
Abstract
Background
Traumatic brain injury (TBI) is associated with increased dementia risk, yet its relationship to Alzheimer's disease (AD) neuropathology is unclear. Prior autopsy studies show inconsistent results, constrained by limited TBI ascertainment, semi-quantitative neuropathology methods, and selection bias.
Objective
To examine whether TBI with loss of consciousness (TBI-LOC) is associated with quantitative measures of tau and amyloid-β42 (Aβ42) pathology in a community-based autopsy cohort.
Methods
The analytic sample included 810 Adult Changes in Thought study brain donors with baseline TBI-LOC ascertainment and quantitative neuropathology data, weighted to represent the full living cohort (n = 5763). Modified Poisson regression estimated rate ratios (RRs) and 95% confidence intervals (CIs) for associations of TBI-LOC with percent-positive AT8 tau immunoreactivity and soluble Aβ42 (GuHCl and RIPA fractions) across brain regions. Models were adjusted for age at death, sex, education, and enrollment cohort; sensitivity analyses included unweighted and quantile models.
Results
Weighted models showed lower frontal tau (RR = 0.50, p = 0.047) and lower soluble Aβ42 in temporal and occipital cortices (p = 0.019–0.037) among donors with baseline TBI-LOC. In unweighted sensitivity analyses, lower temporal RIPA Aβ42 remained significant (p < 0.001), and select inverse associations were observed in duration-stratified analyses, particularly in small >1 h LOC subgroups. Quantile analyses of pathology burden similarly suggest reduced parietal Aβ42 (p = 0.02–0.04); effects were small and driven by sparse subgroups.
Conclusions
TBI-LOC was not associated with greater tau or amyloid burden, and inverse associations likely reflect data sparsity rather than biological protection. These findings suggest that links between TBI and late-life cognitive decline may involve non-AD pathological processes.
Introduction
As the population ages, understanding how traumatic brain injury (TBI) influences risk for late-life dementia has become an urgent priority, yet the neuropathological pathways underlying this association remain uncertain.1,2 Substantial evidence suggests that individuals with a history of TBI are at elevated risk for incident dementia across the lifespan.1,3–7 Though many studies suggest that TBI may accelerate AD or other Alzheimer's disease and related dementias (ADRDs),3,5,8 others report null associations.9,10 It remains unclear the extent to which documented excess risk is mediated by AD-related processes such as tauopathy and amyloidosis.
Autopsy studies investigating TBI and AD/ADRDs have produced heterogeneous results. Some report that TBI increases risk for earlier onset of AD/ADRD pathology11–14 while others, including analyses of population-based cohorts such as the Adult Changes in Thought (ACT) study and Religious Orders Study–Memory and Aging Project (ROS-MAP), have not found greater AD neuropathology among individuals with TBI with loss of consciousness (TBI-LOC).10,15–17 This inconsistency may result from differences in study design, exposure ascertainment, sample selection, and analytic strategy, and underscores the need for population-based autopsy research that incorporates refined brain trauma exposure definitions and quantitative neuropathological measures.
Several methodological gaps have constrained prior work. TBI ascertainment has often relied on single self-report questions or diagnostic codes,3–7 which may underestimate lifetime exposure and lack precision in characterizing severity or duration.10,15 In addition, self-selection into brain donation can introduce systematic bias, 18 and few studies have incorporated selection weights or other methods to account for bias associated with autopsy participation. These limitations make it difficult to determine whether null findings in autopsy studies examining associations of head trauma exposure with AD neuropathology reflect a true absence of association or methodological artifacts. We addressed these limitations in one prior study and still we found no association of TBI with hallmark pathologies of AD, 19 but this study shared another limitation with prior work: it relied exclusively on semi-quantitative staging systems (e.g., Braak, CERAD, Thal) that may be insensitive to detecting subtle differences between older adults with and without a history of TBI.
In the present study, we examined 810 ACT brain donors to test whether TBI with loss of consciousness is associated with quantitative measures of tau and amyloid pathology across multiple regions. TBI exposure was characterized by using several complementary sources, and analyses incorporated weighting methods to account for autopsy selection and reflect the broader ACT cohort. We further examined whether quantitative tau and amyloid-β (Aβ) measures differed by LOC duration to explore whether more prolonged injuries were associated with differing levels of pathology.
Methods
Study sample
The ACT study began in 1994 as a community-based longitudinal cohort study of incident dementia. Adults aged ≥65 years were randomly sampled from a health maintenance organization (Group Health, now Kaiser Permanente Washington) and completed biennial study visits to monitor health and cognition. All enrollees were deemed to be dementia-free at the time of enrollment based on Cognitive Abilities Screening Instrument (CASI) 20 scores ≥85 or clinical consensus, and consent for autopsy was optional and confirmed by next-of-kin at the time of death. As of the November 2022 analytic data freeze, 5763 participants had been enrolled in the study, 971 had come to autopsy, 963 had baseline TBI data, 810 of whom also had available quantitative neuropathology data and were included in the analytic sample (Table 1). Of the 963 autopsied brain donors, 153 were excluded due to technical limitations in tissue availability, staining quality and incomplete assay completion at the time of our analyses.
Description of the study sample, TBI exposure, and standard neuropathology outcomes by baseline TBI with LOC exposure group.
