generation in neuronal apoptosis. Co-localization of TUNEL and NeuN was used to measure apoptosis proteins, and western blot was carried out by double immunofluorescence staining. Immunofluorescence results showed that neuronal apoptosis was significantly increased in the hippocampal CA1 regions of the Aβ1–42 group compared to the control group and HDAC6−/− group. Although there was an increase in neuronal apoptosis in the Aβ1–42 HDAC6−/− group compared to the control group and HDAC6−/− group, it was significantly lower than that in the Aβ1–42 group (Figure 2E, F).
To quantify the expression of related apoptotic proteins, western blot analysis was performed with anti-Bax, anti-Bcl-2, and anti-Caspase-3 antibodies (Figure 2G). The expression levels of Bax and cleaved caspase-3 proteins were significantly increased in the Aβ1–42 group and the Aβ1–42 HDAC6−/− group compared to the control group and HDAC6−/− group. However, the increases in Bax and cleaved caspase-3 protein expression were partially prevented in the Aβ1–42 HDAC6−/− group compared to the Aβ1–42 group (Figure 2I, K). Conversely, the expression level of Bcl-2 protein was significantly decreased in the Aβ1–42 group compared to the control group and HDAC6−/− group. The expression level of Bcl-2 protein in the Aβ1–42 HDAC6−/− group was higher than that in the Aβ1–42 group but lower than the control group and HDAC6−/− group (Figure 2J). These results suggest that the loss of HDAC6 can reduce ROS production, protect against neuronal apoptosis by Aβ1–42-induced in AD mice model.
HDAC6 modulates neuronal apoptosis and ROS levels via Prx2 acetylation
To explore the association between Prx2 acetylation and HDAC6 protein level, immunoprecipitation was used to detect their association in the hippocampus of the Aβ1–42 group and Aβ1–42 HDAC6−/− group. It was observed that the acetylation level of Prx2 was significantly reduced in the Aβ1–42 group compared with Aβ1–42 HDAC6−/− group (Figure 3A, B). These findings suggest that HDAC6, as a deacetylase enzyme, can significantly reduce the level of Prx2 acetylation.
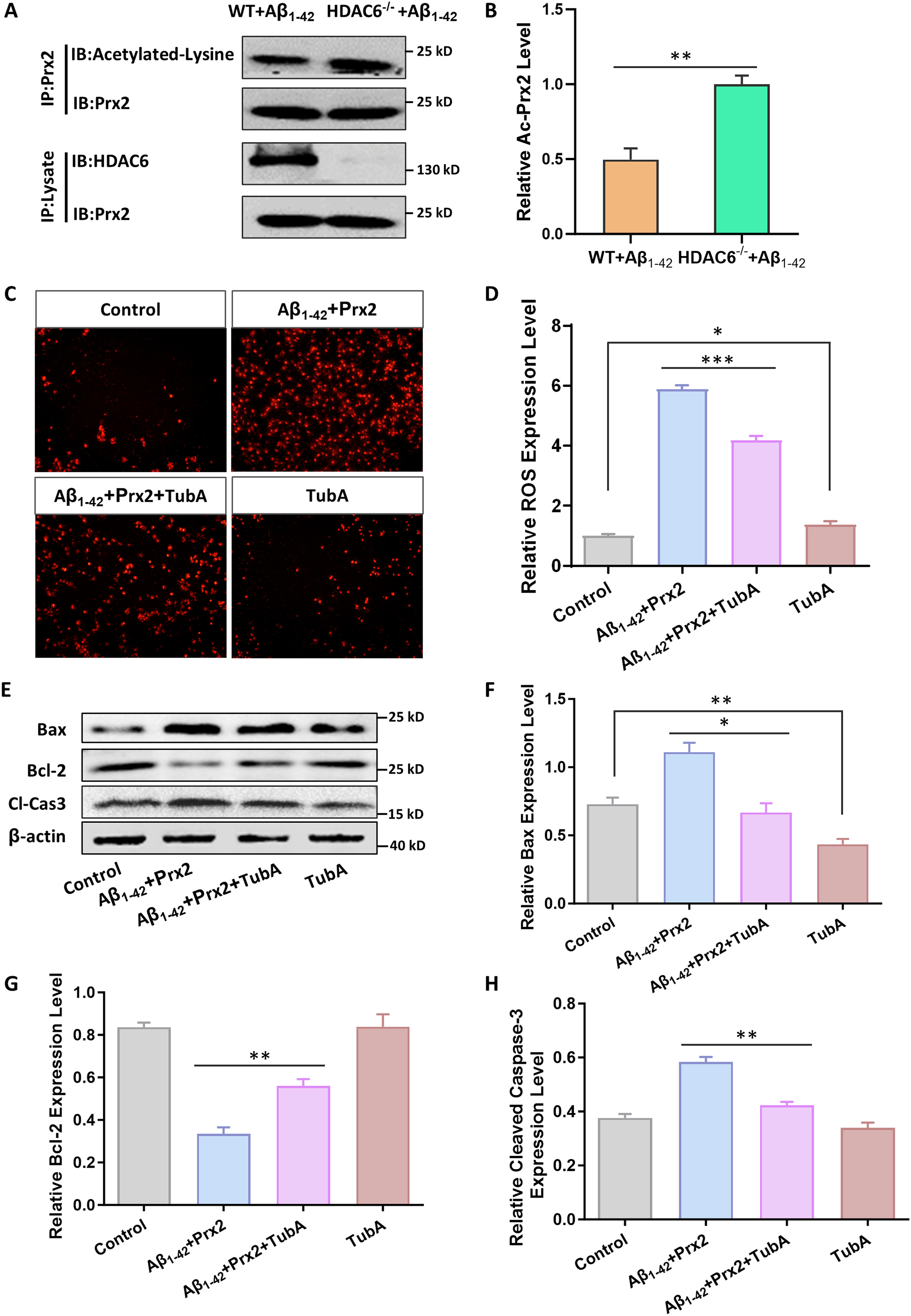
