
Abstract
Background
In cerebral small vessel disease (CSVD), the burden of white matter hyperintensities (WMH) does not fully account for cognitive impairment, suggesting the involvement of intermediary mechanisms.
Objective
We investigated whether a gray matter atrophy network acts as the key mediator linking topologically specific (deep) WMH to multidomain cognitive dysfunction.
Methods
In this retrospective study, 260 patients with CSVD (62 cognitively normal, 125 with mild impairment, 73 with dementia) were included. Cognitive status was assessed neuropsychologically. 3.0 T MRI identified an atrophy network. We then conducted pre-specified mediation analyses and a primary confirmatory analysis using structural equation modeling (SEM) to test whether this atrophy network mediated the effect of deep WMH on cognitive performance.
Results
A 41-region atrophy network was identified
Conclusions
Our findings suggest that in CSVD, a unified network of gray matter atrophy acts as a powerful statistical mediator in the effect of deep white matter injury on cognitive decline. This atrophy pattern may represent a more direct biomarker of the neurodegenerative process underlying cognitive impairment than WMH burden alone.
Keywords
Introduction
Cerebral small vessel disease (CSVD) is the leading cause of vascular cognitive impairment, affecting executive function and processing speed.1,2 However, white matter hyperintensity (WMH) burden correlates poorly with cognitive performance.3,4 This clinical-radiological mismatch implies impairment stems from disrupted vulnerable networks rather than total lesion load. 5 In this network-centric view, secondary neurodegeneration emerges as the crucial link connecting focal insults to global dysfunction.6,7
Remote white matter damage is thought to induce secondary gray matter atrophy via Wallerian degeneration. Indeed, a growing body of evidence has established that WMH burden drives cortical network atrophy and mediates cognitive decline. For example, Habes et al. demonstrated that WMH mediates the effect of age on brain atrophy patterns. 8 Longitudinal studies have further revealed that WMH promotes an Alzheimer-like pattern of neurodegeneration, particularly highlighting the medial temporal lobe's role in mediating memory loss.9,10 Additionally, structural covariance and graph theoretical analyses have shown that WMH-induced network disruptions and specific gray matter atrophy (e.g., insula) are causally linked to executive dysfunction.11,12 Importantly, the presence of such CSVD markers has been shown to act as an active, independent determinant of poor clinical prognosis and limited functional recovery. 13 Beyond these cortical and large-scale network effects, recent studies have begun to delineate the specific microstructural consequences of such atrophy. For instance, Li et al. showed that thalamic atrophy impacts cognition through tract disruption. 14 Yet, the upstream drivers of this atrophy remain unclear in humans. Furthermore, most previous models considered total WMH volume, often neglecting the topographical specificity (e.g., deep versus periventricular) that may drive distinct cascades. A significant limitation is that reliance on advanced quantitative imaging (e.g., DTI) restricts the clinical applicability of these findings.
To bridge this gap, we leveraged routine structural magnetic resonance imaging (MRI) to investigate the pathological cascade in CSVD. Our central hypothesis is that the impact of WMH on cognition is mediated by specific patterns of gray matter atrophy forming a “CSVD-related atrophy network” and that this pathway is differentially driven by deep versus periventricular WMH.
Methods
Study design and participants
In this retrospective, cross-sectional study (2021–2024), we investigated the pathological pathways connecting vascular injury, specific brain atrophy, and cognitive impairment in CSVD (Figure 1). In this retrospective cross-sectional study (2021–2024), we consecutively enrolled 260 patients meeting STRIVE-2 15 and VICCCS 16 criteria for CSVD. Eligible participants, aged 50 years or older, presented with a clinical syndrome compatible with CSVD (e.g., lacunar stroke, vascular parkinsonism, or insidious cognitive decline) and demonstrated at least one core neuroimaging marker of the disease on 3.0 T MRI, such as WMH, lacunes, cerebral microbleeds, or enlarged perivascular spaces. To ensure a homogenous cohort, patients were excluded if they had evidence of large vessel disease (i.e., territorial infarcts or > 50% stenosis of major cranial arteries), potential cardioembolic sources, a clinical diagnosis of another primary neurodegenerative dementia (e.g., Alzheimer's disease), other specific causes of white matter lesions (e.g., multiple sclerosis), major psychiatric disorders, or a history of epilepsy. Participants were subsequently stratified into three groups based on a comprehensive clinical assessment that included a standardized cognitive assessment protocol and an evaluation of functional status using the Activities of Daily Living (ADL) scale. 17 Following established diagnostic criteria, participants were classified as: (i) Cognitively Normal (CSVD-CN, n = 62); (ii) Mild Cognitive Impairment (CSVD-MCI, n = 125); (iii) Dementia (CSVD-Dementia, n = 73).