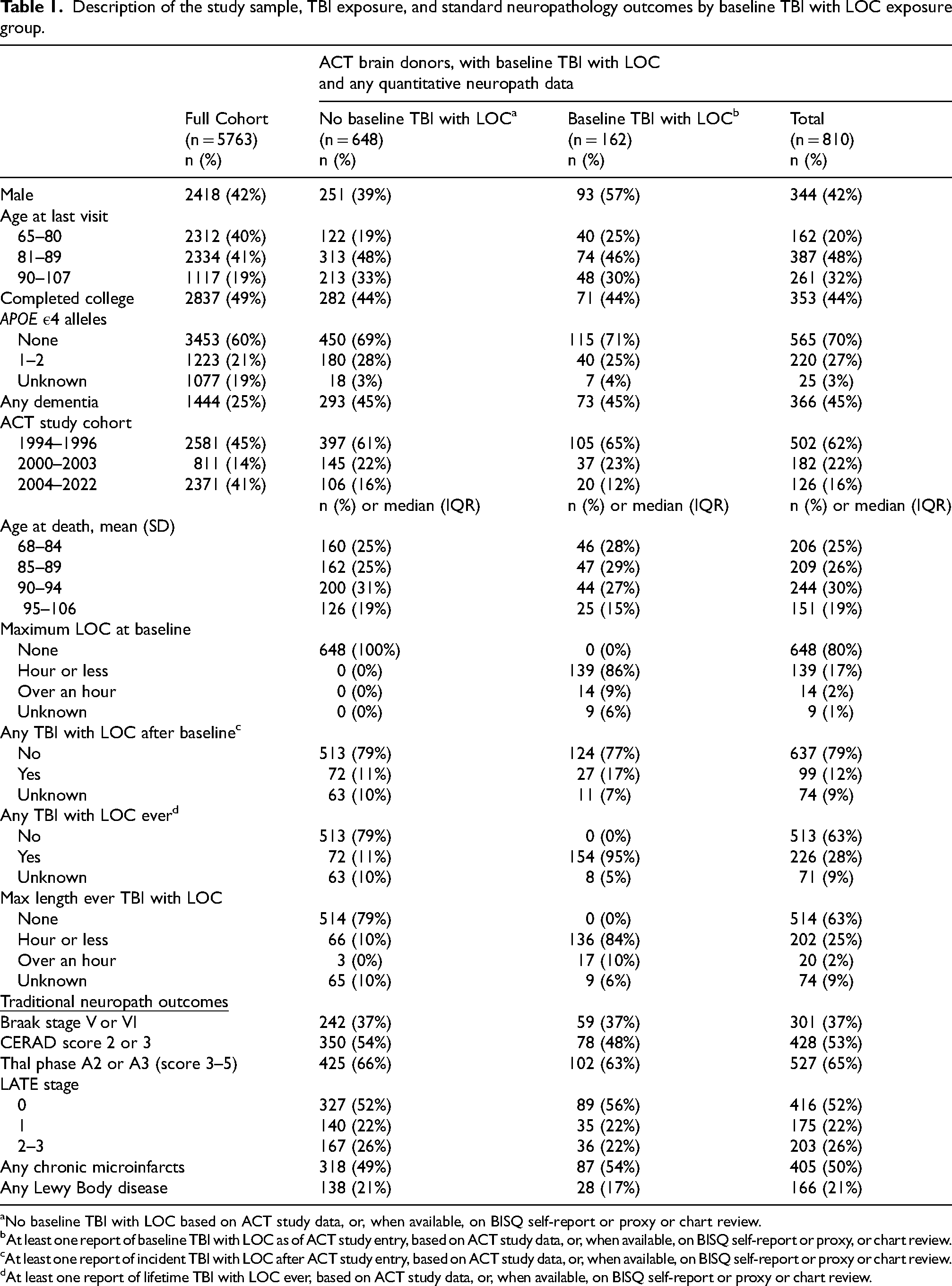
No baseline TBI with LOC based on ACT study data, or, when available, on BISQ self-report or proxy or chart review.
At least one report of baseline TBI with LOC as of ACT study entry, based on ACT study data, or, when available, on BISQ self-report or proxy, or chart review.
At least one report of incident TBI with LOC after ACT study entry, based on ACT study data, or, when available, on BISQ self-report or proxy or chart review.
At least one report of lifetime TBI with LOC ever, based on ACT study data, or, when available, on BISQ self-report or proxy or chart review.
TBI ascertainment
TBI with loss of consciousness (TBI-LOC) was identified using multiple complementary sources, reflecting the evolution of exposure assessment in ACT over time (Supplemental Table 1). At enrollment from 1994 through 2016, participants were asked through a single “legacy” item whether they had ever experienced “an injury so severe that you lost consciousness.” If endorsed, follow-up questions queried type of injury and approximate duration of LOC (≤1 h, >1 h, or unknown). Beginning in 2014, a structured lifetime head trauma interview (the Brain Injury Screening Questionnaire, core BISQ) 21 was administered to a subset of participants and informants. The core BISQ used contextual cues (e.g., falls, motor vehicle crashes, sports) to query possible head trauma events, and for all reported events recorded the presence and duration of LOC and/or altered mental status. For deceased or dementia-diagnosed participants, BISQ data were obtained from close informants. Beginning in 2020, a more condensed 7-item version of the BISQ was given to all ACT participants (BISQ-7). Finally, TBI exposure characterization was supplemented by structured abstraction of Kaiser Permanente Washington medical records, including historical paper charts and later electronic records, as a part of the ACT chart abstraction protocol described previously. 22
All available data on lifetime head trauma were harmonized to create exposure variables. “Baseline TBI-LOC” referred to TBI sustained prior to study entry. “Incident TBI-LOC” includes TBI with LOC occurring after enrollment, and “Lifetime TBI-LOC” combined all TBI with LOC reported at any time (pre/post enrollment). LOC duration was categorized as none, ≤1 h, or >1 h. Because head trauma ascertainment methods shifted over time, including retirement of the legacy item in 2016, cohort and incident TBI-LOC status were included as covariates in regression models to reduce potential bias. Detailed mild TBI without LOC and repetitive head impact exposures were not uniformly available across all decedents and were therefore not used as primary exposure definitions in the present analysis.