HDAC6 modulates neuronal apoptosis and ROS levels via Prx2 acetylation. (A) The immunoprecipitation for association between Prx2 acetylation and HDAC6 protein level in the hippocampus of the Aβ1–42 group and Aβ1–42 HDAC6−/− group. (B) Quantified results of HDAC6 expression and Prx2 acetylation. (C, D) Fluorescence detection of intracellular ROS fluorescence intensity and quantitative statistical analysis of HT22 neurons during treatment of Tub A. (E) HT22 neuronal cells were treated with HDAC6 inhibitor Tub A to detect the expression of neuronal apoptosis proteins (Bax, Bcl-2, cleaved caspase-3) by western blot. (F-H) Quantified results of neuronal apoptosis proteins (Bax, Bcl-2, cleaved caspase-3) by western blot. The data are expressed as mean ± SEM. (n = 3) **p < 0.01, ***p < 0.001.
To investigate the effect of the HDAC6-Prx2 signal pathway on ROS expression and cell apoptosis, we treated HT22 cells with an HDAC6 inhibitor. An AD cell model was established by utilizing Aβ1-42.57,58 Subsequently, Tubastatin A was administered to the cells to suppress HDAC6 expression. As ROS play a pivotal role in neurodegenerative disorders, ROS production was quantified using an ROS assay kit. The findings exhibited a notable rise in ROS production in the Aβ1-42 Prx2 group as opposed to both the control and TubA groups. However, following Tubastatin A treatment, ROS levels decreased significantly (Figure 3C, D). Hence, the use of HDAC6 inhibitors demonstrates a capacity to diminish ROS production. We also extracted proteins from the four groups and examined apoptotic protein expression using western blot analysis (Figure 3E). The results demonstrated that the expression level of Bax and cleaved caspase-3 proteins significantly increased in the Aβ1-42 Prx2 group compared to the control group and the TubA group. However, the increase in Bax and cleaved caspase-3 protein expression was partially prevented by administering TubA (Figure 3F, H). Conversely, the expression level of Bcl-2 protein significantly decreased in the Aβ1-42 Prx2 group compared to the control group and the TubA group. In the Aβ1-42 Prx2 TubA group, the expression level of Bcl-2 protein increased compared to the Aβ1-42 Prx2 group, but was lower than that in the control group and the TubA group (Figure 3G). In conclusion, HDAC6 inhibitors can regulate ROS production and neuronal apoptosis induced by intracellular Prx2 overexpression in an AD cell model.
