
Abstract
Background
Amyloid deposition is a key pathologic hallmark of Alzheimer’s disease (AD). Since cardiac amyloidosis also involves abnormal amyloid accumulation, shared proteinopathy mechanisms may underlie both conditions.
Objective
We investigated whether brain amyloidosis on PET imaging is associated with electrocardiogram (EKG) findings of cardiac involvement by AD pathology.
Methods
We included 191 participants (mean age 66 ± 13.0 years) in our analysis, who underwent amyloid PET imaging and EKG within one year. EKG abnormalities (including low QRS voltage, poor R-wave progression (PRWP), bundle branch block, and sinus bradycardia) were assessed for associations with brain amyloid deposition, quantified in Centiloids, and cognitive function. A subset (n = 164) also underwent 18F-MK6240 tau PET, and the relationship between these EKG abnormalities and tau deposition was also assessed.
Results
PRWP was associated with greater brain amyloid deposition (coefficient = 45 ± 12.15, p < 0.001) and worse cognition on the Clinical Dementia Rating scale (coefficient = 0.22 ± 0.098, p = 0.027). There was a trend for higher likelihood of cortical tau deposition with PRWP (odds ratio = 3.66, p = 0.05). Sinus bradycardia was associated with higher likelihood of cortical tau deposition (odds ratio = 2.81, p = 0.009). Other EKG abnormalities were not significantly associated with brain amyloid/tau deposition or cognition.
Conclusions
PRWP and sinus bradycardia were found to be associated with AD pathology in the brain. These findings warrant further investigation into how cardiac electrophysiologic abnormalities may reflect a possible link between brain and cardiac pathologies or AD-associated cardiac dysfunction.
Keywords
Introduction
Brain amyloidosis is one of the key pathological hallmarks of Alzheimer’s disease (AD), which, together with tau neurofibrillary tangles, adversely alters neuronal function and leads to neuronal death.1,2 The pathogenesis of disease from amyloid accumulation is however not unique to AD, as deposition of amyloid can occur in nearly every organ system throughout the body. For example, cardiac amyloidosis (CA) is the most common cause of restrictive cardiomyopathy in the world3-5 and is most often attributed to extracellular accumulation of transthyretin (TTR), serum amyloid A, and/or light chain-type amyloid. While multiple genetic factors have been implicated in different types of amyloidosis, increasing age is a unifying risk factor among them.1,6,7
Moreover, it has been postulated that amyloid-β (Aβ) pathology in AD may itself be associated with cardiac dysfunction, since Aβ deposition has been shown to be present in AD heart specimens.4,8,9 It is believed that Aβ can enter the systemic circulation via a disrupted blood-brain barrier, a process that allows for the measurement of currently widely-used plasma Aβ biomarkers, depositing within the interstitium and extracellular matrix of cardiac tissue; this can disrupt myocardial architecture and affect cardiomyocyte function, leading to electrical conduction abnormalities on EKG.4,9,10
As accumulation of amyloid often precedes clinical manifestations of the disease on the order of years to decades, diagnostic imaging serves a pivotal role in the evaluation of amyloidosis 7 . Abnormal accumulation of amyloid can be directly imaged using various radiotracers, namely 18F-florbetapir, 18F-florbetaben, 18F-flutemetamol, and 18F-flutafuranol within the brain, and Technetium-99m-PYP to visualize TTR-amyloid within the heart. 6 In the setting of CA, the presence of amyloid may be indirectly inferred using cardiac magnetic resonance imaging (MRI), echocardiography, and electrocardiography (EKG). 6 Low QRS-complex voltage is the most specific finding of CA on EKG, although PRWP, bundle branch block, and sinus bradycardia have also been described.11,12 Beyond amyloid-specific mechanisms, cardiac dysfunction itself, arising from altered myocardial relaxation, microvascular injury, or autonomic imbalance, has been increasingly recognized as a contributor to cognitive decline and neurodegeneration; studies have shown that impaired cardiac output or subclinical left ventricular dysfunction may reduce cerebral perfusion, promoting tau hyperphosphorylation and neurofibrillary tangle formation, even in the absence of overt AD-associated cardiac abnormality.13,14
As deposition of amyloid can result in AD and secondary cardiac involvement, this intuitively supports the idea that the presence of amyloid in one organ system may increase the risk of developing amyloid in another, due to parallel pathogenesis of these proteinopathies. Given the projected increase in the incidence of both AD and CA throughout the world over the coming decades, combined with a recent increase in investigational and FDA-approved pharmacologic treatments for different types of amyloidosis, it is essential to further explore the complex relationships between different variations of this disease. 3 This study sought to examine the relationship between AD and electrophysiologic evidence of cardiac dysfunction on EKGs in individuals with varying levels of brain amyloidosis on PET. Little has been documented regarding the incidence of AD with a concomitant cardiac abnormality in vivo.