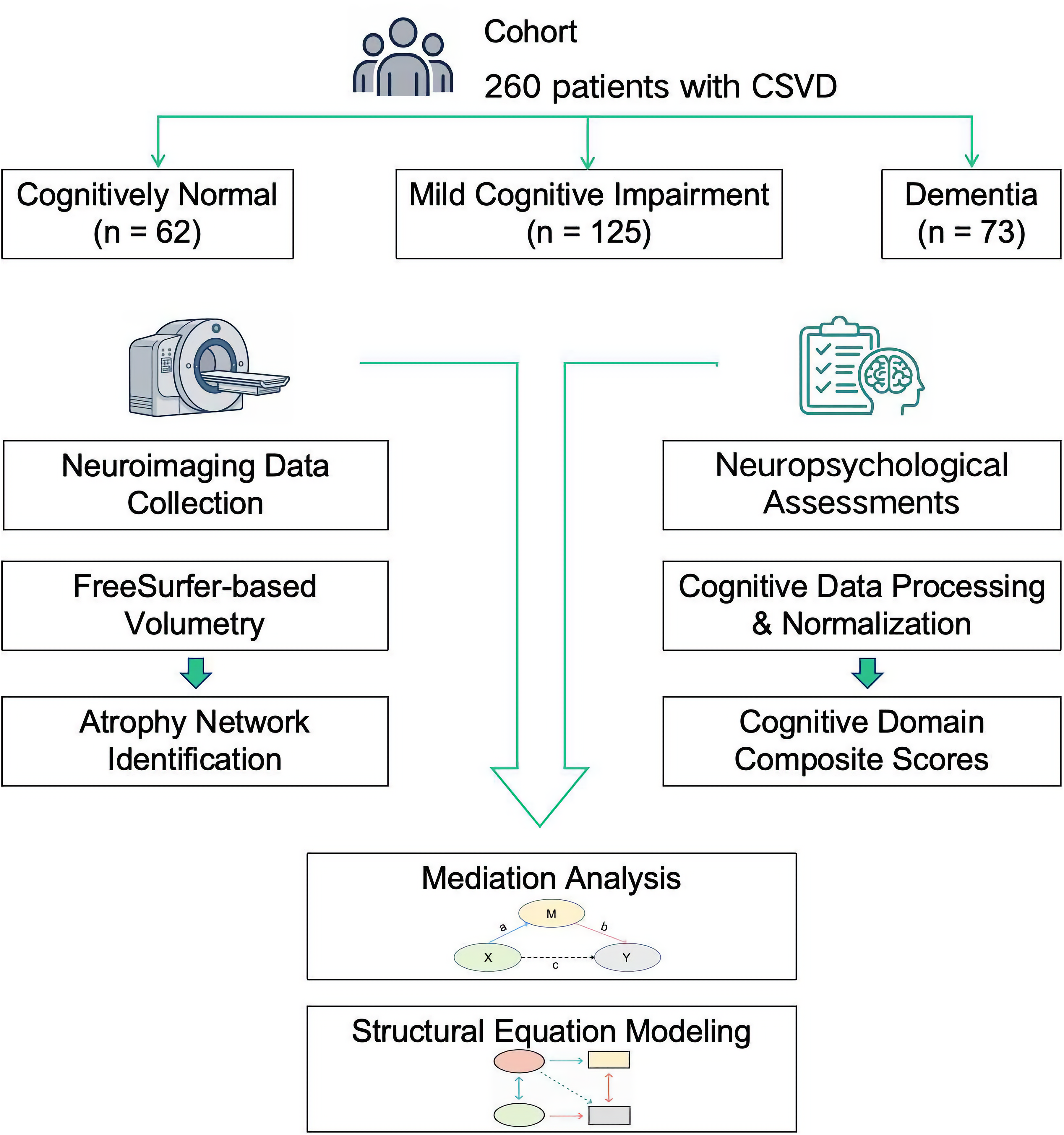
Study design and analytical workflow. A cohort of 260 patients with CSVD was stratified into three groups based on cognitive status (cognitively normal [CN], n = 62; mild cognitive impairment [MCI], n = 125; dementia, n = 73). Neuroimaging data were processed using a FreeSurfer-based pipeline for volumetric analysis and atrophy network identification. Neuropsychological data were processed to derive composite cognitive domain scores. These two data streams were integrated in subsequent mediation analyses and SEM.
Cognitive assessment procedures
All participants underwent 3.0 T MRI and a neuropsychological battery comprising the Mini-Mental State Examination (MMSE), 18 Montreal Cognitive Assessment (MoCA), 19 ADL scale, and the Clock Drawing Test (CDT). 20 Sub-items were mapped to six domains: memory, executive function, language, visuospatial function, attention/working memory, and orientation. To generate domain-specific Z-scores, raw scores were normalized against the CSVD-CN group means and SDs (Z = [raw − mean_CN] / SD_CN) and subsequently averaged within each domain.
Brain MRI scan analysis
MRI scans (T1, T2, FLAIR, DWI, SWI) were acquired on 3.0 T Philips Medical Systems scanners. Detailed sequence parameters are in Supplemental Table 1. MRI data were quantified using two complementary strategies: T1-weighted structural images were processed using FreeSurfer (https://surfer.nmr.mgh.harvard.edu/fswiki/rel7downloads, version 8.0.0). 21 Given that the data were derived from routine clinical scans, we employed the recon-all-clinical pipeline22–25 to extract 262 metrics, including Destrieux-based cortical parcellations, 26 and subcortical subfields.27–30 To ensure the reliability of these automated measurements, all processed data underwent a rigorous, two-step quality control procedure: a systematic review of FreeSurfer's automated QC metrics, followed by visual inspection of all cortical and subcortical segmentations by two independent raters. Participants with significant, uncorrectable segmentation errors were excluded. All structural brain volumes were corrected for total intracranial volume (TIV). WMH and global cortical atrophy (GCA) were visually assessed by two experienced neurologists using the Fazekas scale 31 and the GCA scale, 32 respectively.
Statistical analysis
All statistical analyses were performed using Python (https://www.python.org/, version 3.10), 33 with a two-sided significance threshold of p < 0.05. First, Fisher's exact test compared Fazekas scores (dWMH, PVWMH, total) across cognitive groups. Second, Analysis of covariance (ANCOVA) with age and education as covariates identified atrophy-associated regions among 262 TIV-corrected volumes (FDR q < 0.05). Significant regions were grouped into three composite indices (medial temporal lobe (MTL), thalamus, cortex) by averaging Z-scores normalized to the CSVD-CN group; Third, multiple linear regressions assessed associations between these atrophy indices and cognitive domains, adjusting for covariates. Finally, mediation and structural equation modeling (SEM) were conducted using the semopy library. Mediation analyses tested whether atrophy indices mediated WMH/GCA effects on cognition (Significance was tested using 1000 bootstrap iterations to estimate 95% confidence intervals). For SEM, a latent “CSVD-related Atrophy Network” factor was constructed from the three indices to model the dWMH-cognition pathway, prioritizing executive function and memory. Model fit was evaluated via χ2, CFI, TLI, and RMSEA, comparing a baseline model against alternative structures (Model A: allowing for residual covariance; Model B: including direct paths).
Results
Baseline characteristics of the study cohort
The final cohort consisted of 260 patients with CSVD, stratified into three groups based on cognitive status (CSVD-CN, n = 62; CSVD-MCI, n = 125; CSVD-Dementia, n = 73). Baseline demographic and cognitive characteristics are presented in Table 1. The groups showed significant differences in age, years of education, and all cognitive domain scores (p < 0.001 for all). Our analyses revealed a notable dissociation among WMH subtypes (Supplemental Table 2). A significant association was found between the severity of deep WMH (dWMH) and cognitive status (p = 0.031), with a clear graded increase in burden from the CN to the Dementia group. In contrast, the distributions of periventricular WMH (PVWMH; p = 0.060) and total WMH (p = 0.12) across the groups did not reach statistical significance. Similarly, analysis of global cortical atrophy (GCA) scores for the main cortical regions (frontal, parieto-occipital, and temporal) also showed no significant group differences. However, a more localized measure, the Frontal Horn GCA Score, was significantly associated with cognitive status (p = 0.001), suggesting that periventricular atrophy may be relevant (Supplemental Table 3). Based on these initial findings, dWMH was selected as the primary upstream vascular injury marker for our main pathway analysis. Subsequently, our large-scale screening of 262 regional brain volumes identified a specific network of 41 regions whose atrophy was significantly associated with cognitive status (FDR-corrected q < 0.05; Supplemental Figure 1). The anatomical distribution of this network was highly concentrated, with the vast majority of regions located in the MTL (63.4%) and the thalamus (22.0%), while only a few regions were sparsely distributed across the cerebral cortex (14.6%). The full list of these regions is detailed in Supplemental Table 4. This limbic-thalamic-predominant pattern is visually presented in Figure 2(a) and 2(b). Intergroup comparisons confirmed a clear graded pattern across the key identified variables: progression in clinical staging (from CN to Dementia) was paralleled by increased dWMH severity, pronounced thalamic atrophy, and poorer executive function (Figure 2(c)).