Prx2 K196 acetylation is regulated by HDAC6 and affects the oxidative stress capacity of cells
The effect of HDAC6 on the acetylation level of Prx2, its acetylation site, and antioxidant stress ability were investigated in HT22 cells. Firstly, The HDAC6 plasmid, the Prx2 plasmid and co-transformed Prx2 and HDAC6 plasmids were separately transfected into HT22 cells to observe their relationship by immunoprecipitation. The results confirmed that there is an interaction between HDAC6 and Prx2. Next, further research will focus on the regulatory effect of HDAC6 on Prx2 acetylation. The results showed a significant increase in Prx2 acetylation level in the Prx2 group compared to the Prx2 and HDAC6 group, indicating that HDAC6, as a deacetylase enzyme, can decrease the Prx2 acetylation level. This finding was consistent with that observed in vivo (Figure 4A-C). Next, the acetylation site of Prx2 was detected by creating Prx2 mutant plasmids K196R, mimicking non-acetylated lysine, cloned in a FLAG plasmid and transfected into HT22 cells. The results showed that the K196R group was significantly low expression of Prx2 acetylation compared with the Prx2 group, indicating that K196 was the acetylation site of Prx2 (Figure 4D).
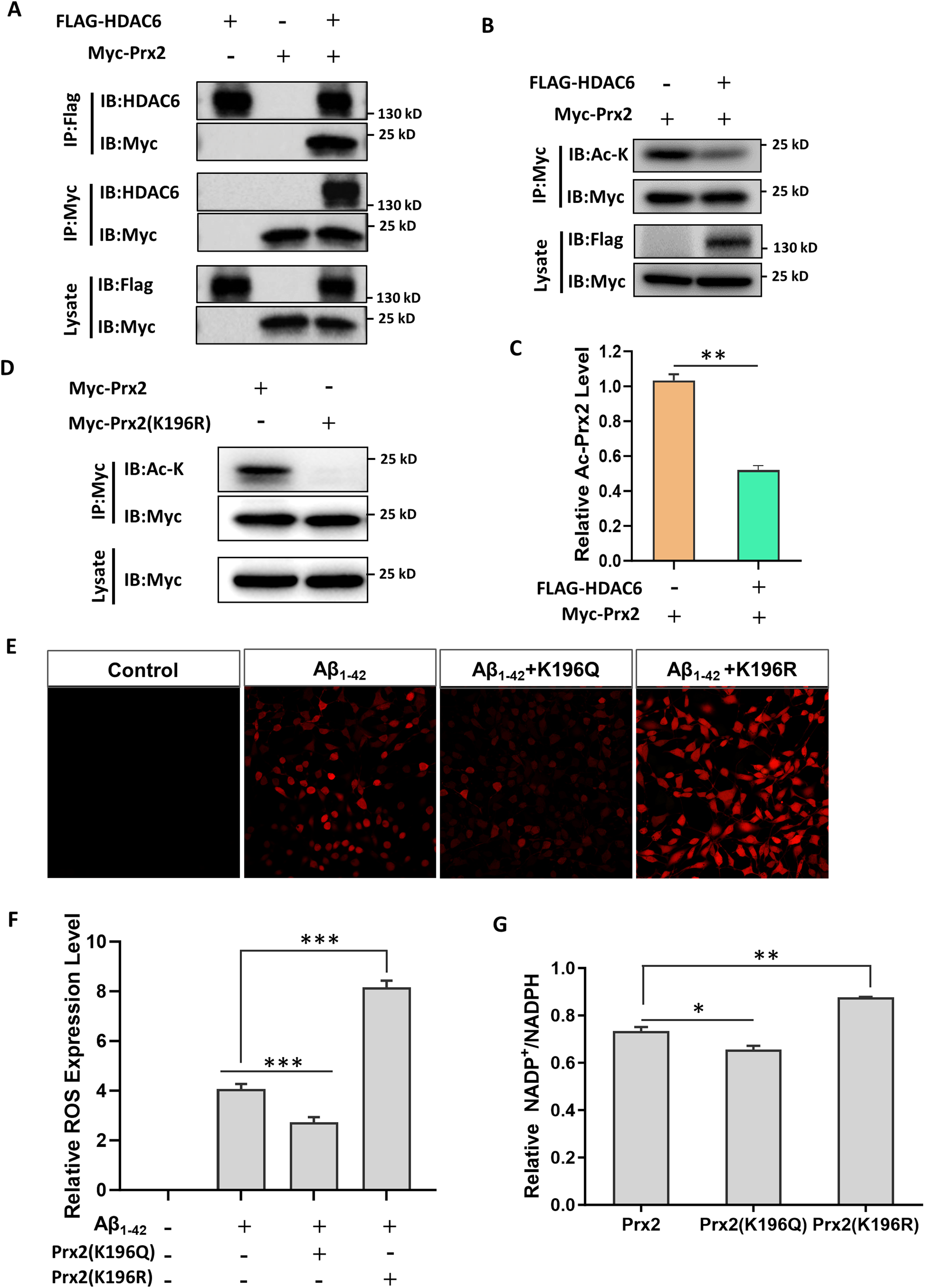
Prx2 K196 acetylation is regulated by HDAC6 and affects the oxidative stress capacity of cells. (A) The immunoprecipitation for interaction between Prx2 and HDAC6 protein level in HT22 cells. (B) The immunoprecipitation for association between Prx2 acetylation and HDAC6 protein level in HT22 cells. (C) Quantified results of HDAC6 expression and Prx2 acetylation. (D) The immunoprecipitation for Prx2 