
Abstract
Background
Although the molecular basis of Alzheimer's disease (AD) is often studied in Caucasians, its genetic basis in Bangladesh remains elusive.
Objective
We explored the association between single-nucleotide variants (SNVs) of Apolipoprotein E (APOE) and other genes for AD risk among Bangladeshis.
Methods
We recruited AD patients and controls aged ≥18 years and conducted next generation sequencing (NGS) to analyze ∼30 Mb of the human exome. The NGS targets ∼99% of the Consensus Coding Sequence and RefSeq annotations, along with the complete mitochondrial genome.
Results
A total of 132 (72 AD: 60 control) age- and sex-matched participants were sequenced. The average age of AD was comparable to that of controls (64.5 ± 11.3 versus 63.6 ± 9.8, p = 0.69), and the gender distribution (p = 0.49) was similar between the study groups. Following NGS analysis 23 SNVs from 16 potential genes were identified. The APOE variant rs429358 was associated with a significantly increased age- and sex- adjusted risk of AD (OR) 8.0 (95% CI: 2.3–27.9, p = 0.001) in the additive model, 10.5 (95% CI: 2.8–39.5, p < 0.001) in the dominant model, and 6.9 (95% CI: 1.8–26.9, p = 0.005) in the heterozygous genotype model, as determined using the Bonferroni adjustment method. The top SNV for predicting AD was the APOE variant rs429358 with a -logP value of 3, followed by BDNF gene variant rs6265 at 1.3, and CR1 gene variant rs1349409945 at 1.2.
Conclusions
The APOE variant rs429358 is linked to an 8-fold increased risk of AD among Bangladeshis.
Introduction
Alzheimer's disease (AD) is an increasing global health concern, with over 55 million people living with dementia; AD accounts for 60–70% of cases.1,2 The prevalence of dementia in South Asia is rising, ∼3% among older adults, due to an ageing population and lifestyle changes, but these figures may be understated due to a lack of awareness and limited diagnostic resources.3,4 As life expectancy increases, global and regional prevalence of AD is expected to double by 2050 highlighting the urgent need for effective public health strategies. AD causes memory loss, confusion, and impaired thinking, significantly affecting daily life, like struggle with decision-making and recognizing loved ones, often exhibiting changes in behavior, such as aggression and depression.4–6 Declining physical abilities can cause dependency on caregivers, strain relationships, and create financial burdens for families, which may ultimately reduce life expectancy.6–9 The average annual treatment costs for AD vary by region10–16; from $1000 in India to £25,000 per patient in the UK,11,12 and $60,000 in the United States.13,14
Genetic analyses can identify individuals at higher risk of AD by detecting specific gene variants linked to an earlier disease onset within certain populations.17–24 Recent genome-wide association studies (GWAS) in the USA,25,26 UK,27,28 and Asia29,30 have identified multiple single nucleotide variants (SNVs) linked to AD, specifically variants near APOE, CR1, BDNF, BIN1, CLU, TREM2, and PICALM remain strongly implicated. Early-onset familial AD is linked to mutations in the APP, PSEN1, and PSEN2 genes, while the APOE ε4 allele is the primary gene associated with increased risk for late-onset AD.14,24 Mutant genes are involved in amyloid processing, lipid metabolism, inflammation, and synaptic function, all crucial to AD development.2,6 Understanding these pathophysiological mechanisms aids targeted therapy development, informs families of risks, and enhances preventive strategies and clinical trial design. Although polygenic risk scores31–35 developed in European ancestries often underperform in South Asian populations, highlighting the necessity for broader more inclusive genomic studies in order to better understand global AD risk. Further, while genetic studies on AD are limited in South Asia,36,37 emerging research highlights unique genetic variants emphasizing the need for ancestry-specific studies that can identify molecular pathways involved in the disease aiding in developing personalized treatments and effective interventions.
There are very few studies38,39 that have observed cognitive status and atrophy scores in AD or dementia prevalence across sociodemographic characteristics among older adults in Bangladesh. To the best of our knowledge, this is the first next-generation sequencing (NGS) study to describe potential genetic variants among the Bangladeshi AD population.