Dementia and Alzheimer's disease ascertainment
Methods for identifying dementia cases have been described previously.23–25 Briefly, participants were screened every two years using the CASI, 20 a 100-point measure of global cognition. Individuals scoring <85 underwent a standardized clinical and neuropsychological assessment, and dementia diagnoses were established at multidisciplinary consensus conferences according to DSM-IV criteria 26 ; probable or possible Alzheimer's disease was defined using NINCDS–ADRDA criteria. 27
Neuropathology
Autopsied brains were processed and evaluated at the University of Washington using standardized ACT protocols. 28 For participants with postmortem intervals less than 8 h, one hemisphere was dissected fresh and frozen; all cases underwent formalin fixation for histopathological assessment. Formalin-fixed tissue from standardized cortical and subcortical regions were embedded in paraffin and sectioned for neuropathologic evaluation. Neuritic plaque density was rated according to the Consortium to Establish a Registry for AD (CERAD) criteria (none, sparse, moderate, frequent), Aβ deposition was rated using Thal phase, and neurofibrillary tangle distribution was staged using the Braak and Braak system (I-II, III-IV, V-VI). A combined measure of AD-NC (ABC score) was derived in accordance with National Institute on Aging-Alzheimer's Association (NIA-AA) guidelines,29,30 with simulated ABC scores applied to older cases collected prior to 2012 to ensure consistency across participants. Limbic-predominant age-related TDP-43 encephalopathy (LATE) was staged per 2019 consensus recommendations. 31
Chronic microinfarcts were identified across 12 standardized cortical and subcortical regions (bilateral middle frontal gyrus, superior and middle temporal gyri, inferior parietal lobule, occipital/calcarine cortex, basal ganglia at the level of the anterior commissure, and thalamus at the level of the subthalamic nucleus) using hematoxylin and eosin staining.
Lewy body disease was assessed using α-synuclein immunohistochemistry in the amygdala, anterior cingulate cortex, and middle frontal cortex, with additional hematoxylin and eosin/Luxol fast blue evaluation of brainstem sections, in accordance with NIA-AA guidelines.
The ACT study recently updated classification systems for both microinfarcts and Lewy body disease and all cases in the present analysis were re-evaluated under the revised criteria. Therefore, these outcomes are not directly comparable to those reported previously.10,19
Quantification
Quantitative assessment of tau pathology was carried out in samples of frontal (middle frontal gyrus), temporal (superior and middle temporal gyrus), parietal (inferior parietal lobule), and occipital (calcarine fissure) cortices; hippocampal subfields including CA1, entorhinal cortex, subiculum, and transentorhinal cortex; and the amygdala. Formalin-fixed paraffin-embedded (FFPE) tissue blocks were sectioned at 5 µm, placed on charged glass slides, and immunolabelled with an antibody to phosphorylated tau (ptau, clone AT8 [Thermo Fisher Scientific Cat# MN1020, RRID:AB_223647]) at 1:1000 dilution, according to established protocols. The slides were then scanned on Aperio AT2 digital scanner (Leica, Vista, CA) at 20X magnification, and the images were uploaded for analysis into HALO software (Indica Labs, Albuquerque, NM, USA, RRID:SCR_018350). For all regions, relevant fields of view were manually traced, and percentage of the tissue area positive for ptau immunostaining was determined using the HALO Area Quantification module (HALO® v.3.4.2986). This approach quantified total AT8-immunoreactive area within manually annotated regions of interest and was intended as a measure of overall regional phospho-tau burden rather than lesion-specific pathology. Accordingly, AT8-positive signal meeting the predefined analysis threshold contributed to the % area metric, irrespective of lesion subtype (e.g., tangles, neurites, glial tau pathology, etc.). A threshold-based segmentation approach was used, with signal thresholds defined based on staining intensity and applied consistently across all cases within each region to ensure comparability. An object classifier was not used, and lesion-specific tau morphologies were not separately quantified at this time. This metric was selected to provide a robust, reproducible, and regionally comparable estimate of total AT8-positive burden, rather than to resolve lesion-specific effects.
Aβ42 concentrations were measured in GuHCl- and RIPA-extracts obtained from 3 adjacent 15 µm-thick FFPE sections of frontal, temporal, parietal, and occipital cortices by Luminex-based immunoassay (Luminex Corporation, Austin, TX, USA). 32 All Aβ42 measurements were generated specifically for the present study using tissue from the same cases and cortical regions included in the quantitative tau analyses. Tissue was processed using established FFPE protein extraction methods with sequential extraction of soluble and insoluble fractions prior to assay. Assays were performed according to the manufacturer's protocol, using plate-specific standard curves generated in duplicate for each 96-well plate with kit-provided recombinant Aβ standards. Quality control procedures included evaluation of standard curve performance, inclusion of replicate samples and internal controls, and monitoring of intra-assay and inter-assay variability (target coefficients of variation <10% and <15%, respectively). Aß42 was quantified by immunoassay to provide analyte-specific biochemical measurements of regional amyloid burden. Although amyloid immunohistochemistry was performed as part of standard neuropathologic evaluation, these semiquantitative assessments were used for neuropathologic staging rather than quantitative regional analysis and were therefore not directly comparable to the regional image-based tau measures used in the present analyses.