and Prx2 deacetylation site (K196R) on acetylation level in HT22 cells. (E) The immunofluorescence for ROS expression in Aβ1–42 group, Aβ1–42 K196Q group, and Aβ1–42 K196R group. (F) Quantification of ROS expression of all groups. (G) The anti-oxidative stress of Prx2, K196Q, and K196R were measured by the NADP/NADPH ratio. The data are expressed as mean ± SEM. (n = 3) *p < 0.05, **p < 0.01, ***p < 0.001.
Subsequently, we transfected the K196Q and K196R plasmids into HT22 cells that had been treated with Aβ1–42 (AD model). This was done to observe the expression of ROS. The results demonstrated that when compared with the control group, the expression of ROS in the other three groups went up to various degrees. In comparison with Aβ1–42 group, Aβ1–42 K196Q group could remarkably decrease the expression level of ROS. In contrast, Aβ1–42 K196R group could significantly elevate the expression of ROS (Figure 4E, F).
Finally, the NADP/NADPH ratio was measured among the cells using Amplite™ Fluorimetric NADP/NADPH Ratio Assay Kit. The ability of anti-oxidative stress is expressed using the NADP/NADPH ratio. A lower ratio indicates stronger anti-oxidative stress. Our study found that the K196Q group had a lower NADP/NADPH ratio compared to the Prx2 group, suggesting that the antioxidant capacity of Prx2 was significantly enhanced after acetylation. On the other hand, the NADP/NADPH ratio of the K196R group increased, indicating a reduced ability to counter oxidative stress compared to the Prx2 group (Figure 4G). Therefore, we conclude that HDAC6 is capable of enhancing ROS production and augmenting oxidative stress by reducing the level of Prx2 acetylation.