Methods
The CARED study
The Community Awareness and Research on Early Dementia (CARED) study is a NGS study recruiting adult AD patients aged ≥18 years at the National Institute of Neurosciences and Hospital (NINS&H) in Dhaka Bangladesh from January 2019 to December 2024. We recruited age- and gender-adjusted study participants who had subjective memory complaints and met the diagnostic criteria for dementia as specified in the Diagnostic and Statistical Manual of Mental Disorders (DSM-5; 5th edition). To mitigate selection bias, we excluded other neurodegenerative dementias and reversible causes of dementia, including brain tumors, infections, head injuries, substance abuse, depression, and malnutrition. The NINS&H is the only referral center for dementia patients in the country. It gathered participants from all eight administrative divisions making the study sample representative of dementia prevalence in the Bangladeshi population. This study was approved by the Institutional Review Board (IRB) committee of NINS&H (IRB/NINS/2024/393) and follows the Declaration of Helsinki. Participants willingly consented to the study, and all data were collected anonymously and securely encrypted.
Study procedure
The CARED study gathered sociodemographic data and DNA samples from AD patients, following standard procedures for evaluation and using DSM-5 criteria for diagnosis, which is described elsewhere. 38 In brief: a neurologist and an interventional radiologist confirmed the diagnosis of AD based on MRI findings, unaware of the subjects’ age or clinical diagnosis, and assigned a final score upon agreement. We used Siemens 1.5 and 3 Tesla MRI systems with appropriate phased array coils, following the MRI protocol. 40 T1-weighted axial, sagittal, coronal, and thin coronal oblique images were analyzed for structural changes. Atrophy was assessed using the medial temporal lobe atrophy (MTA) scale, 41 and Koedam's score. 42 Additionally, a Mini-Mental State Examination (MMSE) 43 was conducted to evaluate the severity of cognitive domains, and the MTA score 41 was assessed using coronal TSE images, which maintain a consistent slice position. To exclude the reversible causes of AD, we screened a wide variety of diseases using blood tests, including e.g., blood glucose, serum creatinine, complete blood count, peripheral blood film, vitamin D, and thyroid function tests (thyroid stimulating hormone, free triiodothyronine 3 and 4 levels), serum albumin, serum glutamic pyruvic transaminase and bilirubin, the Venereal Disease Research Laboratory (VDRL), Treponema Pallidum Hemagglutination Assay (TPHA), vitamin B12, calcium, and parathyroid hormone. A chest X-ray was also undertaken.
Next-generation sequencing analysis44–46
A trained healthcare nurse collected 10 ml peripheral blood samples from patients and control subjects in EDTA-coated vials. Each sample was assigned a unique CARED repository ID and stored at −80°C. Qiagen DNA isolation kits were used to extract high-quality genomic DNA from lymphocytes. An OD260/OD280 ratio above 1.8 was accepted as a quality control measure; samples with lower ratios were re-purified. NGS was performed using Illumina platforms to analyze the protein-coding regions of approximately 30 Mb of the human exome, targeting ∼99% of the areas annotated in Consensus Coding Sequence (CCDS) and Reference Sequence (RefSeq), as well as the complete mitochondrial genome. The mean sequencing depth for the exome was 80–100× with over 90% of target bases covered at a depth of ≥20×. The mitochondrial genome was sequenced at a higher depth of 1000–2000×. In some instances, due to the complexity of the sequence, not all variants in the flanking regions can be reliably analyzed. Genetic variant identification was performed using Genome Analysis Toolkit (GATK) best practices framework. Duplicate reads were identified and removed and base quality score recalibration as well as realignment around indels, were conducted using the Illumina DRAGEN™ Bio-IT platform. Illumina's TruSeq Exome Enrichment Kit was utilized to analyze our sample. Quality control (QC) checks are applied to all Variant Call Format (VCF) files to filter out low-quality variant calls. Additional QC metrics included total number of homozygous and heterozygous calls (SNVs and indels), proportion of common variants, distribution of variants across annotated consequence categories and count of extreme heterozygotes (defined as having an alternate allele proportion ≥0.8). We utilized publicly available databases such as OMIM, GWAS catalog, ClinVar, HGMD, LOVD, DECIPHER (population CNV), SwissVar, and the 1000 Genomes project for variant annotation and clinical interpretation, considering only non-synonymous and splice-site variants to select the candidate gene set. However, synonymous or silent variants that do not change the amino acid sequence were not reported, and Geneyx (v5.12) was utilized for variant annotation, interpretation, and reporting. In this study, we addressed missing data by implementing rigorous quality control at both the sample and variant levels. We excluded samples with call rates below 95% to 98% to ensure data reliability and removed variants with poor call rates or excessive missingness to minimize bias. For retained variants, missing genotypes were either imputed using population-based reference panels or coded as missing, depending on the study design and available resources. This approach ensured that downstream analyses, including association testing, were conducted using high-quality, well-curated genotype data.