Methods
Study design
One hundred and ninety-one individuals were prospectively enrolled between 2021 and 2023 into our ongoing research studies on aging and AD and retrospectively analyzed in our study (Table 1). Our study complied with ethical standards for human research and were approved by the WCM Institutional Review Board (IRB #19-11021064). All subjects provided written informed consent before participation. Each subject underwent screening, detailed interviews, clinical and neurological examinations, and neuropsychological assessments, including the Clinical Dementia Rating (CDR) 15 scale and the Blind Montreal Cognitive Assessment (MoCA-22).16,17 Inclusion criteria included age greater than 50 years, having undergone an amyloid PET scan, and having undergone an EKG within 1 year of the amyloid PET scan. None of the enrolled individuals had a clinical diagnosis of cardiac or systemic amyloidosis.
Baseline demographic information of the cohort.

Baseline demographic information of the cohort.
*CDR data were available for 184 participants
**Tau PET data were available for 164 participants
APOE genotyping
Quantitative polymerase chain reaction was used to determine APOE genotypes using the TaqMan Genotyping Assays (ThermoFisher Scientific, Waltham, MA). Individuals with either one or two APOE ε4 alleles were classified as an APOE ε4 carrier, and those with either one or two ε2 alleles as an APOE ε2 carrier.
MR imaging acquisition
All participants underwent MRI of the brain on a 3-Tesla Siemens scanner (Siemens Healthineers, Erlangen, Germany) equipped with a 64-channel head/neck receiver coil. The imaging protocol included a three-dimensional T1-weighted (T1w) Magnetization-Prepared Rapid Gradient-Echo (MPRAGE) sequence with the following parameters: repetition time (TR) = 2400 ms, echo time (TE) = 2.96 ms, inversion time (TI) = 900 ms, flip angle (FA) = 9°, readout bandwidth (rBW) = 240 Hz/pixel, isotropic voxel size = 0.5 mm, matrix size = 512 × 512 × 416, and total acquisition time = 5.75 minutes.
PET imaging acquisition
Amyloid PET imaging was performed on a Siemens Biograph mCT (64-slice) PET/CT scanner. The PiB radiotracer was synthesized by the WCM core radiochemistry facility. PiB PET imaging data were acquired in list mode 50 to 70 minutes after a rapid bolus injection of approximately 555 MBq of PiB. The images were then reconstructed into a 400 × 400 × 109 matrix, with voxel dimensions of 1 × 1 × 2 mm, in four 5-minute frames. For FBB PET imaging, an intravenous injection of approximately 300 MBq (8.1 mCi) of FBB was administered. Images were acquired from 90 to 110 minutes after injection and reconstructed into four 5-minute frames. The images were reconstructed into a 400 × 400 × 55 matrix, and the voxel size was 1 × 1 × 3 mm.
A subset of participants (n = 164) underwent 18F-MK6240 PET scans to assess tau deposition. Participant preparation involved intravenous catheterization followed by the injection of 185 MBq (5 mCi) ± 20% (maximum volume: 10 mL) of radiotracer, administered as a slow single intravenous bolus over 60 seconds or less (6 s/mL max). 3D imaging began 90 minutes after tracer injection. Imaging of the brain was acquired in 6 × 5-minute frames over a period of 30 minutes.