Prx2 K196 acetylation can mitigate neuronal apoptosis and enhance the cognitive function in mice
To determine whether Prx2 K196 acetylation is related to the Aβ1–42 expression and neuronal apoptosis, as well as to verify the effect of HDAC6-Prx2 pathway on AD mice. Adenosine-associated viruses (AAV) containing K196Q and K196R Prx2 were administered into the cortex of AD mice model. The results demonstrated that, in comparison with Aβ1–42 group, the Aβ1–42 expression of Aβ1–42 K196Q group was remarkably reduced. Conversely, Aβ1–42 K196R group was significantly elevated (Figure 5A, B). Simultaneously, we quantified the expression of neurons to verify that the Aβ1–42 expression is closely associated with neuronal apoptosis. The outcome was opposite to that of Aβ1–42 expression. When compared with the Aβ1–42 group, the expression of neurons in the Aβ1–42 K196Q group increased markedly, whereas in the Aβ1–42 K196R group, it decreased substantially (Figure 5C).
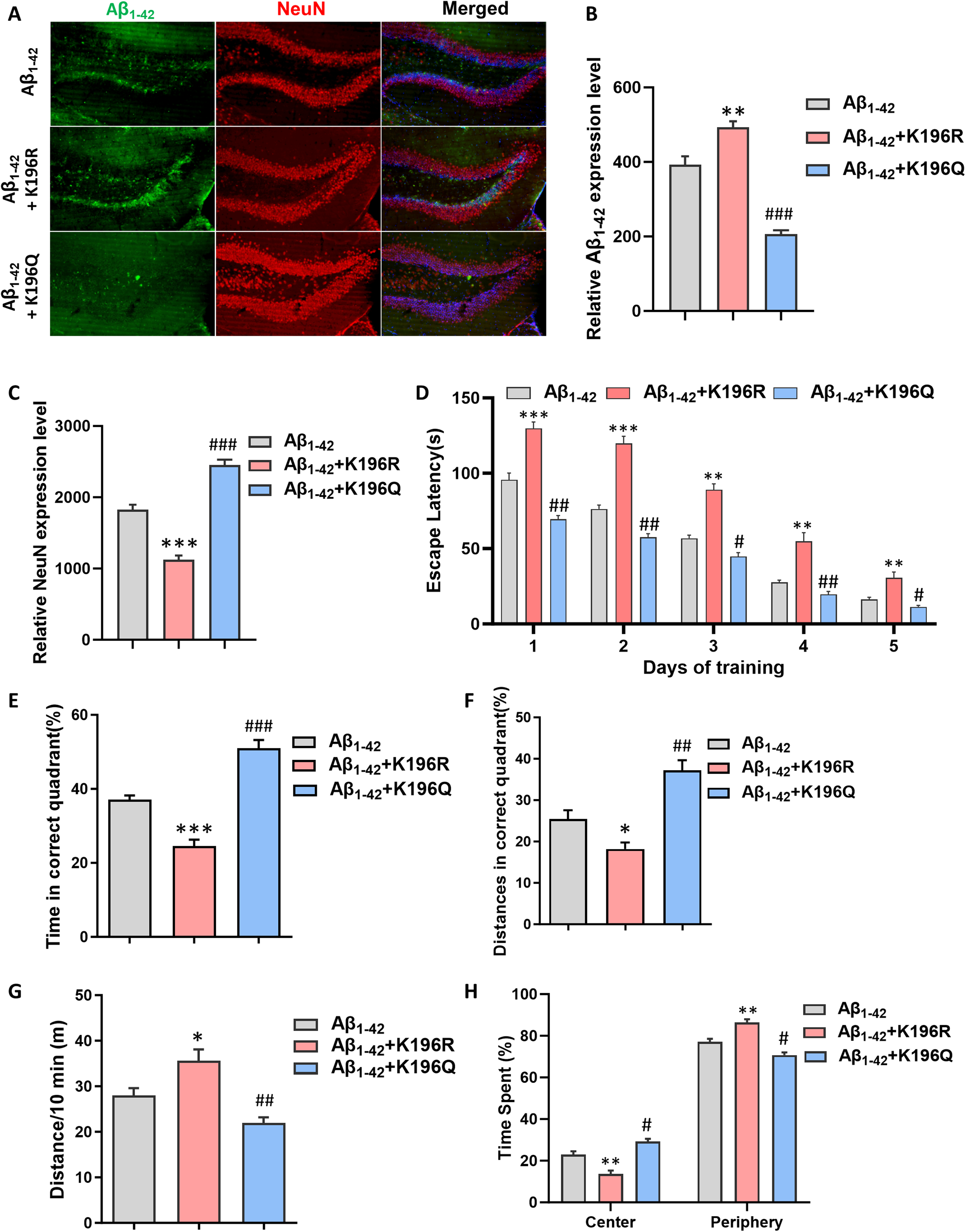
Prx2 K196 acetylation can mitigate neuronal apoptosis and enhance the cognitive function in mice. (A) The immunofluorescence for Aβ1–42 expression and neurons expression in the hippocampal CA1 regions of each group mice. (B) Quantification of Aβ1–42 expression of all groups. (C) Quantification of neurons expression of all groups. (D) Average escape latency to find the hidden platform during 5 consecutive training days for all groups. (E) Swimming time in correct quadrant during the probe test for four groups. (F) Distances in correct quadrant during the probe test for four groups. (G) Distances of all groups in the open field. (H) Time spent in the center and periphery in the open field for all groups. The data are expressed as mean ± SEM. (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001 , #p < 0.05, ##p < 0.01, ###p < 0.001,compared with Aβ1-42 group.