Statistical analysis
We used SPSS v28.0 (IBM Corp., 2021, NY) software for statistical analysis. Continuous variables are presented as means ± SD, and categorical variables are shown as counts and percentages. The chi-square test and Fisher's exact test compared categorical variables, while the Student's t-test compared continuous variables. Furthermore, we utilized four different risk prediction models to identify potential SNVs and allelic effects associated with AD: the additive model, the genotype model, the recessive model, and the dominant model using age and sex- adjusted logistic regression (LR) analysis. In the recessive model, we compared combined major alleles and heterozygous minor alleles against homozygous minor alleles. Conversely, we compared major alleles with combined heterozygous and homozygous minor alleles in the dominant model. Additionally, the -logP values were calculated to provide a logarithmic visualization and ranking of genome-wide variant associations, facilitating the identification of true biological effects while accounting for multiple testing. The additive model was used to compute these -logP values, assuming a linear increase in effect as the number of risk alleles rises from 0 to 2. This model uses a single degree of freedom per allele and generally offers greater statistical power than dominant or recessive models, which can lose power if mis-specified. In next-generation sequencing, where rare variants are common, recessive models are particularly limited because individuals carrying two copies of rare alleles are extremely uncommon. Further, the additive model also partially captures both dominant and recessive effects, making it a robust and efficient choice when the true genetic model is uncertain. Additionally, we employed a genotype model to estimate the independent effects of heterozygotes and homozygotes, using two parameters instead of a single allele-dosage coefficient. This model utilizes two degrees of freedom and does not assume a linear dosage effect; as a result, it offers greater flexibility and can detect dominance deviations or heterosis that additive analysis may overlook. An age- and sex-adjusted LR model was used to estimate odds ratios (ORs) and 95% confidence intervals (CIs). The Bonferroni correction multiple comparison approach was applied by dividing the original unadjusted significance threshold (0.05) by the number of tests (e.g., 0.05 divided by 22 = 0.002). A p-value of <0.05 was accepted as statistically significant.
Results
The CARED study evaluated age- and sex-matched 132 adult Bangladeshi population (72 AD patients and 60 healthy controls). The overall age of AD patients was similar to healthy controls (64.5 ± 11.3 versus 63.6 ± 9.8, p = 0.69), with no difference in gender distribution (p = 0.49). Table 1 demonstrates the baseline characteristics of the study population. We identified 23 single nucleotide variants (SNVs) from 16 potential genes through NGS analysis of patients with AD in Bangladesh. The genes with SNPs of interest were APOE (rs405509, rs429358, rs7412, rs763720372), BDNF (rs6265), COMT (rs4680, rs140215652), CR1 (rs1349409945, rs1344800847, rs757075631), ABCA7 (rs748645497, rs768192987, rs1456598153, rs377020137), SORL1 (rs766557470, rs750421636), MAPT (rs781280791), PSEN1 (rs750080931, rs574671310, rs1474233910), PICALM (rs755346304), HBB (rs33950507), GRN (rs778599933), DCTN1, POLG, HNF1B, KCND3, GBA1, and PKD1.
Basic characteristics of study population (n = 132).