PET image processing
For PET analysis, dynamic PET frames were first realigned to the mean volume 18 and averaged to generate a static image. Each subject’s T1w MPRAGE was processed with the Freesurfer v7.1 standard pipeline 19 and the static PET image was coregistered to the Freesurfer space T1 image using rigid-body transformation and the cost function of normalized mutual information in FMRIB Software Library (FSL). The resulting transformation matrix was saved and subsequently applied to all dynamic frames, aligning the full dynamic PET dataset to the Freesurfer space. SUVR maps were generated using the summed image of the entire acquisition, with the cerebellar cortex as the reference region. 20 An SUVR quantitative threshold of 1.35 was used for the assessment of tau deposition. 21
Amyloid quantification using the Centiloid scale
Centiloid (CL) quantification of amyloid burden in the brain was quantified according to the Global Alzheimer's Association Interactive Network (GAAIN) pipeline. 22 Briefly, this standardization approach utilizes a 100-point scale, in which 0 represents the average amyloid burden in amyloid-negative controls, and 100 represents the average amyloid burden in mild-to-moderate AD patients, although the full range of the scale is not limited. The CL was derived from the SUVR using a tracer-specific linear transformation, expressed for PiB as 23 :
The conversion equation from FBB to PiB was:
SUVR_FBB to PiB = (CTX_SUVR_FBB - 0.5147) / 0.5936
EKG interpretation
EKGs for each patient were analyzed in blinded fashion by both a resident physician, with advanced training in EKG interpretation, and a cardiology fellow physician, with clinical and imaging information removed from the corresponding EKG. Both readers assessed the EKGs independently, and any discrepancies in interpretation were resolved by a senior cardiologist. Each EKG was specifically analyzed for hallmark findings of cardiac amyloidosis, including low QRS voltage (defined as a QRS amplitude ≤5 mm in limb leads or ≤10 mm in precordial leads), PRWP (defined as a lack of increasing R-wave amplitude in precordial leads V1–V4), and conduction abnormalities such as bundle branch block, and sinus bradycardia (heart rate <60 bpm).11,12,24,25
Statistical analysis
All statistical analyses were conducted using STATA software, version 16.1. Multivariate regression models were used. To test whether EKG abnormalities, particularly those suggestive of cardiac amyloidosis, were associated with cerebral amyloid deposition, we used ordinary least squares regression models with the EKG finding (low QRS voltage, PRWP, bundle branch block, sinus bradycardia) as the predictor variable and amyloid deposition, quantified in Centiloids, as the outcome variable. Age, sex, cardiometabolic comorbidities (e.g., hypertension, type 2 diabetes, hyperlipidemia, coronary artery disease, obesity), and APOE genotype were considered as covariates. Hierarchical multivariable ordinary least squares regression models were then constructed as follows: Model 1 included age and sex as covariates. Model 2 included Model 1 covariates with additional adjustment for the cardiometabolic comorbidities listed above. Model 3 further adjusted for APOE ε4 carrier status alone and with APOE ε2 carrier status as an additional covariate. Regression coefficients, 95% confidence intervals (CI), and p-values were reported for each model.
To further evaluate the clinical utility of PRWP as a predictor of amyloid positivity, we performed univariable and multivariable logistic regressions using two separate thresholds: a threshold of >11 Centiloids, as utilized in the TRAILBLAZER-ALZ 2 trial, 26 and a threshold of >18 Centiloids previously published using our data. 23 Odds ratios (OR) and 95% CI were reported for these models.
Then, in the subset with tau PET, we tested whether EKG abnormalities were associated with MTL or cortical tau deposition. To do so, we used logistic regression with the EKG finding (low QRS voltage, PRWP, bundle branch block, sinus bradycardia) as the predictor variable and the presence of tau deposition in the MTL or the cortex as the dichotomous outcome variable. Age, sex, and APOE ε2 or ε4 carrier status were again considered as covariates.
Finally, we tested whether EKG abnormalities were associated with cognition. Using ordinary least squares regression with the EKG finding (low QRS voltage, PRWP, bundle branch block, sinus bradycardia) as the predictor variable and global CDR or MoCA-22 as the outcome variable. Significance was set at p < 0.05.
Results
Baseline demographic variables are shown in Table 1.
Poor R-wave progression is associated with brain amyloidosis
PRWP on EKG was significantly associated with increased brain amyloid deposition, as quantified in Centiloids (regression coefficient = 45.2 ± 12.15, 95% confidence interval [21.3, 69.2], p < 0.001) (Figure 1). This association remained statistically significant in subsequent multivariable ordinary least squares regression models. After adjustment for age and sex (Model 1), PRWP was associated with higher amyloid burden (coefficient = 41.5, p < 0.001). Further adjustment for cardiometabolic comorbidities, including hypertension, hyperlipidemia, coronary artery disease, and obesity (Model 2), strengthened the association (coefficient = 138.0, p < 0.001). The association persisted after additional adjustment for APOE ε4 carrier status (Model 3; coefficient = 145.0, p < 0.001). Adding APOE ε2 carrier status did not significantly change the results (coefficient = 143.0, p < 0.001). Across all models, the direction and magnitude of the association were consistent, supporting a robust relationship between PRWP and brain amyloidosis. The other EKG abnormalities of interest were not associated with brain amyloidosis, including low QRS voltage (p = 0.69), bundle branch block (p = 0.23), and sinus bradycardia (p = 0.41).