Statistical analysis
Modified Poisson regression with robust standard errors 33 was used to estimate rate ratios (RRs) and 95% confidence intervals (CIs) for the association between TBI-LOC and neuropathological outcomes. Separate models were constructed for AT8 tau burden and Aβ42 (GuHCl and RIPA fractions) by brain region and for the standard neuropathological outcomes. All models adjusted for age at death, sex, years of education, and ACT study cohort. For analyses of baseline TBI-LOC, we additionally adjusted for incident TBI-LOC (TBI with LOC occurring after ACT enrollment).
We evaluated four primary TBI exposure definitions: baseline TBI-LOC (yes/no), lifetime TBI-LOC (yes/no), baseline TBI-LOC duration (none, ≤1 h, >1 h), and lifetime TBI-LOC duration (none, ≤1 h, >1 h). Because detailed acute injury severity metrics (e.g., Glasgow Coma Scale scores) were not available for many remote injuries, LOC duration was used as a coarse proxy for injury severity with the >1 h category reflecting a more severely injured group rather than a definitive moderate-to-severe TBI classification. To account for potential bias in autopsy selection, models incorporated inverse probability weights estimated from the full ACT cohort (n = 5763), with bootstrapping to reflect error in weight estimation. Results for the >1 h LOC group were interpreted with caution given sparse data. Sensitivity analyses included unweighted models and the categorization of pathology into 20 quantiles to assess whether findings were driven by skewed distributions or outliers.
In exploratory secondary analyses, we examined associations of incident TBI-LOC with quantitative tau and Aβ42 outcomes. To approximate non-proximal late-life injuries, events reported in the same year as death or the year before death were excluded because only year-level timing was available.
Results
Study sample
The analytic sample included 810 ACT autopsy donors with quantitative neuropathology data and baseline TBI-LOC ascertainment, weighted to represent the full ACT cohort (n = 5763). The mean (SD) age at death was 88.7 (6.6) years, and 42% of donors were male. Approximately 27% carried at least one APOE ε4 allele and 44% had completed college (Table 1). Compared with donors without TBI-LOC, those with baseline TBI-LOC were more often male and somewhat younger and were more likely to have received a dementia diagnosis during life. Educational attainment and APOE ε4 distributions were similar across groups.
Traditional neuropathologic markers of AD, including Braak stage, CERAD neuritic plaque score, and Thal amyloid phase, did not differ by TBI-LOC exposure status. Distributions of LATE stage, chronic microinfarcts, and Lewy body disease were likewise similar across TBI-LOC groups (Table 1).
Quantitative neuropathology outcomes by TBI-LOC
Neuropathological outcomes by baseline TBI-LOC status are presented in Table 1. Quantitative burden of tau (AT8 percent-positive area) across cortical, hippocampal, and amygdala regions are presented in Supplemental Table 2.
In weighted regression models comparing donors with versus without baseline TBI-LOC (Table 2), baseline TBI-LOC was associated with significantly lower frontal tau burden (RR 0.50, 95% CI 0.26–0.90, p = 0.047). Baseline TBI-LOC was also associated with lower soluble Aβ42 concentrations in the temporal (RR 0.65, 95% CI 0.44–0.96, p = 0.037) and occipital (RR 0.71, 95% CI 0.50–0.93, p = 0.028) lobes for the GuHCl fraction, and in the temporal lobe for the RIPA fraction (RR 0.73, 95% CI 0.55–0.93, p = 0.019). No other regional tau or amyloid measures differed significantly. None of these associations were statistically significant in unweighted analyses (Supplemental Table 3) or in quantile analyses (Supplemental Table 4).
Risk of TBI exposure for increased levels of quantitative tau and Aβ42.
