
Abstract
Background
Clonal hematopoiesis (CH) increases with age and elevates the risk of numerous age-associated diseases. However, the association between CH and neurodegenerative diseases has remained unclear.
Objective
We tested the association between the presence of CH and tauopathy in a murine model.
Methods
We established novel models of Tet2 loss-of-function and Jak2 gain-of-function (V617F) CH in CD45.1-expressing PS19 tauopathy mice using unconditioned bone marrow (BM) cell transfer, thereby maintaining brain integrity, clonal expansion, and enabling mutant cell tracking.
Results
In CD45.1-PS19 mice, Tet2-/- cells (CD45.2) started at a fraction of <2% and clonally expanded in all BM cavities and the blood over 5 months. Tet2 mutant and WT immune cells, however, displayed equivalently low capacity to infiltrate the brain of PS19 mice even with advanced tau deposition. While Tet2 mutant microglia displayed elevated IL1-β production, they comprised a small fraction (3.49 ± 1.35%) relative to the high frequency of mutant monocytes in the blood (26.36 ± 4.33%) of aged CD45.1-PS19 mice after clonal expansion. Jak2V617F cells (CD45.2) also expanded clonally in the BM and blood over time and had a modestly increased capacity to infiltrate the brain of aged CD45.1-PS19 mice but their proportion among brain microglia remained low (1.12 ± 0.28%) relative to blood monocytes (28.91 ± 4.66%). Critically, Tet2 or Jak2 CH did not alter tau accumulation in the hippocampus or cortex, nor did they influence brain atrophy.
Conclusions
Our findings suggest that CH mutant cells do not influx the murine tauopathy brain in large proportions and that CH does not modify neurodegeneration or tau accumulation in mice.
Introduction
As people age, somatic mutations accumulate in nearly all tissues. 1 While most of these genetic alterations are benign, certain mutations that arise in hematopoietic stem cells (HSCs) provide proliferative and survival advantages. 2 This leads to their clonal outgrowth and the disproportionate expansion of mutant HSCs, a condition known as clonal hematopoiesis (CH). CH is premalignant and common, affecting 10–20% of individuals over 70, and frequently arises from mutations in Dnmt3a, Tet2, Jak2, Asxl1, or p53.2–6 CH is diagnosed in individuals without hematologic malignancy or cytopenia when the variant allele frequency (VAF) is 2% but has CH mutant cells expand, higher VAFs are reached. 6 Mature progeny deriving from mutant HSCs, like macrophages, retain the mutation which often confers inflammatory phenotypes and re-programming. 7 For example, Tet2 loss-of-function mutations in macrophages impair DNA demethylation and causes the production of pro-inflammatory cytokines, partly through activation of the NLRP3 inflammasome.6,8,9 In contrast, a gain-of-function Jak2 mutation (V617F), activates the JAK-STAT pathway, driving exaggerated myeloproliferation and a distinct inflammatory profile marked by increased IL-1β, interferon signaling, and oxidative stress.10–12 The inflammatory phenotype of CH mutant macrophages, derived from BM progenitors, and recruited to cardiovascular tissues, substantially increases the risk of all-cause mortality and numerous age-related diseases including cardiovascular disease (CVD), coronary artery disease, ischemic heart failure, stroke, and chronic kidney disease among others.4,6,8,13–15
Aging is the strongest risk factor for CH, 2 yet the relationship between CH and age-related neurodegenerative diseases like Alzheimer's disease (AD) and related dementias (ADRDs), including tauopathies, has remained unclear. The possibility that CH mutant immune cells may interact with the brain through cytokine-mediated peripheral-to-central communication or local influx into the brain raises important questions about how CH could modulate AD neuroinflammation, pathology, and disease progression. Surprisingly, recent human epidemiological data suggest that CH protects against AD. 16 Notably, individuals with CH mutations exhibited significantly lower levels of amyloid-β (Aβ) and neurofibrillary tangle burden composed of hyperphosphorylated tau. Unexpectedly, these somatic mutations were not restricted to blood immune cells but were also detected in microglia-enriched brain tissue from known CH carriers. Indeed, amplicon sequencing further revealed that 40–80% of microglia in CH carriers harbored these mutations which correlated with blood VAF, raising the possibility that myeloid cells deriving from mutant hematopoietic clones infiltrate the brain and contribute to the microglia pool in AD. In agreement, CH has been associated with a reduced AD risk and delayed age-at-onset in APOE ε3/ε3 carriers, with similar trends across all other APOE genotypes 17 and large clone CH (VAF>8%) decreased incidence probable dementia. 18 However, other investigations have reached alternate conclusions and nuanced mutation-dependent effects are likely. For example, a community-based cohort study on the UK Biobank found no association between CH and primary neurodegenerative diseases including AD and Parkinson's disease. 19 Moreover, TET2 CH has been associated with a reduction in late onset AD or no association with AD risk depending on the study population and whether individuals with CVD were included in the analysis, a critical variable given the strong positive association between CH and CVD.16,19,20 Non-TET2 CH meanwhile, has been suggested to have no influence on AD 20 whereas another study demonstrated that ASXL1, but not DNMT3A, CH heightens susceptibility to AD. 19 Complicating matters, integrated bidirectional associations are also possible. For example, the prevalence of CH mutations may be enriched in people with AD and APOE ε3/ε3 but not ε4 carriers. 21 There is a clear need to further assess the association between CH and AD in human cohorts.