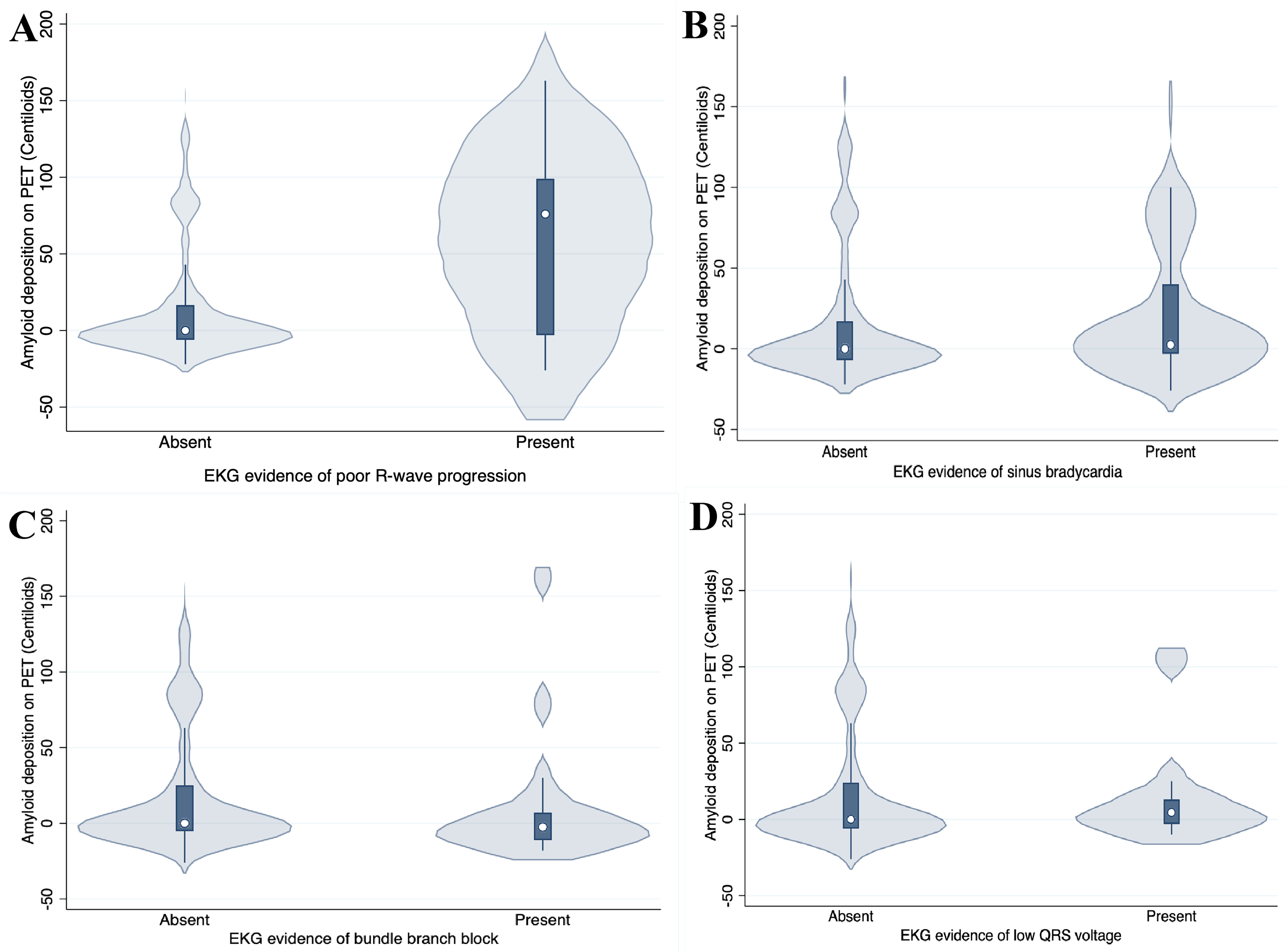
Association between EKG abnormalities and amyloid deposition on PET. Violin plots show Centiloid values in participants with and without EKG evidence of (A) poor R-~>-->wave progression; (B) sinus bradycardia; (C) bundle branch block; and (D) low QRS voltage. White dots indicate the median; dark boxes represent interquartile ranges.