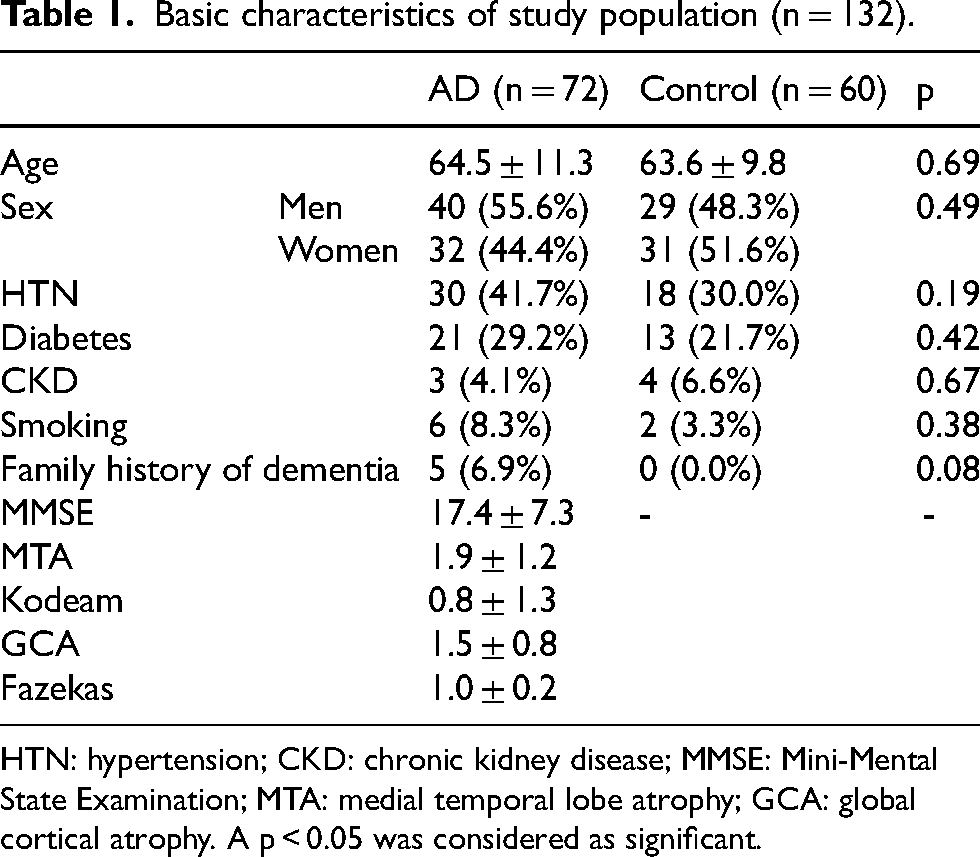
HTN: hypertension; CKD: chronic kidney disease; MMSE: Mini-Mental State Examination; MTA: medial temporal lobe atrophy; GCA: global cortical atrophy. A p < 0.05 was considered as significant.
Logistic regression analysis adjusted for age and sex demonstrated that the APOE variants at rs429358 significantly increased the risk of AD, as shown in Table 2. The odds ratio (OR) for rs429358, adjusted for age and sex, was 8.0 (95% CI: 2.3–27.9, p = 0.001) in the additive model, 10.5 (95% CI: 2.8–39.5, p < 0.001) in the dominant model, and 6.9 (95% CI: 1.8–26.9, p = 0.005) in the heterozygous genotype model. The p-values reported are Bonferroni-adjusted for the significance of the rs429358 additive, dominant, and heterozygous genotype models. Additionally, the CR1 gene rs1349409945 variant demonstrated an OR of 2.7 (95% CI: 0.9–8.0, p = 0.06) in the additive model, 7.6 (95% CI: 0.9–65.2, p = 0.06) in the dominant and recessive model, and 7.5 (95% CI: 0.9–64.2, p = 0.06) in the heterozygous genotype model. The p-values shown in Table 2 were adjusted with the Bonferroni correction method. Additionally, the Hardy-Weinberg (HW) equilibrium found significant p-values for rs429358 (p < 0.001), rs7412 (p = 0.01), rs1349409945 (p < 0.001), and rs377020137 (p < 0.001) (Supplemental Figure 1).
Age and sex- adjusted risk prediction models observed the potentials SNPs of AD among Bangladeshi population.