Experiments in murine models can provide among the most direct causal and mechanistic evidence associating risk factors with disease pathology and recent studies have aimed to establish CH in AD mice. In 5xFAD mice, a model of Aβ driven AD, BM depletion with gamma irradiation followed by replacement of the entire hematopoietic compartment with CH mutant BM, Tet2−/− but not Dnmt3a mutant cells reduced Aβ plaque burden and cognitive decline in animals injected weekly with lipopolysaccharide. 20 These findings point to novel emerging mechanisms linking CH and AD and highlight the role of environmental inflammatory triggers. However, the influence of mutant cells as they clonally expand and at lower VAFs that more closely reflect human biology remains unclear. While irradiation-based models offer a tractable platform for studying hematopoietic contributions to neurodegeneration, irradiation also causes profound geno- and neurotoxic effects, compromises the blood-brain barrier (BBB), and leads to artifactual neuroinflammation.22–28 Moreover, the effect of CH on tau deposition and tauopathy has not been explored. Thus, to establish causal associations between CH and tau associated neurodegeneration and investigate underlying mechanisms, murine models of CH in tauopathy that maintain brain health and immune cell dynamics are needed.
Here, we established a new murine model of CH in tauopathy that uses unconditioned hematopoietic cell transfer to instill CH mutations into a fraction of circulating immune cells and allows for the congenic (CD45.1/2) tracking of mutant and non-mutant endogenous cells. By transferring CD45.2-expressing CH mutant bone marrow cells into unconditioned naïve CD45.1-PS19 mice, a murine model of tauopathy and neurodegeneration, 29 we establish CH and mutant cell clonal expansion while maintaining peripheral-to-central brain immune cell dynamics, and discover that Tet2−/− or Jak2V617F CH does not alter tau burden or neurodegeneration in PS19 mice.
Methods
Animals
Wild-type CD45.1 JAXBoy (PtprcK302E; Strain 033076) and PS19 (P301S Tg; Strain 008169) mice were purchased from the Jackson Laboratory and bred in-house to generate CD45.1+ PS19 transgenic offspring expressing mutant human P301S tau. Mx1-Cre (B6.Cg-Tg(Mx1-cre)1Cgn/J; Strain#003556), Jak2+/flox (B6N.129S6(SJL)-Jak2tm1.1Ble/AmlyJ; Strain#031658), Tet2flox (B6;129S-Tet2tm1.1Iaai/J; Strain#017573) and Tet2+/− (B6(Cg)-Tet2tm1.2Rao/J; Strain#023359) mice were purchased from the Jackson Laboratory and bred in-house. Jak2+/flox and Tet2flox mice were crossed to Mx1-Cre to generate Jak2V617Ffl/fltMx1-Cre or Tet2fl/flMx1-Cre mice. All animal protocols were approved by the Animal Review Committee at the Icahn School of Medicine at Mount Sinai (Protocol numbers: IPROTO202300000060, PROTO202100023, IPROTO202400000058) and followed relevant ethical regulations. All experimental procedures were conducted in accordance with guidelines set by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai. All mice used were cohoused and on a C57BL/6J background. Mice were randomly assigned to interventions and the sex of the animals in each experiment is reported in the figure legends. Mice were provided ad libitum access to food and water in a light- and temperature-controlled vivarium in a standard 12-h light/dark cycle (light Zeitgeber (ZT) 0–12 and dark ZT 12–24) at 23°C and 65% humidity.