We also performed logistic regression analyses using two different Centiloid thresholds for amyloid positivity. Using the threshold of >11 CL (based on TRAILBLAZER-ALZ 2), PRWP was associated with a higher likelihood of amyloid positivity (OR = 6.06, 95% CI [1.68, 28.47], p = 0.009), which remained significant after adjusting for age and sex (OR = 4.91, 95% CI [1.32, 23.57], p = 0.025). Using a threshold of >18 CL, the association was even more pronounced (OR = 8.24, 95% CI [2.28, 38.88], p = 0.003), and again persisted after adjustment for age and sex (OR = 6.77, 95% CI [1.82, 32.69], p = 0.007).
Poor R-wave progression is weakly associated with cortical tau deposition
PRWP was marginally associated with a higher likelihood of cortical tau deposition on PET (odds ratio = 3.65, 95% confidence interval [1.00, 13.41], p = 0.05) (Figure 2). However, the association weakened after adjusting for age (odds ratio = 3.36, 95% confidence interval [0.91, 12.44], p = 0.069), suggesting that age may partially confound this relationship. In contrast, no significant association was observed between PRWP and tau deposition in the MTL (odds ratio = 2.22, 95% confidence interval [0.57, 8.61], p = 0.25).
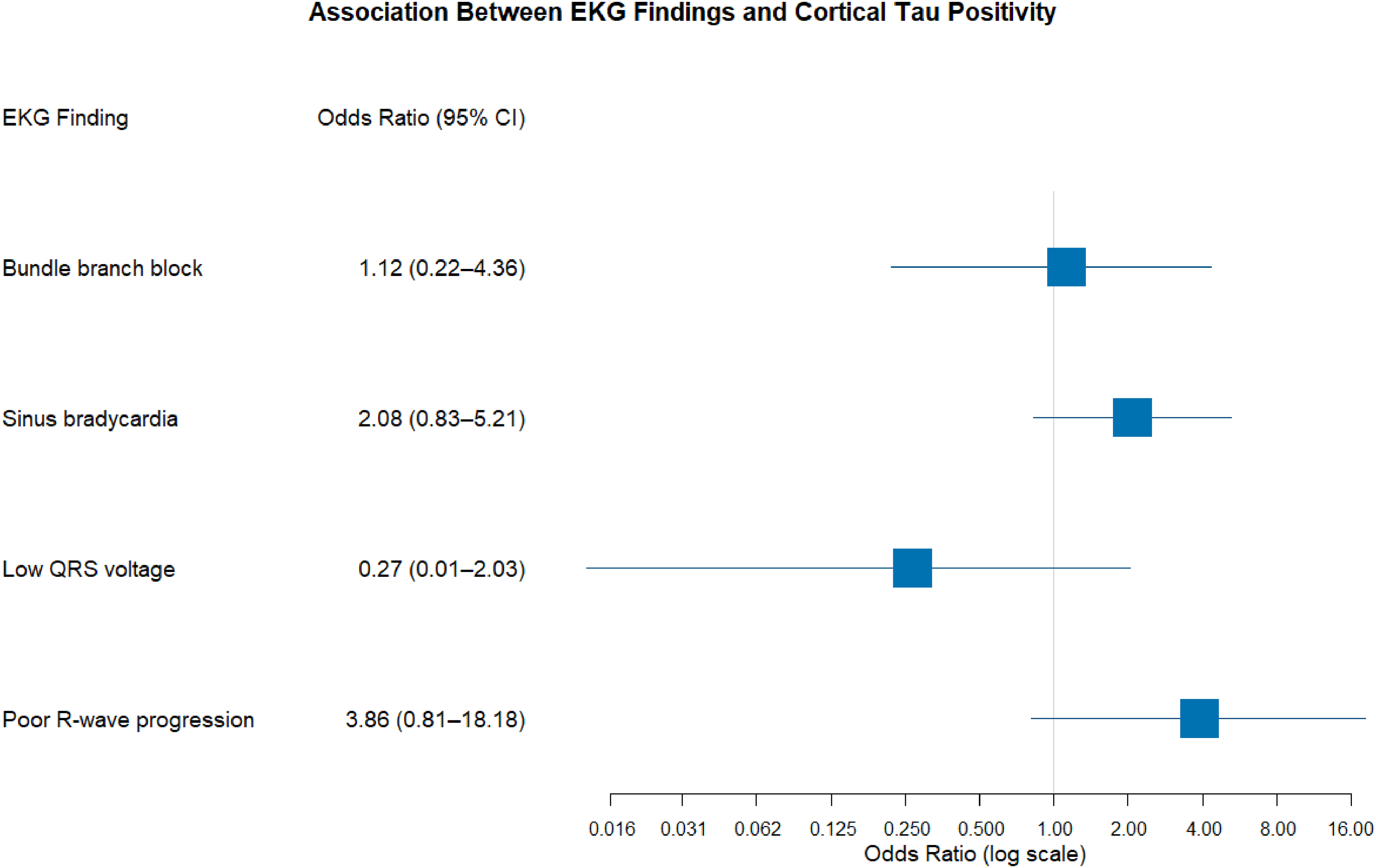
Forest plot showing the association between specific EKG findings and cortical tau positivity on PET. Each blue square represents the odds ratio (OR) point estimate, with horizontal lines indicating the 95% confidence interval (CI). OR > 1 suggests increased likelihood of tau positivity; OR < 1 suggests decreased likelihood. Log scale used for the odds ratio axis.
Sinus bradycardia is associated with cortical tau deposition
Sinus bradycardia was significantly associated with a higher likelihood of cortical tau deposition on PET (odds ratio = 2.81, 95% confidence interval [1.29, 6.15], p = 0.009) (Figure 2). This association persisted after adjusting for age (odds ratio = 2.76, 95% confidence interval [1.26, 6.07], p = 0.01) and APOE ε2 carrier status (odds ratio = 2.68, 95% confidence interval [1.18, 6.08], p = 0.018), but weakened after adjusting for APOE ε4 carrier status (odds ratio = 2.04, 95% confidence interval [0.80, 5.17], p = 0.134). No significant association was observed between sinus bradycardia and MTL tau deposition (p = 0.46). In addition, other EKG abnormalities, including low QRS voltage (p = 0.47 for cortical tau, p = 0.99 for MTL tau) and bundle branch block (p = 0.49 for both cortical and MTL tau) showed no significant associations with tau deposition.
Poor R-wave progression and cognition
PRWP was associated with higher global CDR scores (coefficient = 0.22 ± 0.098, p = 0.027). This association remained significant after adjusting for age (coefficient = 0.20 ± 0.098, t = 2.05, p = 0.04), but weakened with adjustment for APOE ε2 and ε4 carrier status (coefficient = 0.19 ± 0.097, t = 1.92, p = 0.056). In contrast, PRWP was not associated with MoCA-22 scores (coefficient = −0.02 ± 1.14, 95% CI [−2.27, 2.23], p = 0.99). Other EKG abnormalities, including low QRS voltage (p = 0.44), bundle branch block (p = 0.93), and sinus bradycardia (p = 0.82) showed no significant associations with global CDR scores. Sinus bradycardia was however significantly associated with higher MoCA-22 scores (p = 0.007). In contrast, low QRS voltage (p = 0.32) and bundle branch block (p = 0.56) showed no significant associations with MoCA-22 performance.
Discussion
The study was driven by the hypothesis that amyloid accumulation in the brain in AD may occur in parallel with amyloid accumulation in the heart, leading to subclinical EKG abnormalities. Alternatively, cardiac abnormalities seen on EKG may emerge as a response to brain amyloidosis in AD. Either hypothesis would support the use of EKG as a low-cost, screening tool for AD-associated cardiac dysfunction. Given the rising prevalence of both conditions in our aging population and emerging amyloid-targeting therapies, understanding mechanisms leading to both could allow for early intervention.