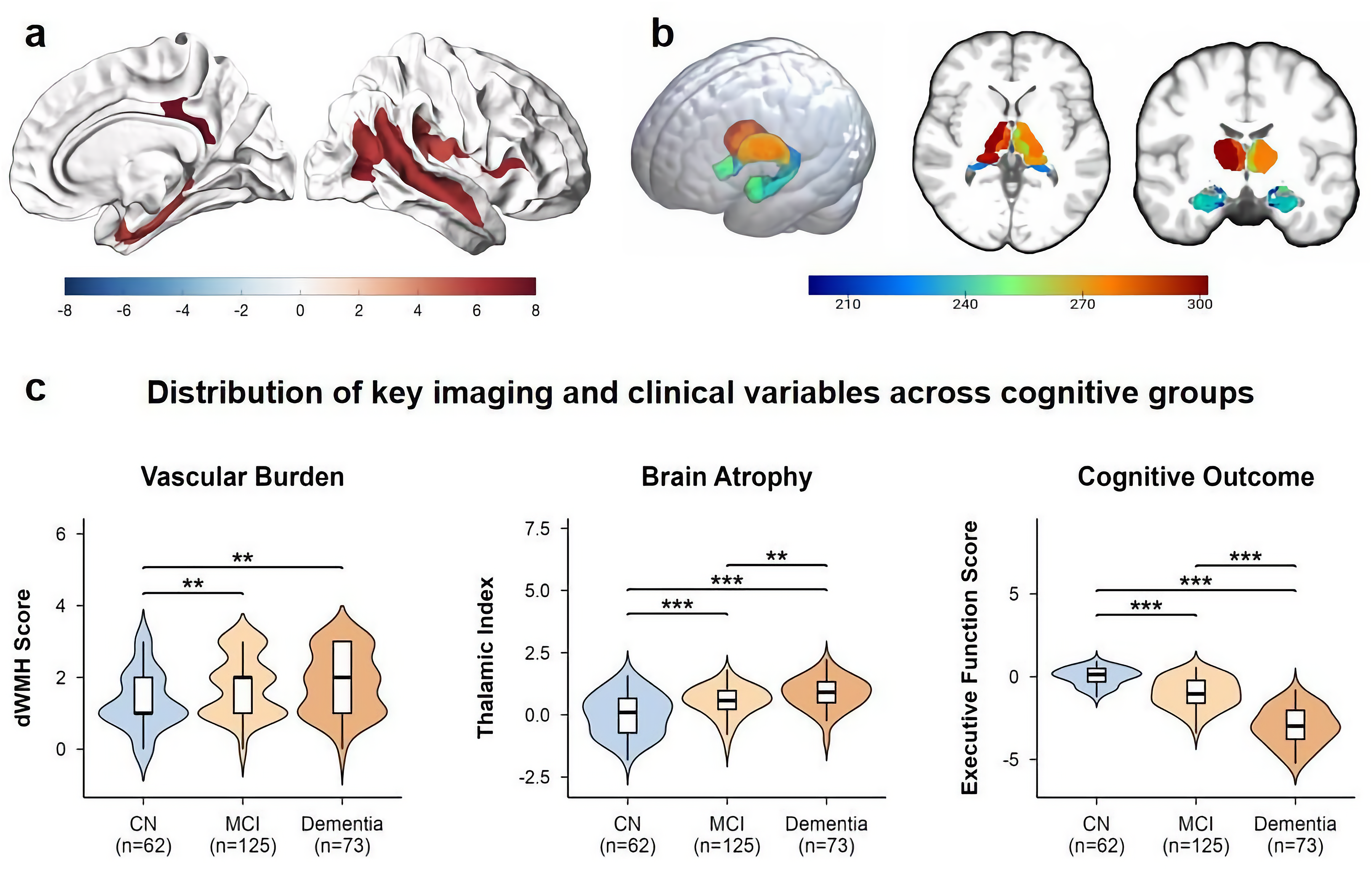
Spatial distribution of the CSVD-related atrophy network and its association with clinical variables. (a) Surface rendering showing cortical regions of the 41-region atrophy network significantly associated with cognitive status (FDR-corrected q < 0.05). (b) Volumetric rendering of subcortical structures within the network, including the thalamus and amygdala/hippocampus. (c) Violin plots showing the distribution of deep white matter hyperintensity (dWMH) scores, the Thalamic Atrophy Index, and the Executive Function Z-score across the three cognitive groups (CN, MCI, Dementia). Boxplots within the violins represent the median and interquartile range. **p < 0.01, ***p < 0.001.
Baseline demographic, clinical, and cognitive characteristics of study participants.