Subsequently, we carried out the water maze test and the open field test on three groups of mice respectively. Contrary to the Aβ1–42 K196R group, Aβ1–42 K196Q group had better performance in the behavioral tests than Aβ1–42 group (Figure 5D-H). The aforementioned findings demonstrate that Prx2 acetylation is capable of mitigating Aβ1–42 expression and decreasing neuronal death, consequently enhancing the learning and memory capabilities of mice and alleviating their state of anxiety.
Discussion
In this study, we showed a novel mechanism with therapeutic implications for treating AD. We established the AD model by injecting Aβ1–42 into WT mice and HDAC6 knockout mice and investigate whether HDAC6 protein contributed to neuronal apoptosis and cognitive impairment in AD. 40 We improved the neurological impairment of AD mice with cognitive dysfunction by overexpressing K196Q-mutated Prx2 in AD model mice. we discovered that Prx2 is a novel substrate of HDAC6, and HDAC6 regulates Prx2 antioxidant capacity through modulating its acetylation at K196 site.
Although the previous research had indicated a correlation between HDAC6 protein and AD, no definitive conclusions have been drawn.59,60 By observing the occurrence of HDAC6-Prx2 acetylation modification, we found that the HDAC6-Prx2 pathway likely plays a role in the progression of AD, an idea supported by our findings that reducing HDAC6 levels decreased neuronal apoptosis and cognitive impairment. Moreover, when HDAC6 was deficient, Prx2 acetylation levels increased, which decreased ROS production and mitigated AD-induced oxidative stress, slowing the progression of AD. As a continuation of our work on HDAC6 protein and Prx2 acetylation, we aimed to investigate further the relationship between HDAC6-Prx2 and AD. Given the role of HDAC6 in regulating the promoter regions of memory-associated genes and its impact on neuronal apoptosis and cognition function, our study showed that HDAC6 inhibition led to Prx2 acetylation, thus providing a protective effect.