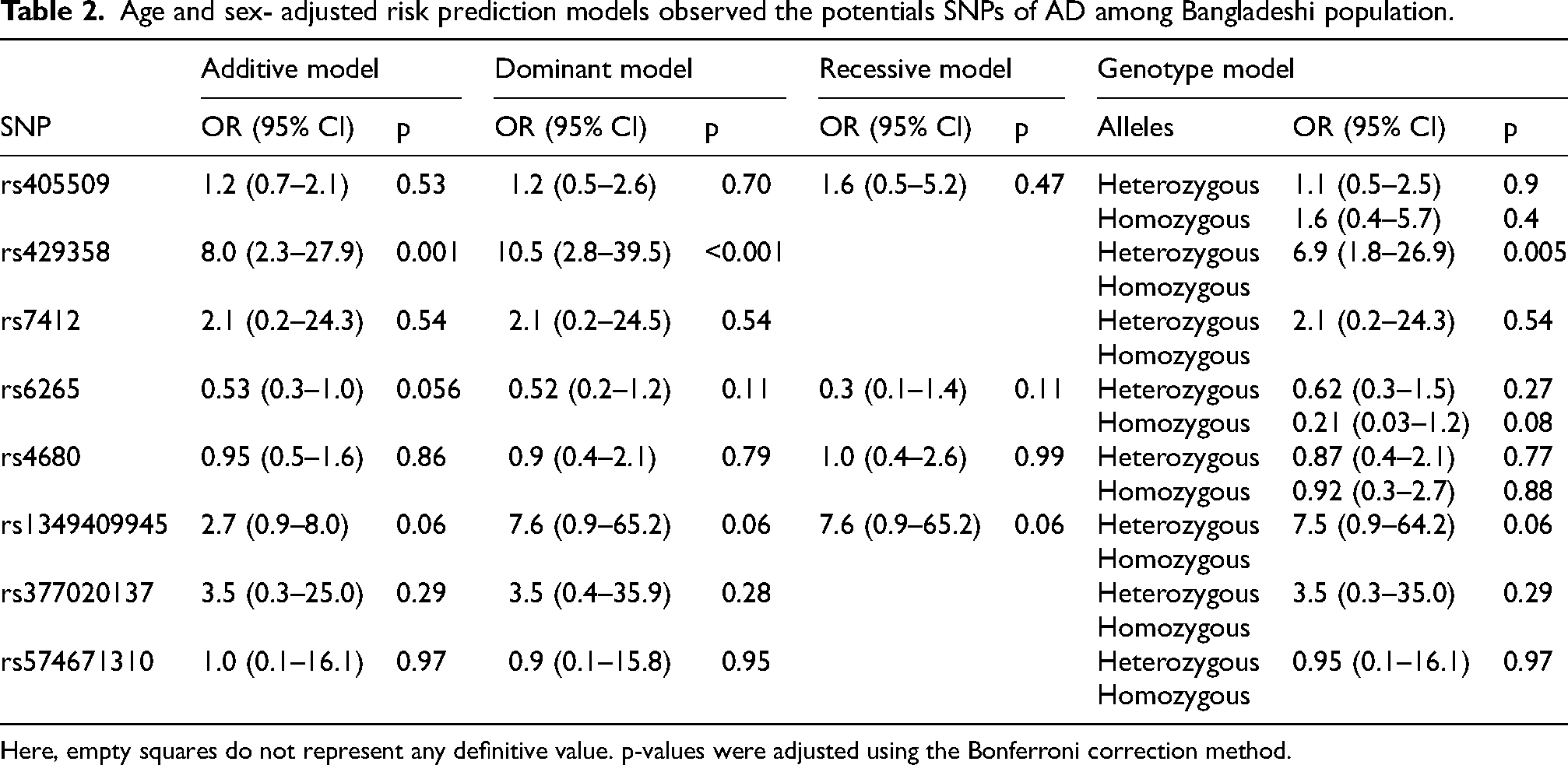
Here, empty squares do not represent any definitive value. p-values were adjusted using the Bonferroni correction method.
The APOE gene comes in different versions (three alleles: ε2, ε3, and ε4; six genotypes: ε2/ε2, ε2/ε3, ε2/ε4, ε3/ε3, ε3/ε4, and ε4/ε4), with ε3 allele and ε3/ε3 genotype being the most common (Table 3). The APOE ε4 allele is strongly associated with increased age- and sex-adjusted risk of AD (OR 19.9, 95% CI 5.2–132.7; p < 0.001), while the ε2 allele may offer some protection. Figure 1 demonstrates the -logP value of potential SNVs for predicting AD, with a higher -logP value indicating the most significant SNVs. The APOE variant rs429358 had the highest -logP value of 3 (p = 0.001), followed by the BDNF variant rs6265 at 1.3 (p = 0.05) and the CR1 variant rs1349409945 at 1.2 (p = 0.06).