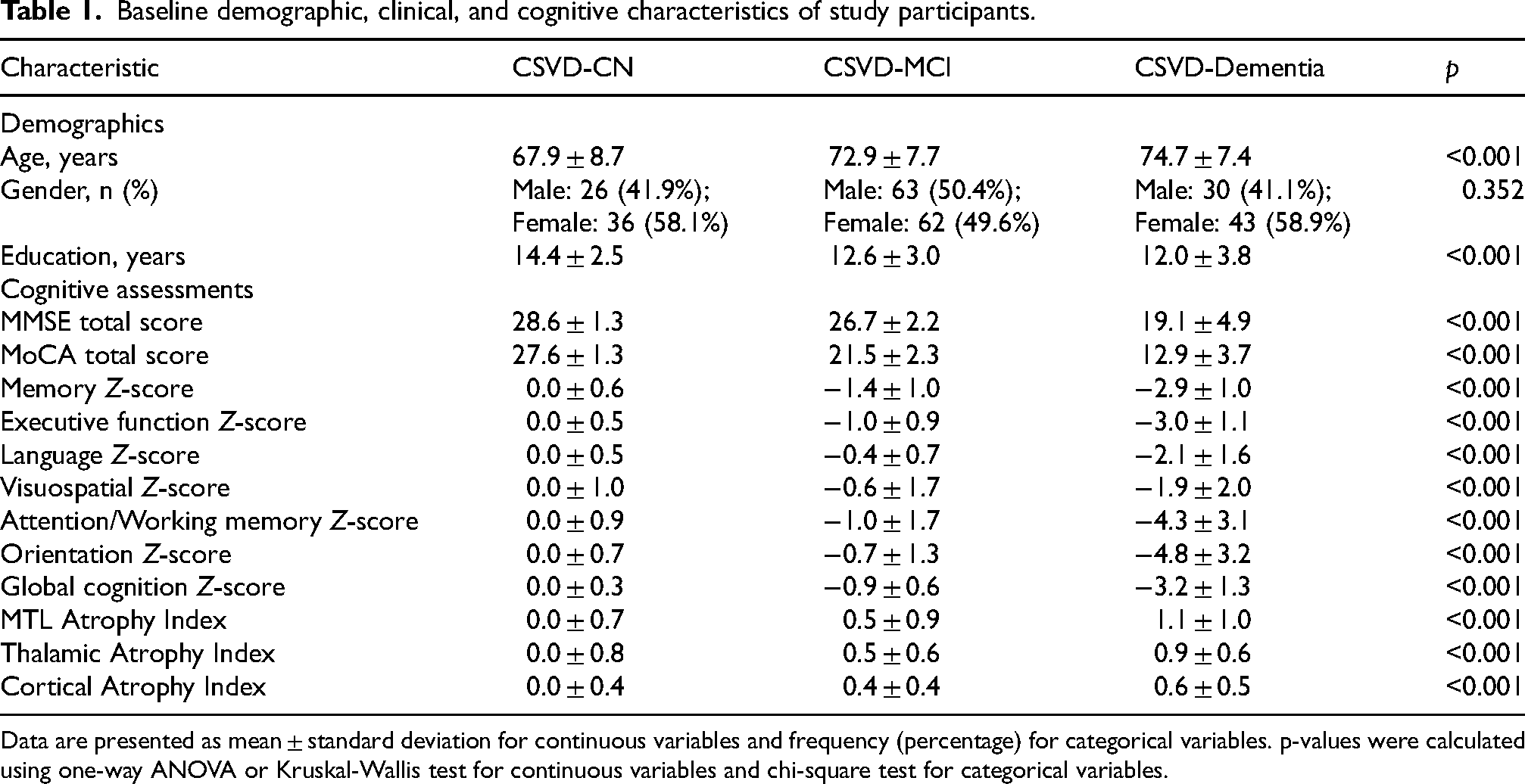
Data are presented as mean ± standard deviation for continuous variables and frequency (percentage) for categorical variables. p-values were calculated using one-way ANOVA or Kruskal-Wallis test for continuous variables and chi-square test for categorical variables.
Multiple linear regression analysis revealed the independent associations between different atrophy patterns and specific cognitive domains. After adjusting for age and years of education, both MTL atrophy and thalamic atrophy independently predicted lower global cognitive performance (β = −0.45, p < 0.001; β = −0.38, p = 0.006, respectively). For memory function, the atrophy of the MTL, thalamus, and cortical atrophy each showed independent predictive effects. Similarly, for executive function, both MTL and thalamic atrophy were likewise key independent correlates (Figure 3). Notably, the predictive effects of different atrophy patterns exhibited distinct domain specificity; for instance, thalamic atrophy was the sole significant predictor of poorer visuospatial performance (β = −0.48, p = 0.012). The complete regression analysis results for all cognitive domains are provided in Supplemental Table 5.
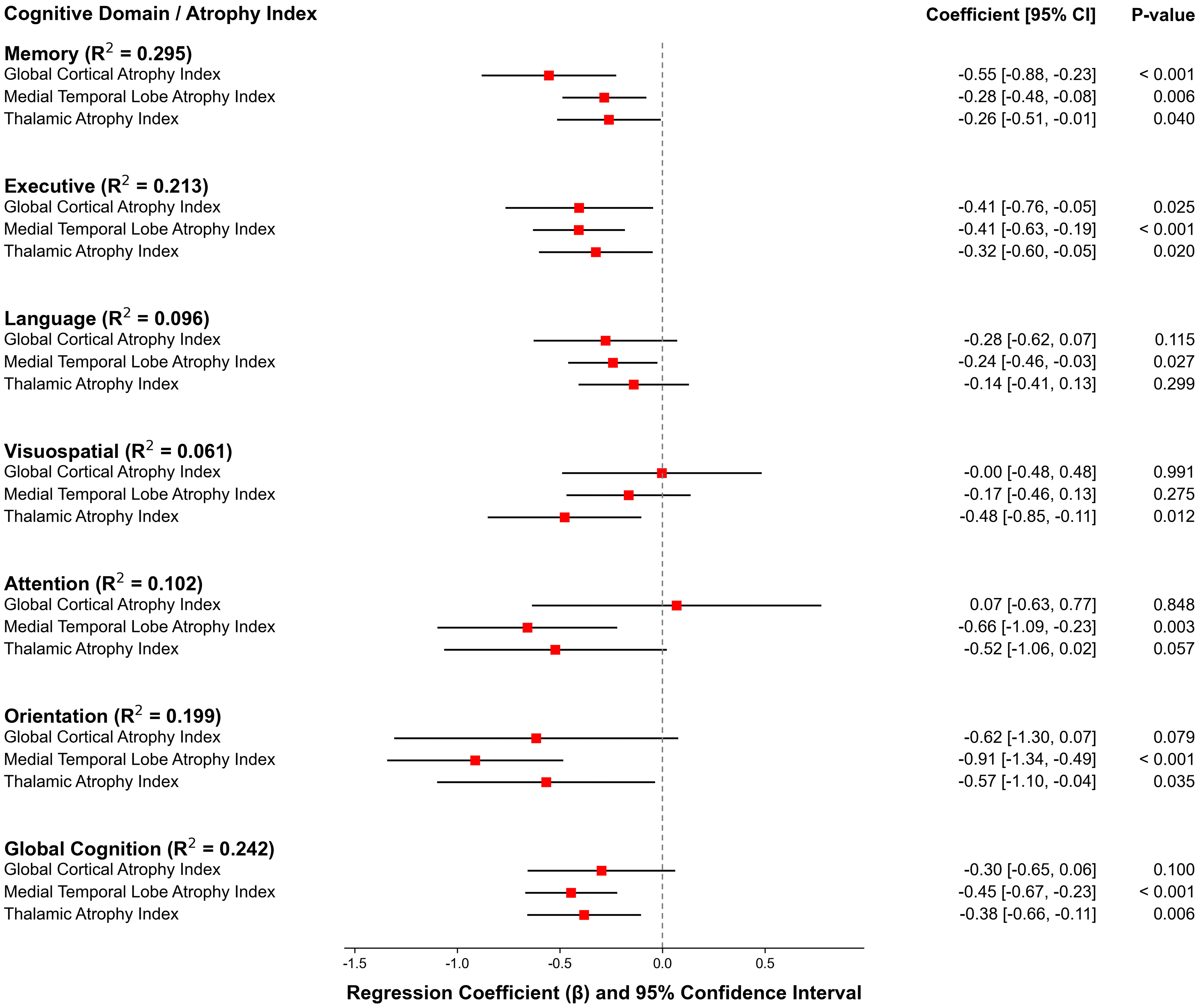
Associations between regional atrophy indices and cognitive domains. Forest plot displaying standardized regression coefficients (β) from multiple linear regression models. For each model, a cognitive domain Z-score was the dependent variable, regressed simultaneously on the three atrophy indices (Global Cortical, Medial Temporal Lobe, Thalamic). Squares represent the β coefficients and horizontal lines indicate the 95% confidence intervals. All models were adjusted for age and education. The adjusted R2 for each full model is reported on the left.