The hippocampus plays a key role in learning, memory and anxiety regulation. Studies have shown a clear relationship between anxiety, depression, and cognitive decline in AD. Anxiety can be the initial psychological response to cognitive impairment, while depression can further exacerbate cognitive impairment.61,62 HDAC6 can be involved in the regulation of emotions. HDAC6 deficiency can improve the behavioral performance of Parkinson's disease (PD) mice. The results showed that the metabolic levels of DA and 5-HT were significantly affected by HDAC6 deficiency, which could significantly regulate the mood of PD mice, thereby improving the behavioral performance of mice. 15 Numerous studies have illustrated the detrimental effect of HDAC protein disorders and acetylation/deacetylation levels on cognitive function. 20 HDAC6 expression was notably elevated in mild cognitive impairment, mild to moderate AD, and severe AD in comparison to other HDACs. 63 Cognitive deficits in AD mouse models can be alleviated through HDAC6 inhibition or reduced HDAC6 activity.21,55,64 Studies have indicated that HDAC6 inhibition, such as Tubastatin A or MPT0G211, could enhance tau hyperphosphorylation and aggregation, and improve learning and memory function in AD.19,65 Among these inhibitors, we selected TubA because it not only specifically inhibits HDAC6 but also protects cells against oxidative stress-induced death in a dose-dependent manner, thereby suggesting a potential link between HDAC6 deacetylase activity and oxidative stress in vitro.66,67 As oxidative stress is closely associated with human neurological disorders, including AD, exploring the HDAC6 protein mechanism could be an innovative and promising approach for the treatment of this disease.
The anti-oxidative stress peroxidase enzyme, Prx, has been investigated in neurodegenerative diseases,25,68 but the role of the Prx subtype in AD pathogenesis remains ambiguous. Modifications of Prx2, such as phosphorylation and oxidation, have been studied in PD models, where inhibition of S-nitrosylation of Prx2 helped protect against oxidative stress-induced neuronal cell death through sulfiredoxin reductase regulation. 69 Furthermore, Tubastatin A increased acetylation of Prx1 and Prx2, impaired ROS generation, and attenuated dopaminergic neurotoxicity in PD. However, phosphorylated Prx2 decreased enzymatic activity and resulted in reduced ROS elimination capacity in PD.70,71 Researchers had demonstrated that HDAC6 inhibition-induced increased acetylation of Prx1 helped recover from Aβ-induced impaired axonal transport, mitigate elevated ROS and Ca2+ levels, and modified mitochondrial axonal transport. 72 In this study, we chose Prx2 since it is localized in the neurons’ cytoplasm, unlike Prx1, which is mainly distributed in oligodendrocytes and microglia. It is therefore of great interest to explore whether and how the HDAC6-Prx2 pathway regulates oxidative stress in AD. Additionally, Prx5 has been proved to control AβO-induced hippocampal neuronal death by combating oxidative stress. 73
Oxidative stress, which leads to ROS generation and decreased antioxidant enzyme capacity, is implicated in AD progression.74,75 Additionally, it can damage DNA, proteins, and lipids by generating peroxides and free radicals, while inducing the apoptotic process by activating apoptotic proteins. Apoptosis and axonal regeneration reduction reliably indicate apoptosis. NADPH-dependent reducing systems play a crucial role in oxidative stress.76,77 The NADP/NADPH ratio, as a substrate or cofactor of antioxidant enzymes, can reflects the oxidative stress state of cells. Its imbalance may lead to oxidative or reductive stress.78,79 Thus, our study aimed to examine the effects of the HDAC6-Prx2 pathway on AD-induced oxidative stress by measuring both ROS generation and the NADP/NADPH ratio.
To summarize, our results indicate that HDAC6 deficiency can increase Prx2 K196 acetylation levels and ameliorate learning and memory deficits in AD mouse models (Figure 6). By inhibiting HDAC6 to lower the acetylation level of Prx2 K196, oxidative stress generation is reduced, anti-oxidative stress capacity is enhanced, and neuronal apoptosis is diminished. Targeting Prx2 and HDAC6 with precision offers a new approach to neuronal protection therapy for AD.