Our findings point to an association between PRWP on EKG and greater brain amyloid deposition, providing evidence of early changes in the heart in association with brain amyloidosis. Additionally, both PRWP and sinus bradycardia were associated with a higher likelihood of cortical tau deposition, which suggests that cardiac electrophysiologic abnormalities may reflect or be a response to AD pathology in the brain.8,27,28 Notably, no significant associations were observed between brain amyloid/tau deposition and other EKG abnormalities, including low QRS voltage and bundle branch block.
The significant association between PRWP and brain amyloid deposition aligns with prior research suggesting a link between cardiac dysfunction, possibly related to amyloid deposition, and AD. For example, Troncone et al. detected Aβ40 and Aβ42 amyloid within the endomyocardium of AD patients using immunoblot and ELISA analysis, while Elia et al. identified Aβ amyloid in the endomyocardium of a mouse model of AD.4,29 The presence of amyloid within the endomyocardium suggests a direct infiltrative process in which Aβ deposits in the interstitium and extracellular matrix of cardiac tissue, disrupting cardiomyocyte function; specifically, the replacement of myocardium with non-conductive amyloid fibrils in the anterior or septal regions can lead to the loss of R-wave amplitude, manifesting as PRWP on the EKG.12,29 30 The systemic circulation of Aβ, likely via a disrupted blood-brain barrier, underscores that AD should perhaps be viewed not as a brain-isolated event, but as a systemic proteinopathy that we can currently monitor with plasma biomarkers and measurable cardiac electrophysiologic consequences.4,10,29,30 The persistence of the association between PRWP and amyloid deposition after adjustments for age, sex, APOE status, and comorbidities underscores a robust link between this cardiac finding and brain amyloidosis. Furthermore, the association with worse global CDR scores indicates that PRWP may occur with more advanced disease, potentially mediated by amyloid deposition or other cardiac abnormalities.
The associations of PRWP and sinus bradycardia with cortical tau deposition are novel findings that suggest a potential link between cardiac electrophysiologic abnormalities and tau pathology. PRWP showed a marginally significant association with cortical tau deposition, whereas sinus bradycardia was significantly associated with cortical tau deposition. These findings may reflect a shared mechanism involving autonomic nervous system (ANS) dysfunction, as tau accumulation in key regions such as the insula, anterior cingulate, and brainstem is known to disrupt central autonomic control, potentially manifesting as cardiac abnormalities like bradycardia.31,32 Literature supports this hypothesis, demonstrating that tau pathology in autonomic regulatory regions is associated with reduced heart rate variability (HRV), a marker of ANS dysfunction, in AD patients.33-3437 Additionally, big tau deposition has recently been shown in the heart, with hyperphosphorylated tau aggregates present in heart specimens of people with heart failure (HF) and AD 38 . Reduced HRV has also been linked to cognitive decline and abnormal amyloid/tau ratios, suggesting a complex interplay between cardiac and neurodegenerative processes. 39 The lack of statistical significance association with MTL tau deposition for both PRWP and sinus bradycardia indicates that these EKG abnormalities may be specifically related to cortical tau pathology, possibly due to the regional distribution of tau in areas critical for autonomic regulation.
Altogether, findings show that autonomic dysfunction driven by cortical and brainstem tau pathology may serve as a primary mechanism underlying the observed EKG abnormalities, rather than direct cardiac amyloid infiltration. Tau accumulation within the insula, anterior cingulate, and medullary autonomic nuclei can impair sympathetic and parasympathetic regulation, producing bradyarrhythmia and conduction changes detectable on EKGs.34,40,41 This interpretation aligns with evidence linking tau burden in autonomic control centers to heart rate variability reduction and cardiac rhythm instability in AD.
EKG is a rapid, low-cost, and low-risk tool for detecting cardiac abnormalities, making it valuable for identifying early signs of cardiac dysfunction in AD patients. PRWP was included as a primary outcome because it serves as a sensitive, though nonspecific, marker of altered ventricular conduction that can precede the overt restrictive cardiomyopathy seen with cardiac amyloidosis. As Aβ circulates systemically and deposits within the myocardium, it may induce cytotoxicity through apoptosis and intracytoplasmic calcium dysregulation, creating a proarrhythmogenic state.12,27 The disruption of cardiomyocytes by these Aβ fibrils may impair the transmission of electrical impulses, leading to arrhythmias, such as PRWP and sinus bradycardia.12,42,12,42,43 Similarly, mitochondria-mediated toxicity has been also shown to be induced in cardiac cells by Aβ.27,28 PRWP, as an early and sensitive marker of conduction anomalies, may reflect amyloid infiltration in conduction pathways, while sinus bradycardia may reflect amyloid and/or tau deposition in the sinoatrial node.12,29 The absence of a significant association with low QRS voltage, despite its historical specificity for CA, may be due to its lower sensitivity, as only approximately 25% of CA patients exhibit this finding, as noted by Martinez-Naharro et al. 44 The non-significant associations with bundle branch block (p > 0.05) may also reflect its lower sensitivity to early amyloid deposition or its association with more common, non-AD-related etiologies.