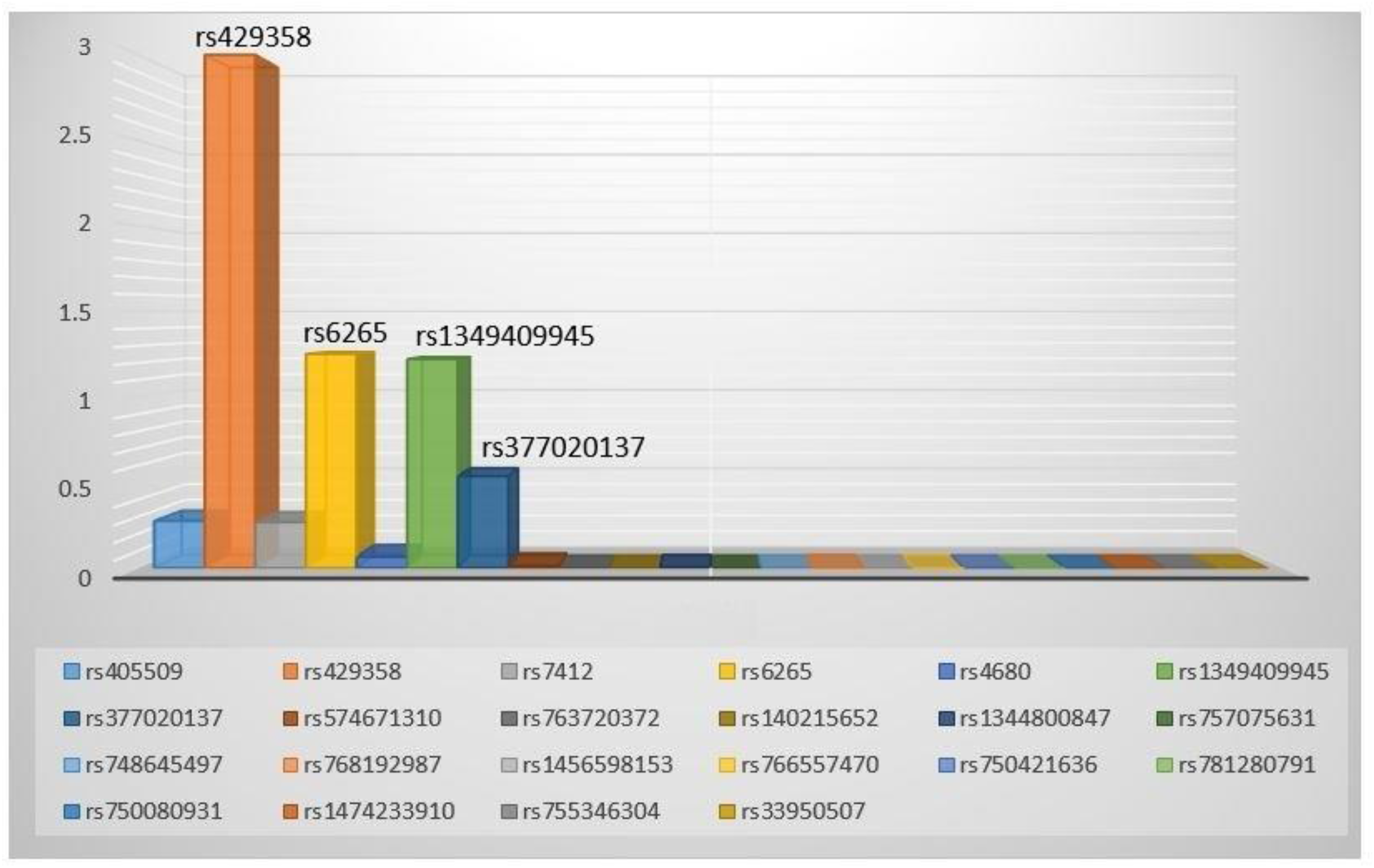
The -logP value of potential SNPs demonstrating the potentials of AD among Bangladeshi population.
Distribution of APOE genotypes and variation in APOE allele frequencies among cases and control.