Pathophysiological pathways: From specific channels to an integrate
Initial mediation analyses of pre-specified pathways network model
To investigate the underlying mechanistic pathways, we first conducted a series of pre-specified mediation analyses (Figure 4; Supplemental Table 6). These models revealed that thalamic atrophy fully mediated the effect of deep WMH on executive function (indirect effect: β = −0.11, 95% CI [-0.19 to −0.04]; Figure 4(a)). The robustness of this thalamic pathway was supported by a similar full mediation when using third ventricle enlargement as an alternative upstream injury marker (indirect effect: β = −0.21, 95% CI [-0.34 to −0.11]; Figure 4(d)). We observed the same full mediation pattern for the effect of PVWMH on executive function via thalamic atrophy (indirect effect: β = −0.15, 95% CI [-0.26 to −0.07]; Figure 4(b)). In contrast, other specified pathways demonstrated different characteristics. The mediation effect of MTL atrophy on the relationship between deep WMH and executive function was not significant (Figure 4(c)). Furthermore, cortical atrophy only partially mediated the effect of frontal horn enlargement on global cognition, as a significant direct path remained (β = −0.16, p < 0.05; Figure 4(e)). These initial single-pathway models, while informative, suggested a complex interplay among atrophy patterns that could not be fully captured by separate analyses. The combination of full, partial, and non-significant mediations indicated that these regional atrophy patterns might not be independent events but rather co-varying components of a single underlying pathology. Therefore, to formally test this hypothesis and assess whether these pathways operate within a unified degenerative network, we subsequently employed SEM.
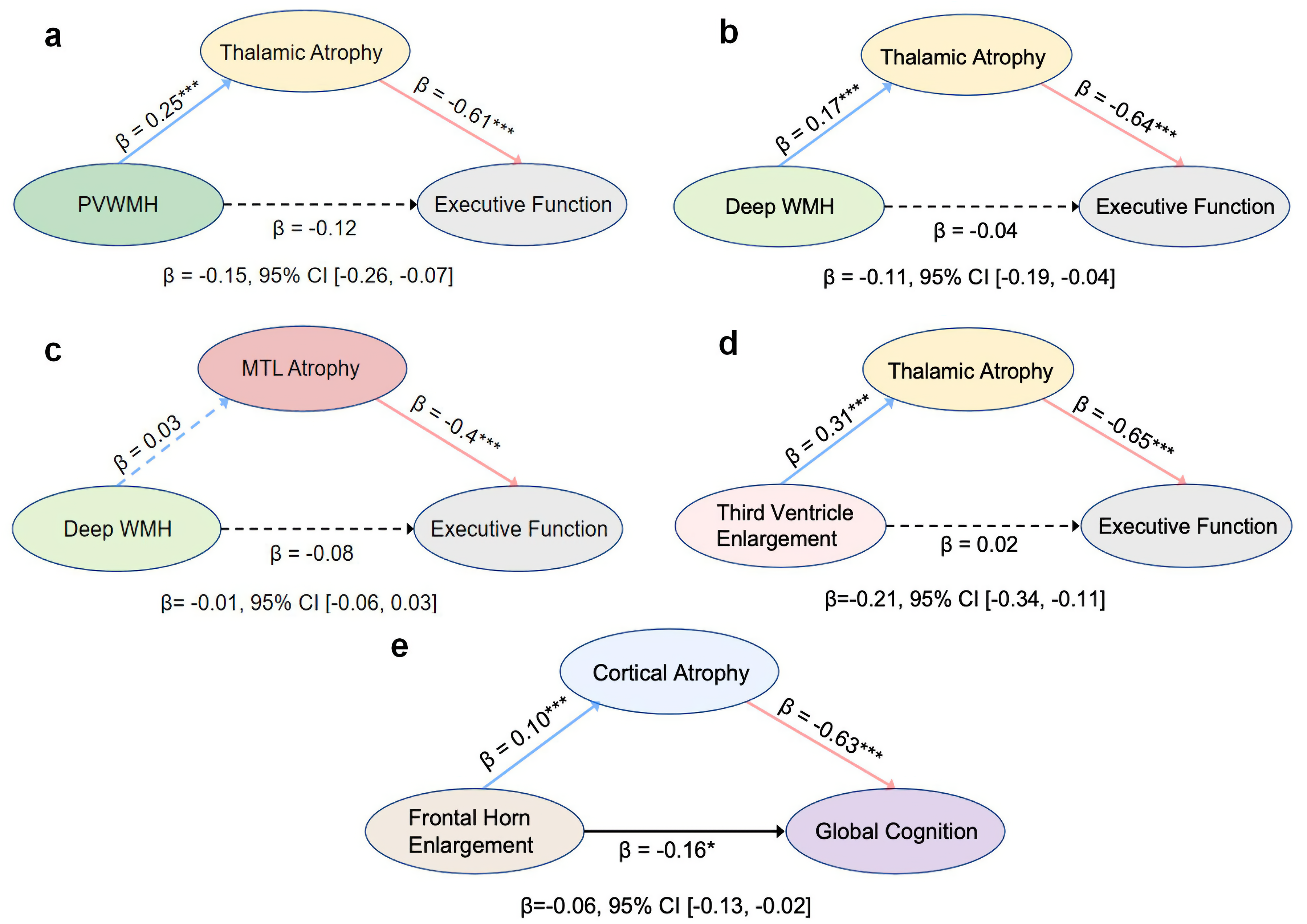
Path diagrams of pre-specified mediation models. Standardized regression coefficients (β) are shown for each path in five separate mediation analyses, all adjusted for age and education. Solid lines indicate significant paths (p < 0.05) and dashed lines indicate non-significant paths. The total indirect effect for each model, with its 95% bootstrap confidence interval (CI), is reported below the diagram. The analyses examined the indirect effect of (a) periventricular WMH on executive function via thalamic atrophy, (b) deep WMH on executive function via thalamic atrophy, (c) deep WMH on executive function via MTL atrophy, (d) third ventricle enlargement on executive function via thalamic atrophy, and (e) frontal horn enlargement on global cognition via cortical atrophy. (*p < 0.05; **p < 0.01; ***p < 0.001).