The non-specific nature of PRWP and sinus bradycardia complicates their use as diagnostic markers for AD-associated cardiac abnormality, as these findings can arise from various etiologies. Current nuclear medicine techniques, such as Technetium-99m-PYP imaging, are specific to TTR-type CA and do not detect Aβ40 or Aβ42 amyloid, limiting their utility in this context.7,12,45 Therefore, alternative diagnostic modalities, such as echocardiography or cardiac MRI, should be considered to evaluate cardiac abnormalities in AD patients with these EKG findings.45,46 The potential link between cortical tau pathology, ANS dysfunction, and cardiac rhythm abnormalities suggests that EKG could serve as a low-cost, screening tool to identify patients at risk for AD-related pathology, warranting further neuroimaging or cognitive assessment. 47
While our study focused on EKG-derived markers, we acknowledge that EKG abnormalities alone cannot establish the presence or mechanism of cardiac involvement in AD and should be viewed as hypothesis-generating. Confirmation with complementary cardiac phenotyping will be critical in future studies. For example, echocardiography and cardiac magnetic resonance imaging can detect myocardial infiltration, wall thickness changes, strain abnormalities, and tissue characterization not captured by EKG, while circulating biomarkers may provide additional insight into cardiomyocyte injury and amyloid-related toxicity.45,48 Moreover, autonomic metrics such as heart rate variability may offer a quantitative assessment of neurocardiac dysfunction and may help link cortical and brainstem tau pathology with cardiac electrophysiologic changes.35,36,45,49 Integrating EKG with multimodal cardiac imaging, biomarkers, and autonomic assessments will be essential to clarify mechanisms and guide subsequent studies of AD-associated cardiac dysfunction.11,50,51
This retrospective analysis has several limitations. First, it focused solely on EKG findings, without incorporating other diagnostic modalities for AD-associated cardiac abnormality as noted, such as serum biomarkers, echocardiography, cardiac MRI, or endomyocardial biopsy. Second, the study was limited to Aβ amyloid and did not evaluate other amyloid variants, such as TTR, serum amyloid A, or light chain amyloid. Third, the analysis did not directly assess ANS dysfunction or HRV, which could mediate the observed associations between EKG abnormalities and cortical tau pathology. Future prospective studies should integrate multimodal imaging, serum biomarkers, and HRV assessments to further elucidate the relationship between cardiac abnormality, AD, and ANS dysfunction. Pathological studies confirming Aβ and tau deposition in cardiac tissue would also strengthen the mechanistic understanding of these associations.
Conclusion
In conclusion, our findings highlight significant associations between PRWP, sinus bradycardia, and AD pathologies, including brain amyloidosis, cortical tau deposition, and cognitive impairment. These associations may be mediated by amyloid infiltration in the heart and tau-related ANS dysfunction, suggesting a complex interplay between cardiac and neurodegenerative processes. Further research is needed to validate these findings and explore their clinical implications for the diagnosis and management of AD and AD-associated cardiac abnormalities.
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Ethical Considerations
This study received ethical approval from the Weill Cornell medicine IRB (approval #19-11021064) on August 11, 2025.
Consent to Participate
All participants provided written informed consent to participate.
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by the following grants: National Institutes of Health/National Institute on Aging (R01 AG068398, R01 AG080011, R01 AG085972, U01 AG082845) awarded to G.C. and NIH R01 NS104127 and Pennsylvania Department of Health Collaborative Research on Alzheimer's Disease (PA Cure) Grant, awarded to S.F.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Gloria C. Chiang receives research funds from Minoryx Therapeutics, consulting fees from Life Molecular Imaging and Alnylam Pharmaceuticals, and speaker honoraria from Efficient CME and PeerView CME.
Data availability statement
De-identified data are available upon request to the corresponding author.