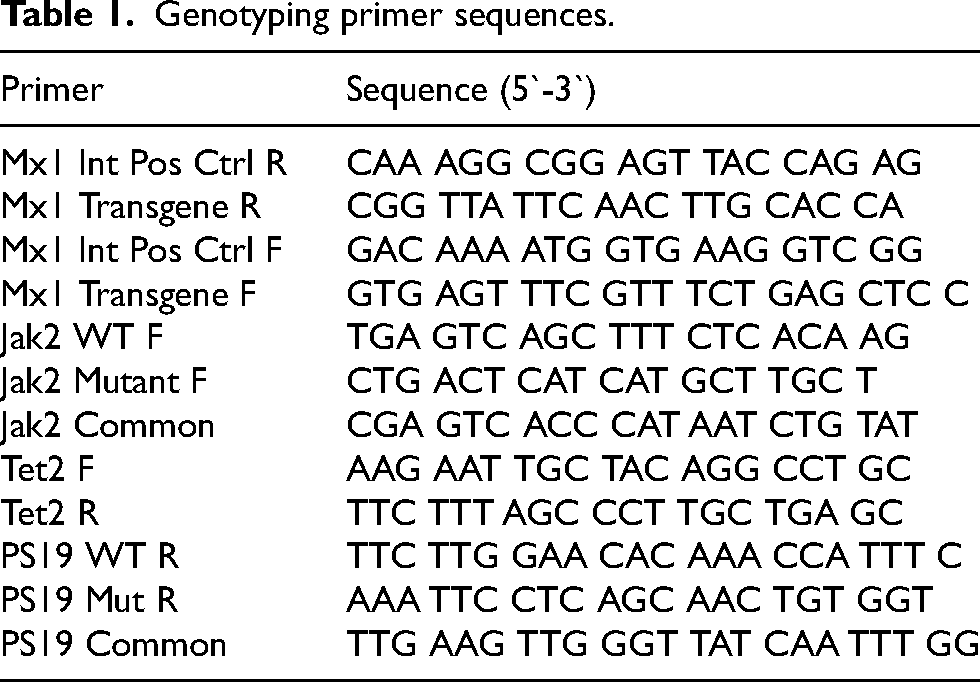
Genotyping primer sequences.
In vivo interventions
Unconditioned bone marrow cell transfer. An adoptive bone marrow transfer was used to model the progression of CH in PS19 mice. To induce Cre-mediated recombination in Jak2V617Ffl/flMx1-Cre or Tet2fl/flMx1-Cre donors, mice received intraperitoneal (i.p.) injections of 200 μg/mouse/day polyinosinic:polycytidylic acid (Poly:IC, Sigma, 42424-50-0) three times 48 h apart. As controls, C57BL/6 (WT) mice received the same regiment. One week later, bone marrow cells were harvested from all donor mice and enumerated using a hemocytometer. 3-month-old unconditioned naïve CD45.1-PS19 recipients were anaesthetized with isoflurane (3% in 97% oxygen) before receiving a total of 1.5 × 107 CD45.2+ Jak2V617Ffl/flMx1-Cre or Tet2fl/flMx1-Cre or C57BL/6, split into 3 doses of 5 × 106 cells delivered in consecutive days via the retro-orbital sinus.
Cheek bleeds for expansion assessment. Sequential cheek bleeds were performed every 2–4 weeks as indicated to monitor BM cell engraftment and expansion in PS19 recipient mice. Mice were anesthetized with 3% isoflurane prior to blood collection. A 25-gauge needle was used to obtain a 50 µL sample from the cheek vein. Samples were immediately processed for flow cytometry to evaluate cellular expansion based on CD45.1 and CD45.2 expression.