Confirmatory SEM: Identification of a unified atrophy network
To resolve the complexity observed in the initial analyses, we performed a SEM analysis to formally compare competing network configurations. This rigorous model comparison yielded a robust result (Supplemental Table 7). A model incorporating a single latent factor representing a CSVD-related atrophy network with a covariance between the residuals of executive function and memory (Model A) was the only model to achieve an excellent fit to the data (Δχ2 (18) = 19.97, p = 0.334; CFI = 0.996; TLI = 0.994; RMSEA = 0.021). This model was significantly superior to a baseline model without the residual covariance (Δχ2 (1) = 31.70, p < 0.001) and was more parsimonious than more complex models that included non-significant direct pathways from WMH to cognition (Δχ2 (2) = 0.10, p = 0.951).
The final, optimal model is depicted in Figure 5. It revealed that dWMH burden was a significant predictor of the CSVD-related atrophy network (β = 0.145, p < 0.05). This atrophy network, in turn, acted as a full and powerful mediator, strongly predicting declines in both executive function (β = −0.64, p < 0.001) and memory (β = −0.572, p < 0.001). The model also quantified a significant residual covariance between executive function and memory (r = 0.28, p < 0.001), confirming their coupled decline. The CSVD-related atrophy network was strongly and significantly indicated by thalamic (standardized factor loading = 0.742), MTL (standardized factor loading = 0.787), and cortical atrophy (standardized factor loading = 0.534). Detailed parameter estimates for the final model (Model A) are provided in Supplemental Table 8.
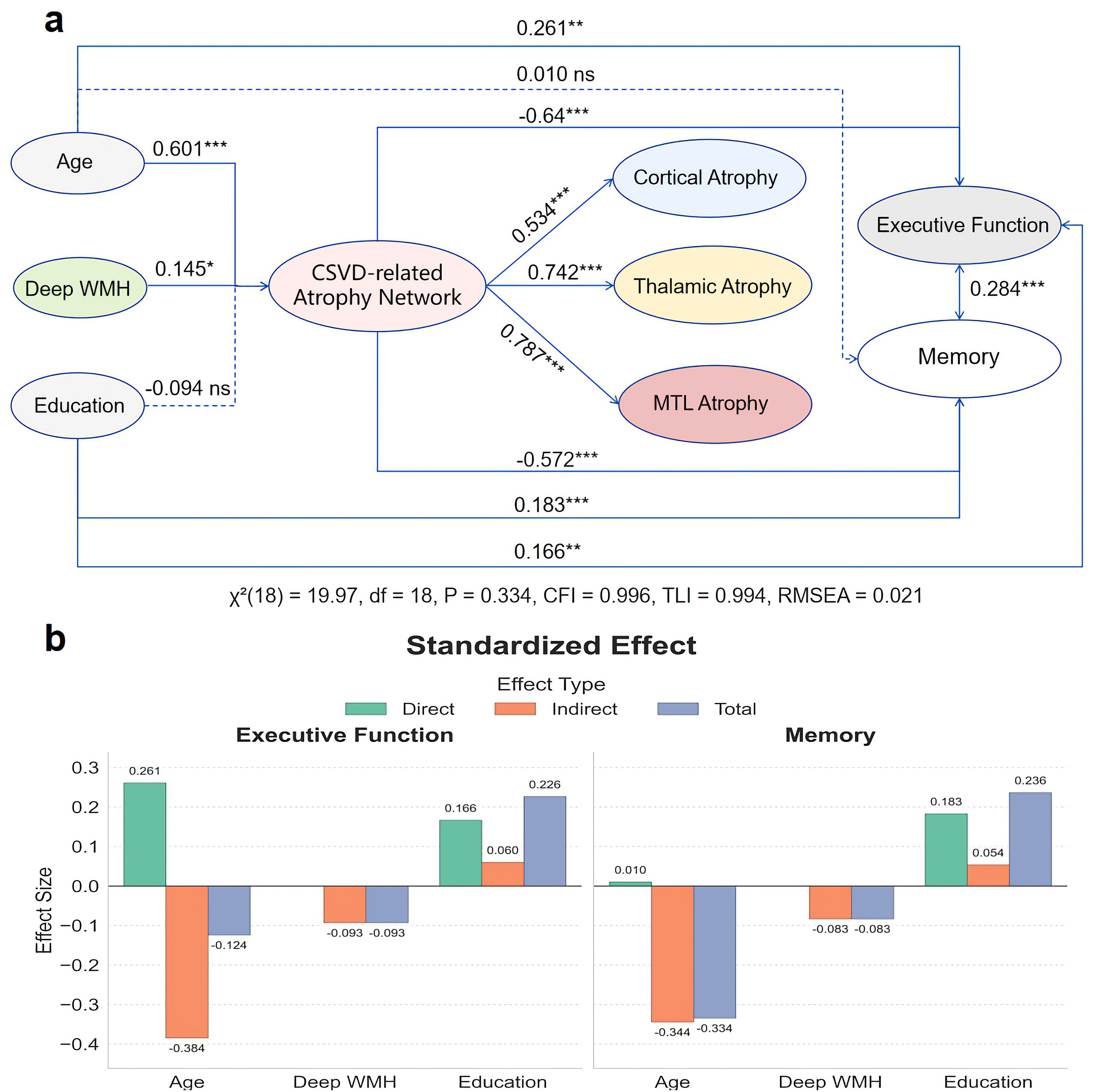
Final structural equation model (SEM) of a unified atrophy network. (a) Path diagram for the optimal SEM (Model A). A latent factor (“CSVD-related Atrophy Network”) links deep WMH to cognitive outcomes. Path values represent standardized coefficients (β). Solid lines denote significant paths (p < 0.05), dashed lines denote non-significant paths. Goodness-of-fit indices are reported below. (b) Bar charts decomposing the total standardized effects of Age, Deep WMH, and Education on Executive Function and Memory into direct and indirect components, as derived from the final model.