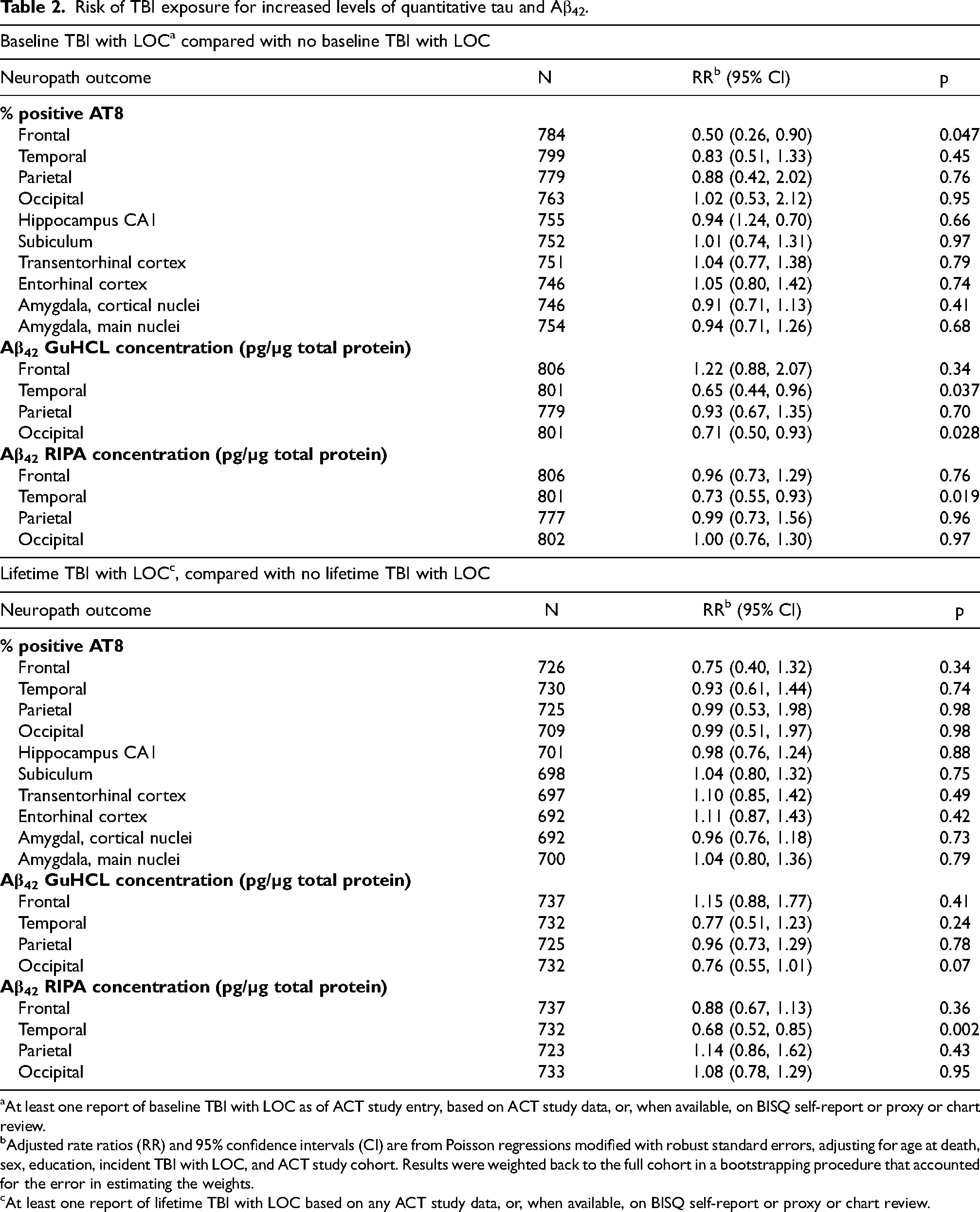
At least one report of baseline TBI with LOC as of ACT study entry, based on ACT study data, or, when available, on BISQ self-report or proxy or chart review.
Adjusted rate ratios (RR) and 95% confidence intervals (CI) are from Poisson regressions modified with robust standard errors, adjusting for age at death, sex, education, incident TBI with LOC, and ACT study cohort. Results were weighted back to the full cohort in a bootstrapping procedure that accounted for the error in estimating the weights.
At least one report of lifetime TBI with LOC based on any ACT study data, or, when available, on BISQ self-report or proxy or chart review.
When considering lifetime TBI-LOC (pre- or post-enrollment; Table 2), no significant associations with tau burden were observed. Soluble Aβ42 levels again showed an inverse association: temporal RIPA Aβ42 was significantly lower in donors with lifetime TBI-LOC (RR 0.68, 95% CI 0.52–0.85, p = 0.002). These findings were consistent across weighted and unweighted analyses (Supplemental Table 3). In quantile analysis, temporal Aβ42 was not statistically significant, though effects on parietal Aβ42 were (Supplemental Table 4).
Quantitative neuropathology outcomes by TBI-LOC duration
Duration-stratified analyses were limited by small sample sizes, especially for the >1 h LOC at baseline subgroup (n = 14; Table 3). Donors with baseline LOC ≤1 h had lower frontal tau (RR 0.48, 95% CI 0.24–0.98, p = 0.046) and lower temporal Aβ42 (GuHCl: RR 0.59, 95% CI 0.39–0.90, p = 0.012; RIPA: RR 0.70, 95% CI 0.51–0.90, p = 0.012). Compared to the no-TBI with LOC group, the >1 h subgroup showed markedly low risk for frontal tau (RR 0.12, 95% CI 0.03–0.57, p = 0.003) and with reduced frontal RIPA Aβ42 (RR 0.44, 95% CI 0.27–0.82, p = 0.004). This subgroup, however, included virtually no individuals with high tau burden; >90% had <0.5% frontal AT8, and only one case exceeded that threshold (maximum 1.02%), compared with a maximum of 35.95% in the no-TBI group. Given the small and highly skewed >1 h group, these estimates should be interpreted with caution. Notably, despite their low measured tau burden, individuals in the baseline LOC > 1 h subgroup had a higher prevalence of dementia (64% versus 45% in other groups), including both AD and multiple-etiology dementias. Unweighted analyses were similar to the primary results (Supplemental Table 5), but the only significant finding based on TBI severity quantiles was reduced temporal tau for TBI with LOC >1 h (p = 0.04).
Risk of select neuropathological endpoints by TBI exposure severity (duration of LOC).