Cell collection. At sacrifice, mice were anesthetized using 3% isoflurane and peripheral venous blood was drawn through retro-orbital bleeding. Red blood cells were eliminated using RBC lysis buffer (BioLegend, Catalog #420302). Cardiac puncture was performed and mice were perfused with 10–20 mL PBS. the bone with PBS from a 25-gauge needle, then mechanically dissociated by passing through a 19-gauge needle to generate a single-cell suspension. Remaining red blood cells were subsequently lysed with RBC lysis buffer. The brain and skull were excised, minced and digested with 450 U ml−1 collegenase I, 125 U ml−1 collagenase XI, 60 U ml−1 DNase I and 60 U ml−1 hyaluronidase in PBS for 30 min at 37C. Samples were passed through a 100 μm cell strainer. Brains were further processed by mixing with 30% percol layered on top of 70% percol. The percol gradient was centrifuged at 500 g for 30 min with the brake off. The cell fraction was collected and washed with PBS before downstream applications.
Flow cytometry. Single-cell suspensions were stained with antibody cocktail in MACS buffer (0.5% BSA and 2 mM EDTA in PBS) for 30 min at 4C in the dark. To differentiate between live and dead cells, the cell suspensions were then stained with live/Dead blue (Thermo fisher) at a dilution of 1:1000 in PBS at 4C for 30 min. Monoclonal antibodies were used for flow cytometry analyses at a dilution of 1:700 (Table 2). CountBright™ Absolute Counting Beads (Invitrogen Reference #C36950) were used to determine absolute cell counts. Data were collected on an Cytek Aurora cytometer with Spectroflow software and analyzed using FlowJo.
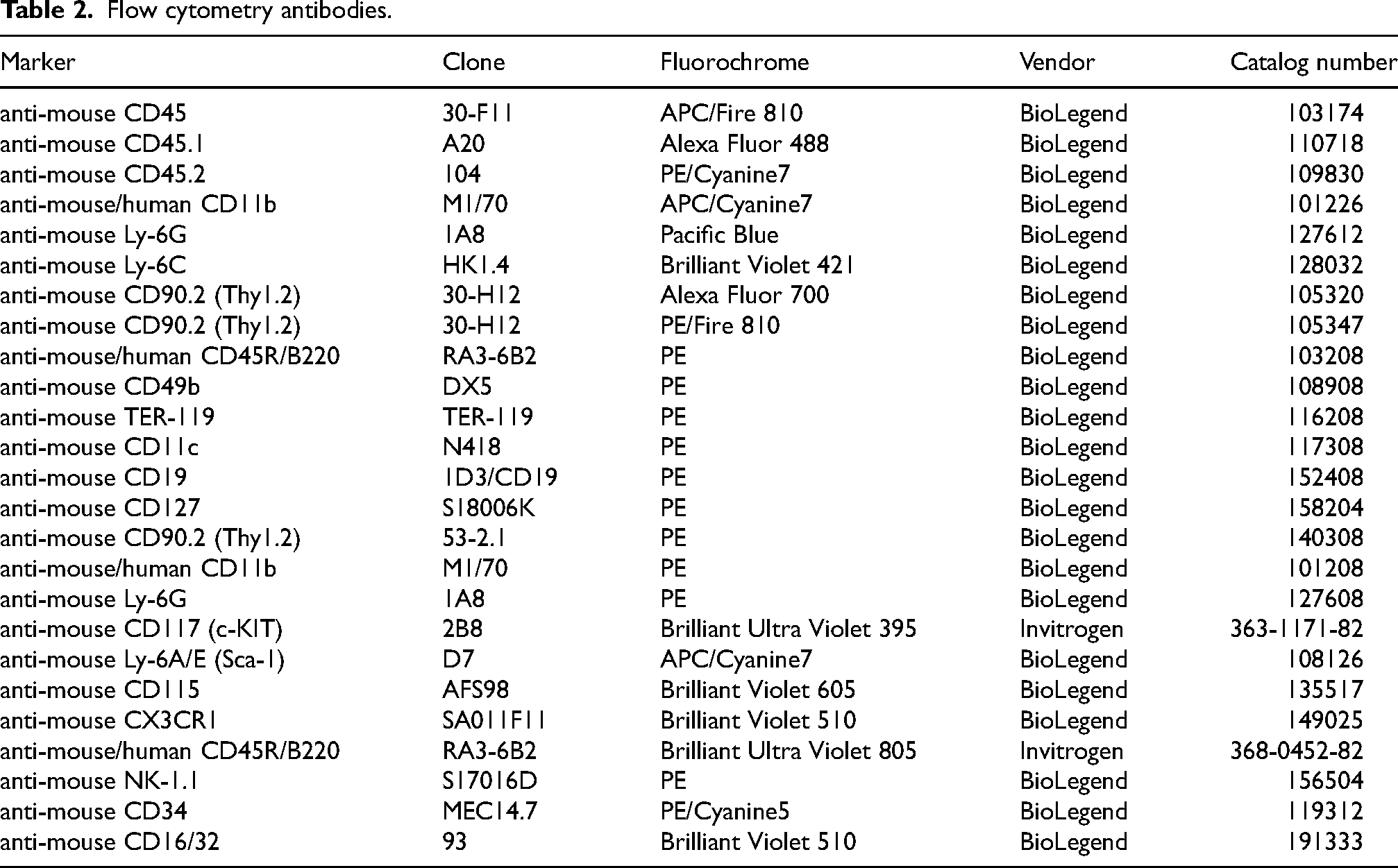
Flow cytometry antibodies.