Discussion
This study employed a multi-level analytical strategy, culminating in a confirmatory network modeling approach, to provide a comprehensive mechanistic explanation for the well-documented discrepancy between WMH burden and cognitive performance in CSVD. Our central finding, derived from a rigorous comparison of competing structural models, is that a unified CSVD-related atrophy network—encompassing cortical, medial temporal, and thalamic regions-acts as the full mediator of the relationship between deep WMH and multi-domain cognitive impairment. While our initial exploratory analyses identified a specific thalamic-executive pathway, our final integrated model situates this observation within a more comprehensive, network-level framework that significantly advances our understanding of CSVD pathophysiology. Notably, while the total effect of PVWMH on cognitive status was at a borderline significance level (p = 0.060), its indirect effect on executive function via thalamic atrophy was robustly significant. This is consistent with modern mediation frameworks, which do not require a significant total effect to probe for a significant indirect pathway. This finding reinforces our central thesis: the cognitive impact of WMH is not determined by the total lesion burden itself, but is instead channeled through the specific disruption of critical downstream neural networks. It suggests that the resulting network atrophy serves as a more direct and sensitive measure of the underlying neurodegenerative process than the upstream vascular markers.
Our whole-brain, unbiased screening revealed that structural changes most relevant to the cognitive status gradient are highly concentrated in the limbic system and thalamus, providing strong in vivo evidence for the selective vulnerability hypothesis in CSVD. 34 This finding challenges a view of CSVD focused primarily on white matter burden and is consistent with the emerging conceptualization of CSVD as a network disorder. 35 Deep gray matter structures, such as the MTL and thalamus, may be particularly susceptible to CSVD-related chronic hypoperfusion, blood-brain barrier dysfunction, and inflammatory responses, owing to their high metabolic rate, rich vascular supply, and pivotal role as hubs in whole-brain networks.36–38 Indeed, previous research has demonstrated that the medial temporal region exhibits selective vulnerability to cerebral hypoperfusion, 39 while reduced thalamic perfusion has been strongly linked to clinical disability in white matter diseases such as multiple sclerosis. 40 Therefore, our findings underscore that CSVD is a disease that preferentially targets specific gray matter network nodes, with its clinical phenotype being determined by the structural and functional integrity of these critical hubs.
Further insight into the nature of this atrophy network is provided by its specific anatomical composition. It is notable that the involved cortical regions—such as the posterior cingulate gyrus, the superior temporal sulcus, and the insula—are not primary sensorimotor areas but are well-established hubs within large-scale cognitive networks, including Default Mode and Salience networks. The preferential vulnerability of these highly connected, metabolically active hubs is a recurring theme in neurodegenerative diseases. 41 In the context of CSVD, their atrophy is likely a dual consequence of chronic hypoperfusion in watershed vascular territories and disconnection from underlying white matter tracts damaged by the disease. 42 The convergence of atrophy on these specific cortical nodes, alongside the thalamus and MTL, suggests that our “CSVD-related atrophy network” factor is not merely a measure of diffuse brain shrinkage, but rather a specific index of the degradation of the brain's core cognitive architecture.
The thalamus, a core hub for information integration with widespread cortical projections, is considered a prime target for secondary neurodegeneration in CSVD. This vulnerability is highlighted by recent findings that thalamic volume loss is closely linked to disrupted microstructural integrity of connecting white matter tracts, even in the absence of direct focal lesions. 14 This mechanism has direct pathological evidence from multiple sclerosis, which demonstrates that the extent of thalamic atrophy correlates significantly with the total burden of distal cerebral white matter lesions, rather than solely with focal thalamic pathology. 43 Similarly, neuroimaging studies in CSVD have established that microstructural damage to specific thalamo-prefrontal white matter tracts is strongly associated with executive dysfunction.42,44 The core contribution of this study lies in its in vivo validation of a pathological cascade that links upstream vascular injury to downstream executive dysfunction, confirmed through mediation analysis. Our model provides strong support for the hypothesis that the detrimental effects of both deep WMH and periventricular WMH on executive function are largely mediated by thalamic atrophy. This finding provides crucial human in vivo evidence for secondary neurodegeneration hypothesis, 45 which posits that remote white matter lesions can lead to atrophy and dysfunction in anatomically connected gray matter nodes via Wallerian degeneration. 46 Our data strongly support the notion that the cognitive impact of WMH extends far beyond the mere disconnection of white matter tracts; more critically, it involves anterograde or retrograde neuronal degeneration in connected gray matter nuclei. 45 Specifically, deep white matter regions are rich in critical projection fibers connecting the thalamus and the prefrontal cortex. 47 Damage to these fibers likely disrupts the integrity of the thalamo-frontal circuit, ultimately causing thalamic neuronal loss and volume atrophy, which may represent the most direct pathological mechanism for executive dysfunction in CSVD patients. 14 Scaling up from localized circuits to whole-brain topology, this perspective strongly aligns with recent graph theoretical and structural covariance network studies, which demonstrated that WMH drives propagating gray matter atrophy across large-scale networks, leading to topological disruptions that directly impair executive functions.11,12 By situating thalamic atrophy within a broader network-level failure, our findings reinforce the concept that localized white matter damage disrupts the global structural covariance of the brain.
Our findings are highly complementary to the recent work by Li et al. 14 which focused on the downstream mechanisms of thalamic atrophy. Their study, using advanced diffusion imaging, elegantly showed that thalamic atrophy exerts its cognitive effects by disrupting the microstructural integrity of its efferent white matter connections. Our study addresses the antecedent, upstream question of what drives this initial thalamic atrophy. Synthesizing these findings provides, for the first time, in vivo evidence for a multi-stage pathological sequence in CSVD. This sequence is initiated by upstream vascular injury (e.g., deep WMH) which induces secondary thalamic atrophy—our primary finding. This atrophy, in turn, leads to the downstream disruption of thalamocortical white matter integrity, the mechanism identified by Li et al. as the critical link to clinical executive dysfunction. This integrated model reinforces the concept of CSVD as a network disease, where focal vascular insults trigger a sequence of remote structural and connective failures that culminate in cognitive impairment.