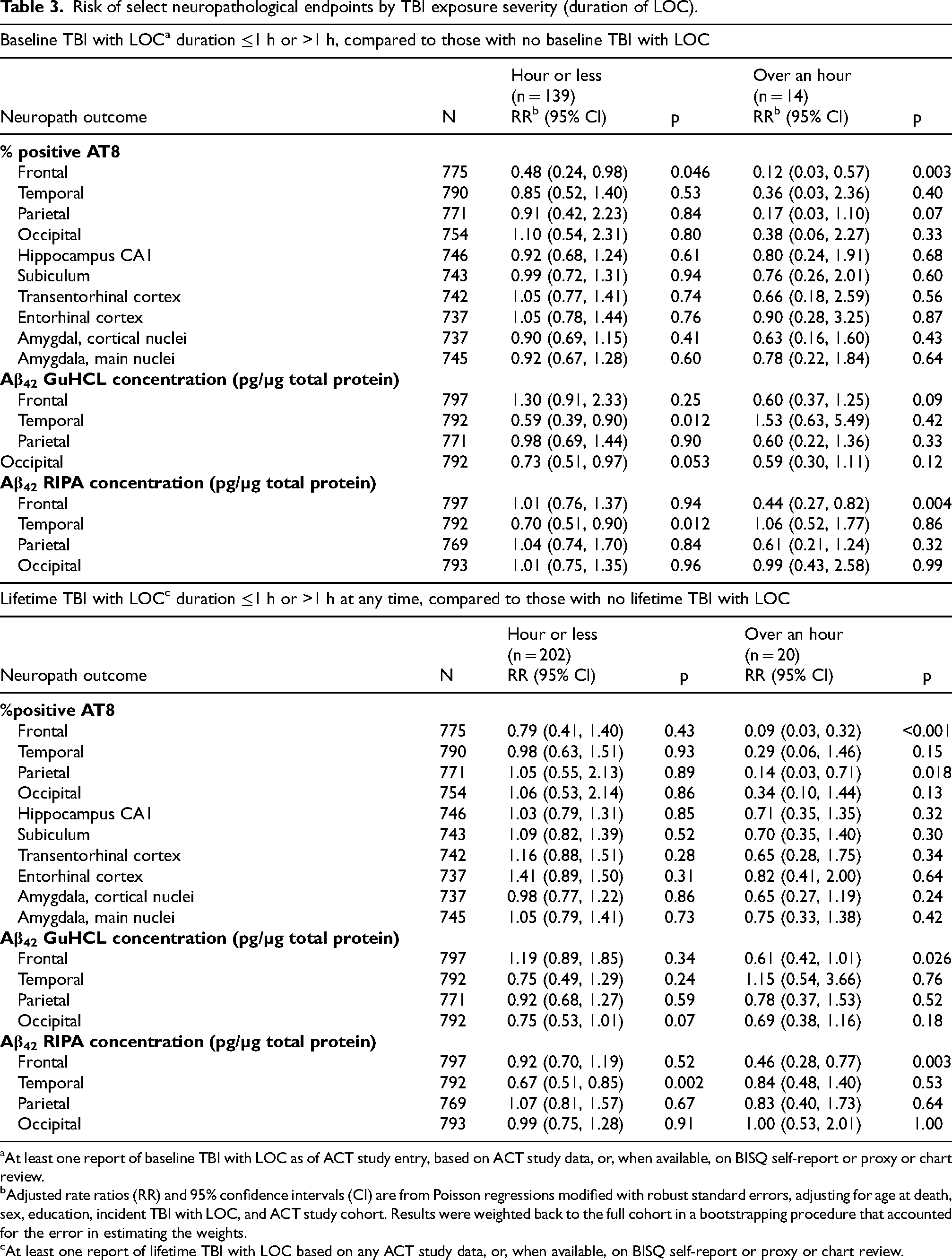
At least one report of baseline TBI with LOC as of ACT study entry, based on ACT study data, or, when available, on BISQ self-report or proxy or chart review.
Adjusted rate ratios (RR) and 95% confidence intervals (CI) are from Poisson regressions modified with robust standard errors, adjusting for age at death, sex, education, incident TBI with LOC, and ACT study cohort. Results were weighted back to the full cohort in a bootstrapping procedure that accounted for the error in estimating the weights.
At least one report of lifetime TBI with LOC based on any ACT study data, or, when available, on BISQ self-report or proxy or chart review.
When stratified by duration of maximum LOC duration at any point (Table 3), ≤1 h LOC was associated with lower temporal RIPA Aβ42 (RR 0.67, 95% CI 0.51–0.85, p = 0.002), but no tau or GuHCl Aβ42 differences. The >1 h LOC group (n = 20) again showed lower frontal tau (RR 0.09, 95% CI 0.03–0.32, p < 0.001), lower parietal tau (RR 0.14, 95% CI 0.03–0.71, p = 0.018), and reduced frontal Aβ42 (GuHCl: RR 0.61, p = 0.026; RIPA: RR 0.46, p = 0.003). Unweighted analyses (Supplemental Table 5) revealed significant inverse associations, for lower frontal tau for >1 h (p < 0.001) LOC. Significant reductions in Aβ42 were also observed, including temporal RIPA Aβ42 for ≤ 1 h LOC (p = < 0.001), and frontal (GuHCl and RIPA fractions) for > 1 h LOC (p < 0.03). Quantile analyses showed similar patterns for parietal Aβ42.