Primary microglia culture. The brains of 3-day-old pups were isolated in sterile PBS and washed with HBSS before incubating in 0.25% trypsin at 37°C for 1 h. Cells were pelleted, strained, and washed. Cells were plated in DMEM with 5% FBS and 50 ng ml−1 GM-CSF. Medium was changed one day later and every other day thereafter. After one week, 500,000 cells were split into treatment wells, allowed to re-adhere over 2 days, then exposed to 10 ng/mL LPS or PBS control for 24 h. After stimulation, cell culture supernatant was collected and flash frozen for cytokine measurement. Cells were then washed once with PBS and homogenized and harvested by scraping and dissociating in Trizol for gene expression profiling.
Histology and molecular imaging
Volumetric analysis. The left hemi-brain of each mouse was fixed with formalin for 7 days at 4C and then placed in 30% sucrose at 4C overnight. Tissues were embedded in Tissue-Tek OCT compound (Sakura Finetek). Samples were rapidly frozen in 2-methylbutane (Fisher Scientific) chilled with dry ice. Coronal sections were cut at 20 μm and mounted. Hippocampal and lateral ventricle volume was determined by collecting serial sections, starting from the initial appearance of the hippocampus and continuing until the end of the hippocampus was reached. Images were collected on a Keyence BZ-X microscope. Hippocampus and lateral ventricle were manually traced using ImageJ. The volume was calculated using the formula: volume = (sum of area) x (distance between sections).
Immunofluorescence. Serial frozen sections (20 um) were prepared as described above. Frozen brain sections were rehydrated for 5 min in PBS at room temperature. Antigen retrieval was performed with a commercial kit (Fisher Scentific, BDB550524) at 95C for 20 min. Sections were permeabilized for 10 min with 0.25% PBS-TritonX (PBSX) and then blocked with 10% normal goat serum in 1% BSA (MP Biomedicals, 160069) and 0.5% saponin (Sigma, SAE0073) in PBS for 1 h at room temperature. Primary antibody incubation was performed overnight at 4°C in 1% BSA (MP Biomedicals, 160069) and 0.5% saponin (Sigma, SAE0073) in PBS using the following antibodies: anti-AT8-biotin (Fisher Scientific, MN1020b; 1:200), anti-CD45.2-biotin (Biolegend 109804; 1:100). Systems, 234308). After washing three times for 5 min each in 0.1% PBS-Tween, sections were incubated with the secondary antibody Streptavidin DyLight 647 (Vector Laboratories, SA-5649-1; 1:500) for 2 h at room temperature in the dark. Slides were then washed 3 times in 0.1% PBS-Tween and incubated with DAPI (1:1000 in PBS, Invitrogen, PI62247) for 10 min at RT in the dark, followed by another 3 washes in 0.1% PBS-Tween. Slides were mounted with DAKO Fluorescence Mounting Medium (S302380-2), cover slipped and sealed using nail polish. Images were collected on a Keyence BZ-X microscope using a 20X objective with standardized brightness and exposure settings. Areas covered by antibody-fluorophores and their numbers were analyzed with ImageJ.