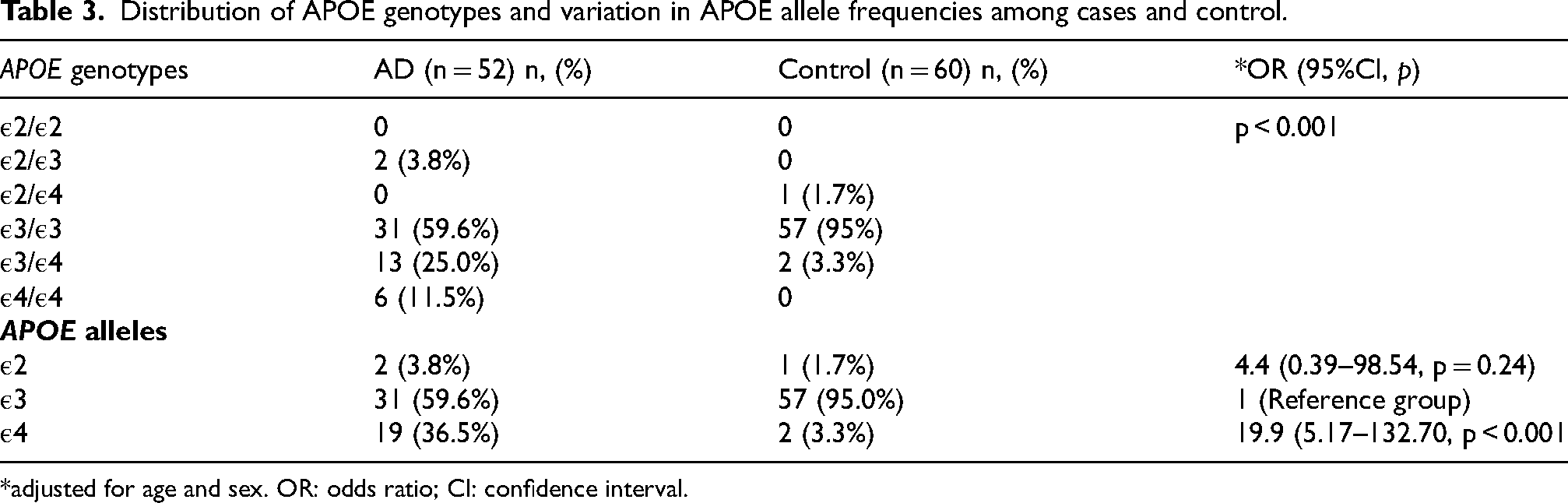
*adjusted for age and sex. OR: odds ratio; CI: confidence interval.
Discussion
We present the first Bangladeshi genetic study on AD and show that APOE variants at rs429358 are associated with ∼8 times higher risk of disease in the additive, dominant, and heterozygous genotype models. The -logP value also identified APOE variant rs429358 as the top SNV for predicting AD, followed by BDNF variant rs6265 and CR1 variant rs1349409945.
The APOE gene plays an important role in lipid metabolism and neuronal repair while the rs429358 variant is central to defining the APOE ε4 allele, a major genetic contributor to Alzheimer's disease, supporting our study findings.19,20,47 Further, individuals with one copy of the ε4 allele have about a 3-fold increased risk, while homozygous carriers have ∼12-fold higher risk than those with the ε3/ε3 genotype.22,24,48,49 The pathogenicity of the APOE rs429358 variant is linked to its decreased ability to clear amyloid-beta peptides and its impact on tau pathology, neuro-inflammation, and synaptic dysfunction, ultimately contributing to neurodegeneration.19,24,50 A comprehensive study by Genin and colleagues 47 confirmed a dose-dependent effect of APOE ε4 on the susceptibility to AD, indicating a significant risk factor for both late- and early-onset AD. Several existing studies, including findings from the Alzheimer's Disease Neuroimaging Initiative (ADNI), 17 indicate that carriers of the APOE ε4 variant experience more significant amyloid accumulation, hippocampal atrophy, and earlier cognitive decline.18–20 Nevertheless, recent literature has observed a link between APOE ε4, microglial activation, and the integrity of the blood-brain barrier, which may worsen AD pathology.49–52 In a small Saudi Arabian study, Abdi and colleagues 18 found that the APOE rs429358 variant showed a ∼9 times higher frequency of AD than controls, in concordance with our study findings. Further, in a recent study, Kulminski and colleagues 53 found that the risk of AD is influenced by the interaction between rs7412 and rs429358, along with several tissue-specific polymorphisms in the 19q13.3 region, which are not only driven by common evolutionary forces.