Incident TBI-LOC
In exploratory secondary analyses, we examined incident TBI-LOC occurring after ACT enrollment while excluding injuries considered proximal to death (same year as death or the year before death). In these unweighted analyses, incident TBI-LOC was associated with higher AT8 burden in the frontal cortex (RR 2.18, 95% CI 1.18–4.05), parietal cortex (RR 2.06, 95% CI 1.11–3.84), subiculum (RR 1.41, 95% CI 1.07–1.86), transentorhinal cortex (RR 1.50, 95% CI 1.11–2.02), and entorhinal cortex (RR 1.40, 95% CI 1.06–1.85) (Supplemental Table 6). Frontal Aß42 in the RIPA fraction was lower (RR 0.65, 95% CI 0.46–0.92), while other Aβ42 measures were not significantly associated. Given the exploratory nature of these analyses, and the inability to establish whether pathology preceded injury or followed it, these findings should be interpreted cautiously.
Traditional outcomes
In additional analyses examining traditional neuropathologic outcomes, lifetime TBI-LOC was associated with a higher risk of chronic microinfarcts in weighted models (RR = 1.28; 95% CI 1.05–1.58; p = 0.018), while no associations were observed for Braak stage, CERAD score, Thal phase, LATE stage, or Lewy body disease (Supplemental Table 7). Analyses stratified by LOC duration (≤1 h versus >1 h) showed similar directional patterns but were limited by small sample sizes with TBI-LOC > hour.
Summary
Across multiple definitions of exposure and quantitative neuropathological indices of AD, TBI with LOC was not associated with increased tau or amyloid pathology. Instead, a consistent, though weak, pattern of inverse associations emerged: lower frontal tau and lower soluble Aβ42 (temporal, occipital, and frontal regions) in those with TBI-LOC. These findings were consistent across weighted and unweighted analyses but were still influenced by small, sparse subgroups, particularly those with >1 h LOC, where inverse associations with frontal and parietal tau and amyloid reached statistical significance. Clinical dementia prevalence was higher in these longer-LOC groups despite their lower measured tau and amyloid pathology (Tables 1 and 3); lifetime TBI-LOC was associated with chronic microinfarcts
Discussion
In analyses considering baseline TBI-LOC, lifetime TBI-LOC, incident TBI-LOC, and TBI severity (per LOC duration), TBI with LOC was not associated with increased tau or amyloid pathology. Instead, a consistent, though tenuous pattern of inverse associations emerged: lower frontal tau and lower soluble Aβ42 (temporal, occipital, and frontal regions) in those with TBI-LOC. In unweighted and quantile analyses, several of these inverse associations remained statistically significant, including findings involving frontal tau and selected regional Aß42 measures (Supplemental Tables 4 and 5). These findings were sensitive to analytic weighting and heavily influenced by small, sparse subgroups, especially those with >1 h LOC. Clinical dementia prevalence was higher in these longer-LOC groups despite their lower quantitative indices of AD pathology (Tables 1 and 3).
While prior studies have reported associations between traumatic brain injury and Alzheimer-type neuropathology,34,35 other studies, including earlier ACT analyses, found no association between TBI-LOC and AD pathology.10,15–17,19 Our present analyses, using quantitative measures of tau and amyloid and weighting back to the parent ACT cohort, extend this literature by showing no increased burden of hallmark AD pathologies, even when measured using rigorous quantitative methods, with TBI-LOC. Although our AT8 metric reflects aggregate regional phospho-tau burden rather than lesion-specific pathology, it nevertheless provides a reproducible measure of whether overall tau immunoreactivity differs by TBI-LOC exposure; in this cohort, we did not observe evidence of increased burden. These findings underscore the importance of sample selection and exposure ascertainment in observed associations.
The inverse associations in our models should be interpreted with caution. The apparent protective effects of TBI-LOC on tau and Aβ42 burden were present in both weighted and unweighted analyses, though strongest in small subgroups with limited pathology range, particularly among those with >1 h LOC duration. In quantile analyses, significant reductions were also observed for parietal Aβ42 (GuHCl and RIPA fractions), reinforcing that these apparent inverse effects are influenced by sparse data and the absence of high-pathology outliers, rather than true biological protective effects of TBI. Notably, despite lower measured tau burden, the subgroup with LOC >1 h had a higher prevalence of clinical dementia (∼64% versus ∼45% in other groups). This paradox, higher dementia prevalence despite lower tau and amyloid, emphasizes the importance of considering other pathological processes that may be primary and/or synergistic drivers of clinical dementia. Indeed, prior autopsy studies have suggested increased risk of microinfarcts, Lewy body pathology, or arteriolosclerosis with TBI.10,15
In exploratory secondary analyses of incident TBI-LOC occurring after ACT enrollment, excluding injuries considered proximal to death, we observed higher AT8 burden in several cortical and medial temporal regions (Supplemental Table 6). These findings are intriguing but should not be interpreted as evidence that late-life TBI accelerates neurodegenerative pathology. Because exact injury dates were unavailable and only year-level timing could be used, even “non-proximal” injuries may still have occurred relatively close to death. More importantly, these analyses cannot determine whether elevated tau burden preceded injury and increased vulnerability to falls or other TBI mechanisms, reflected post-injury changes, or both. Accordingly, we view these findings as hypothesis-generating and supportive of future longitudinal biomarker-based studies designed specifically to address temporality.
For completeness, we also examined the effects of TBI-LOC on traditional neuropathological outcomes. Lifetime TBI-LOC was associated with the presence of chronic microinfarcts, the expected direction (Supplemental Table 7) suggesting potential vascular sequelae of TBI in a subset of individuals. This is a new finding, perhaps reflecting the update to ACT assessment methods for microinfarcts.