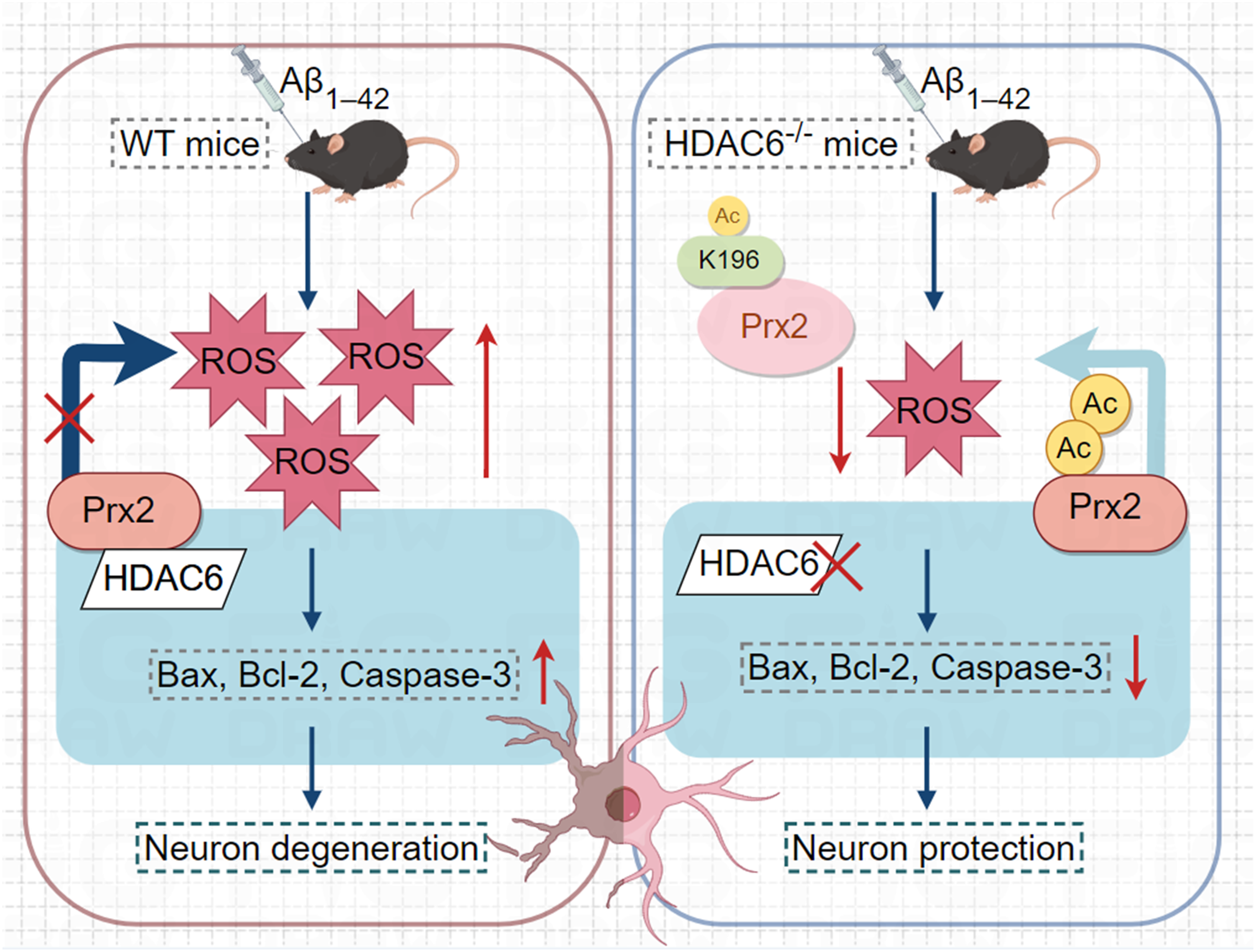
Mechanism of action of HDAC6 and Prx2 K196 acetylation in AD mouse models.
Footnotes
Acknowledgements
We appreciate the help from Institute of Stroke Research and Department of Neurology, Xuzhou Medical University.
Ethical considerations
All the animal experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the laboratory animal ethical committee of Xuzhou Medical University.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Fundamental Research Funds for the Central Universities (Grant No. 2018BSCXA12), Postgraduate Research & Practice Innovation Program of Jiangsu Province (Grant No. KYCX18_1931).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The original contributons presented in the study are included in the article material; further inquiries can be directed to the corresponding authors.