The BDNF gene rs6265 variant, known as the Val66Met polymorphism,21,54 reduces the secretion of neurotrophic factors, resulting in impaired cognition, energy balance, and cardiovascular health, which are essential for the survival of neurons and the growth of new neurons and synapses. Abanmy and colleagues 21 found that the BDNF gene rs6265 variant is significantly associated with cognitive performance in the elderly control group and AD patients. A recent meta-analysis 55 indicated that the A allele of rs6265 may increase AD risk in Caucasian females and female late-onset AD patients but not in Asians. Contrarily, Li and colleagues 56 found a gender-specific association of rs6265 with AD, noting a distinct risk trend for females regarding its effect on AD endophenotypes among the Chinese population, supporting our study findings. Additionally, Voineskos and colleagues 57 conducted a study using high-resolution MRI and diffusion tensor imaging, finding that the BDNF gene variant rs6265 increases the risk to brain structures and age-dependent cognitive function impairment, particularly in the early stages of AD.
Despite Lambert and colleagues 23 first linking complement Component Receptor 1 gene (CR1) to an increased risk of AD, Brouwers and colleagues 58 identified four specific SNVs of CR1 (rs646817, rs1746659, rs11803956, and rs12034383) among the European AD population. Nevertheless, Biffi and colleagues 59 discovered the CR1 rs6656401 variant as a potential SNV among Americans AD due to vascular amyloid deposition in the brain. Additionally, Zhu and coworkers 60 described that the CR1 gene rs1349409945 variant encodes a type-I transmembrane glycoprotein, a significant risk gene for late-onset AD. Although we found a quasi-significant association of the BDNF gene rs6265 variant and CR1 gene rs1349409945 variant with AD, the novelty is we first described the potential association of rs6265 and rs1349409945 among the Bangladeshi AD population.
As with any study, several limitations need to be noted. Our results are based on a small sample size; however, this is the first NGS study among the Bangladeshi population to find the APOE rs429358 variant strongly associated with AD. Given the rarity and limited evidence from a Bangladeshi perspective on AD the current study's findings provide valuable insights into the literature and reduce outcome bias. Insofar as the SNP frequencies in the APOE locus are biased toward population-specific haplotypes, 52 indicating that ancestral background may influence disease phenotypes, our study results can be utilized for more specific risk assessment based on population descent. Although association testing results in the APOE region differ by models adjusting for the ε2/ε3/ε4 genotype, future GWAS may uncover new findings in existing evidence. Although our findings are robust, they are limited to the analyzed sample and may not accurately reflect fetal chromosomal structure in cases of confined placental mosaicism or maternal cell contamination. Insofar as all genetic tests have limitations in sensitivity and specificity, it's important to note that DNA analysis has an error rate of ∼2%, which should be considered with the patient's clinical history and other diagnostic findings for informed decision-making. Furthermore, the quasi-significant association between the BDNF rs6265 variant and the CR1 rs1349409945 variant in AD may be attributed to the small sample size, so larger studies are needed to validate our findings. Finally, the APOE ε4 allele frequencies in our control population were comparable to those in the Indian Bengali (∼5.5%), 61 North Indian (4.7%), 62 and Chinese (7.3%) 63 populations. We observed a high odds ratio for APOE ε2; however, the wide confidence interval and non-significant p-value suggest uncertainty due to the limited data. We recommend a large-scale study to confirm these findings.
Conclusion
The APOE variant rs429358 increases AD risk by 8-times in the Bangladeshi population.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261419066 - Supplemental material for Next-generation sequencing study identifies the first genes associated with Alzheimer's disease in Bangladeshi population: The CARED study
Supplemental material, sj-docx-1-alz-10.1177_13872877261419066 for Next-generation sequencing study identifies the first genes associated with Alzheimer's disease in Bangladeshi population: The CARED study by Maliha Hakim, Redoy Ranjan, Gie Ken-Dror, Md. Abdullah Yusuf, Mohammad Nur Uddin, Mim Tanzila Mamun, Alif Al Mamun, Ghulam Kawnayn and Pankaj Sharma in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
We thank Incepta Pharmaceuticals Ltd for their support in conducting the research project. We are also grateful to our patients for their support and contribution.
Ethical considerations
The institutional review board clearance was obtained from the National Institute of Neurosciences and Hospital (NINS&H) in Dhaka, Bangladesh, Bangladesh (IRB/NINS/2024/393).
Consent to participate
An informed written consent was obtained from the patients, and the data were encrypted.
Consent for publication
Not applicable.
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data are not publicly available due to privacy or ethical restrictions; however, the data supporting current study findings are available on reasonable requests from the corresponding author, Professor Dr Maliha Hakim, who holds the data and responds to external requests for data access.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.