Our initial mediation analyses suggested a degree of pathway specificity, particularly given the non-significant MTL-memory pathway. However, our final, optimal SEM model provides a more nuanced explanation. The model reveals that atrophy of the thalamus, MTL, and cortex are not independent events but rather a co-varying indicator of a single, underlying process of network-level degeneration. The failure of the isolated MTL-memory pathway likely reflects the reality that the cognitive impact of CSVD is not neatly segregated into independent channels. Instead, the degradation of this entire cortico-subcortical network collectively impairs multiple cognitive domains, a finding consistent with the contemporary view of CSVD as a systems-level disorder that disrupts large-scale brain networks. 48 Crucially, the robust association we observed between this unified atrophy network and memory decline underscores the prominent role of its key nodes; particularly, the involvement of the MTL is consistent with longitudinal evidence showing that increased WMH burden promotes an Alzheimer-like pattern of neurodegeneration.9,10 Furthermore, similar to findings highlighting that WMH mediates age-related brain atrophy, 8 our model confirms that this unified atrophy network is a pivotal intermediary in age- and vascular-related cognitive trajectories.
From a clinical perspective, these findings have significant translational implications. The demonstration that a unified atrophy network fully mediates the effect of WMH firmly shifts the clinical focus from the upstream radiological marker (WMH) to the downstream neurodegenerative consequence (atrophy). This suggests that monitoring the integrity of this cortico-subcortical network may offer a more direct biomarker of cognitive risk than quantifying WMH burden alone. The presence of significant WMH on an MRI report should therefore be interpreted as a powerful warning sign for probable, widespread atrophy, prompting a more thorough assessment of multi-domain cognitive functions. This proactive approach is essential because, as emphasized in recent clinical outcome studies, CSVD markers act as active and independent determinants of poor prognosis. 13 Thus, early detection and management of WMH could be critical for preventing secondary network degradation and subsequent functional decline. Furthermore, the strong coupling identified between executive and memory decline provides a model-based explanation for the common clinical presentation of multi-domain impairment. This underscores that effective therapeutic strategies may need to be multi-faceted, not only targeting upstream vascular risk factors but also exploring neuroprotective interventions aimed at preserving the structural integrity of this entire vulnerable brain network. 49
The deliberate use of clinical semi-quantitative visual scales (e.g., Fazekas, GCA) instead of automated algorithms represents a core strength of this study, enhancing its translational value. While previous seminal studies have elegantly established that total WMH burden drives localized cortical thinning and mathematical network disruptions, our study builds upon this foundation by demonstrating that deep WMH specifically drives a unified, clinically measurable cortico-subcortical atrophy network—captured holistically via SEM. By design, our study bridges research with practice by linking these complex, secondary neurodegenerative processes to tangible imaging features seen on routine MRI. Crucially, this provides an immediately actionable framework: a specific visual rating (e.g., deep WMH grade 2–3) should be interpreted not as an isolated finding but as a powerful indicator of risk for underlying network atrophy, prompting targeted screening of cognitive functions. This grounding in real-world tools offers a cognitive model for interpreting CSVD neuroimaging that can be readily adopted by clinicians, facilitating the translation of our findings into patient care.
Our study has several strengths. A primary one is the application of a multi-level analytical framework that progressed from examining individual pathways to testing an integrated network with a confirmatory SEM. In contrast to analyzing single mediation pathways, the SEM approach allowed us to model a latent “atrophy network” from multiple regional indices and formally test its role within competing theoretical structures. This provides a more robust and holistic assessment of the proposed pathological cascade. Another strength is our use of clinically-based visual rating scales for WMH, applied to MRI data acquired during routine clinical workflow, which enhances the translational relevance and generalizability of our findings to routine clinical practice. The primary limitation is the cross-sectional design, which precludes definitive causal inference from our data. The mediation model was therefore not intended to establish causality de novo, but rather to assess the consistency of our in vivo data with the secondary neurodegeneration hypothesis. This hypothesis is built upon a biologically plausible temporal sequence where upstream vascular injury is understood to precede downstream neuronal atrophy in connected regions, a process mechanistically described by Wallerian degeneration.46,50 This directionality is supported by prior evidence, including post-mortem and computational studies demonstrating that thalamic atrophy is associated with remote, rather than local, white matter injury.6,43 Furthermore, the potential for residual confounding from unmeasured variables, such as specific vascular risk factors, cannot be excluded. Consequently, our findings should be interpreted as providing strong statistical support for this pathological cascade, suggesting network atrophy is a critical mediator. Confirmation of this temporal sequence necessitates future longitudinal research, and a more comprehensive pathophysiological picture could be achieved by integrating multimodal imaging techniques. 51
Supplemental Material
sj-docx-1-alz-10.1177_13872877261457126 - Supplemental material for Deep white matter injury and cognitive decline in cerebral small vessel disease: Mediation by a unified atrophy network
Supplemental material, sj-docx-1-alz-10.1177_13872877261457126 for Deep white matter injury and cognitive decline in cerebral small vessel disease: Mediation by a unified atrophy network by Chengzhe Wang, Chen Zhang, Hailong Li, Litao Wang, Xuhui Chen, Cong Wang, Yan Zhang, Zhifei Zhang, Mina A, Jie Wang, Jinfeng Wu, Yongan Sun and Ailian Du in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
We sincerely thank the patients and their families for their time and dedication to this study. Their participation was crucial to advancing the scientific understanding of cerebral small vessel disease. The computations in this paper were run on the Siyuan-1 cluster supported by the Center for High Performance Computing at Shanghai Jiao Tong University.
Ethical considerations
The study protocol was approved by the Human Research Ethics Committee of Peking University First Hospital (2022Y317-002) and Tongren Hospital, Shanghai Jiaotong University School of Medicine (2022-098-01). All procedures were conducted in accordance with the principles of the Declaration of Helsinki.
Consent to participate
Patient consent was waived due to the retrospective nature of the study and the use of de-identified data.
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the Shanghai Jiao Tong University Medical Engineering Cross Research Funds (grant number YG2024ZD27), the National Natural Science Foundation of China (grant numbers 82571751, 81971181, 12231018, 82401397).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request. They are not publicly accessible due to ethical and privacy protocols.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.