Our study has several strengths. We leveraged a large, well-characterized, community-based autopsy cohort with detailed quantitative neuropathological measures, multiple sources of TBI ascertainment, and methods to weight results from the autopsy back to the broader ACT population. These features increase generalizability and minimize bias from selective participation in brain donation. By directly modeling quantitative pathology, we were able to test for subtle regional differences that may not have been apparent with traditional staging.
Limitations should also be noted. First, the quantitative neuropathology data used herein used complementary but distinct modalities to quantify tau and Aβ, with AT8 measured by digital pathology and Aβ42 measured by regional immunoassay. As a result, these analyses could not evaluate direct spatial co-localization of tau and Aβ pathology within the same tissue sections or specific lesion contexts. Second, because AT8 burden was quantified as total percent area positive rather than by lesion-specific object classification, these analyses could not distinguish whether observed associations were driven by specific phospho-tau lesion types. Thus, shifts in particular lesion populations may have been obscured by this aggregate burden metric. Third, small sample sizes, particularly in subgroups with LOC >1 h, limited statistical power and stability of estimates. Despite integrating retrospective self-report, proxy interview, and medical record abstraction, TBI exposure misclassification remains possible. In addition, because TBI history was derived from multiple sources with incomplete or estimated timing information, the exact number of unique TBI-LOC events per participant could not be reliably determined across all sources, limiting evaluation of recurrent or cumulative TBI-LOC burden. Furthermore, because the primary exposure was defined as TBI with LOC, these analyses do not capture the full spectrum of mild TBI without LOC or RHI, and LOC duration served only as a coarse proxy for injury severity for remote injuries lacking detailed acute clinical data.
Our analyses focused on hallmark AD-related markers; we did not systematically evaluate other candidate TBI-related neuropathologies such as axonal injury, perivascular or interface astroglial change, or chronic traumatic encephalopathy. Finally, because ACT enrolled adults aged ≥ 65 years who were dementia free at baseline, individuals with more severe TBI who died earlier or developed dementia before enrollment may be unrepresented in this cohort, potentially leading to underestimation of later-life neuropathologic or clinical risks associated with earlier-life TBI.
Clinical implications
Current results build upon prior reports that AD-related neuropathology is not elevated among individuals with TBI-LOC. When considered in context of substantial evidence that TBI is a risk factor for cognitive decline and dementia, these findings suggest that the pathways through which remote TBI contributes to later-life impairment and decline may not operate primarily through tau or amyloid accumulation, but rather through alternative mechanisms such as vascular injury, axonal degeneration, or other non-AD processes. The paradox observed in small subgroups (higher dementia prevalence despite lower measured tau and amyloid) further underscores the likelihood that additional neuropathological substrates contribute to post-traumatic neurodegeneration. Clarifying these mechanisms will be essential for identifying biomarkers for post-TBI dementia diagnosis, monitoring, and novel therapeutic targets that are relevant to TBI survivors who remain at risk for late-life cognitive decline even in the absence of hallmark AD pathology.
Conclusion
In this community-based autopsy cohort of 810 donors from the ACT study, TBI with loss of consciousness was not associated with greater quantitative burdens of tau or amyloid pathology across cortical and limbic regions. In weighted and unweighted analyses, donors with baseline TBI-LOC showed lower frontal tau and lower soluble Aβ42 in selected cortical regions, with additional reductions in parietal Aβ42 observed in quantile models. These inverse associations were small in magnitude and largely driven by sparse subgroups with limited pathology range. Taken together, these findings suggest that TBI-LOC does not accelerate hallmark AD-related processes in late life. Instead, the cognitive risks associated with TBI may arise through alternative mechanisms, vascular, axonal, or inflammatory, that warrant further investigation using rigorous quantitative methods. Future research integrating comprehensive data on lifetime head trauma exposure, clinically accessible imaging and biomarker data, and broader neuropathologic assessments will be essential for delineating the pathways linking TBI to late-life neurodegenerative vulnerability.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261457138 - Supplemental material for Traumatic brain injury and late-life Alzheimer's disease neuropathology: Quantitative investigation in the Adult Changes in Thought study
Supplemental material, sj-docx-1-alz-10.1177_13872877261457138 for Traumatic brain injury and late-life Alzheimer's disease neuropathology: Quantitative investigation in the Adult Changes in Thought study by Enna Selmanovic, Laura E. Gibbons, Rebecca D. Folkerth, C. Dirk Keene, Amber Nolan, Paul Crane, Nadia Postupna, Cecilia S. Lee, Caitlin S. Latimer, Jeanelle Ariza-Torres, Patrick R. Hof and Kristen Dams-O'Connor in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
Ethical considerations
All study procedures were approved by institutional review boards at Kaiser Permanente Washington and the University of Washington.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was originally funded by the National Institute on Aging (grant number U19AG066567). Data collection for this work was additionally supported, in part, by prior funding from the National Institute on Aging (grant number U01AG006781). All statements in this report, including its findings and conclusions, are solely those of the authors and do not necessarily represent the views of the National Institute on Aging or the National Institutes of Health.
Additional research support was graciously provided by the National Institute of Aging (grant numbers R01AG061028, R01AG060942, P30AG066514), the National Institute of Neurological Disorders and Stroke/ National Institute of Child Health and Human Development (grant number 1U01NS086625), the National Institute of Neurological Disorders and Stroke (RF1NS115268, RF1NS128961,U01NS137484), the Research Training in the Neuroscience of Aging (T32AG049688) and the Department of Defense (grant numbers W81XWH-17-1-0330; W81XWH-22-1-0999).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.