Molecular assays
Cytokine quantification by ELISA. Blood was collected into tubes containing 5 mM EDTA and centrifuged at 2000 g for 10 min to separate plasma, which was carefully collected for analysis. The resulting supernatant was frozen for further analysis. IL-1β levels in plasma and cell culture supernatant were quantified using an ELISA kit from R&D Systems (IL-1β: MHSLB00).
qPCR. Total RNA was extracted using Trizol (Thermo Fisher) following the manufacturer's protocol. Briefly, cell culture samples were scraped in 0.8 mL of Trizol and dissociated with a 25-guage needle on ice, followed by the addition of 160 μL of a chloroform:isoamyl alcohol (49:1) mixture and vortexed. The samples were then centrifuged at >20,000 g for 30 min at 4°C, after which the aqueous phase was carefully transferred to a fresh tube containing 500 μL of 100% molecular grade isopropanol. Samples were mixed by flicking/inverting and were frozen at −80°C overnight. The next day, samples were thawed and centrifuged again at >20,000 g for 30 min at 4°C. The supernatant was discarded, and the RNA pellet was washed twice with 1 mL of 80% (v/v) molecular-grade ethanol, each time followed by centrifugation for 10 min. The pellet was then air-dried for 20 min at room temperature and resuspended in approximately 12 μL of molecular-grade water. RNA concentration and purity were assessed using a Nanodrop spectrophotometer. Complementary DNA (cDNA) was synthesized from 1 μg of total RNA per sample using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real-time TaqMan PCR was performed using the following FAM-labelled TaqMan primers (Thermo Fisher Scientific): Il1b (Mm00434228, Nlrp3 (Mm00840904), Aim2 (Ec07035002), all normalized to Actb (VIC-labelled, Mm00607939). Samples were run in duplicates and average delta Cq values for each replicate were obtained using Design and Analysis 2 software (Applied Boiosystems).
Statistics
Results are presented as mean ± s.e.m. Statistical tests were performed using GraphPad Prism 8 and included unpaired two-tailed Student's t-tests (two groups) and nonparametric Mann-Whitney U-tests (when Gaussian distribution was not assumed). For multiple comparisons, one- or two-way ANOVA, or nonparametric Kruskall-Wallis test, followed by Tukey's multiple-comparisons test for one-way-, and two-way ANOVA, and Dunn's multiple comparisons test for Kuskall-Wallis test were used. For ANOVA analyses exact p values are reported. p < 0.05 was considered to be statistically significant. Power calculations were used to determine the numbers of animals required, based on standard deviations from previous studies in mice.
Results
Establishment of Tet2 CH in PS19 mice by unconditioned hematopoietic cell transfer
Because TET2 is among the most frequently mutated genes in CH, we assessed how Tet2 loss-of-function CH impacts the progression of phospho-tau mediated neurodegeneration in PS19 mice, an established model of tauopathy. PS19 mice were chosen because CH in tauopathy had not been previously investigated, tau accumulation closely aligns with cognitive impairment, and unlike beta-Amyloid models, PS19 mice develop neurodegeneration and neuronal loss. To do so, we relied on an established strategy of transferring BM cells into naïve unconditioned recipient mice that did not undergo chemical or irradiation BM depletion 30 and allowed us to track and identify CH mutant cells by CD45.1/2 (Figure 1A). First, we bred CD45.1-expressing mice with PS19 mice to generate tauopathy mice in which all leukocytes, including microglia, express CD45.1 instead of CD45.2. To establish Tet2 CH with control over when gene knockdown is induced, Tet2fl/flMx1-Cre mice (CD45.2) were i.p. injected with Poly(I:C) which efficiently repressed Tet2 in blood leukocytes (Supplemental Figure 1A). Alongside, Mx1-Cre mice (CD45.2) were also injected with Poly(I:C) and served as Tet2+/+ (wildtype, WT) controls. Seven days later, bones were collected from the Tet2fl/flMx1-Cre and Mx1-Cre mice and a total of 1.5 × 107 bone marrow cells (BMCs), split over three consecutive days, were i.v. injected into unconditioned, naïve, 3-month-old CD45.1-PS19 mice. This strategy generated CD45.1-PS19 mice harboring engrafted CD45.2 Tet2−/− or WT cells which we refer to as PS19Tet2−/− and PS19WT, respectively. In line with prior reports using a similar unconditioned transfer strategy, 30 2 weeks after cell transfer the proportion of donor CD45.2 cells amongst blood monocytes and neutrophils was equivalent between PS19Tet2−/− and PS19WT at 4.69 ± 0.8% and 5.12 ± 0.7%, respectively (Figure 1B and Supplemental Figure 1B). Over time and as expected, the fraction of CD45.2 Tet2−/− blood monocytes and neutrophils in PS19Tet2−/− mice steadily increased reaching 26.36 ± 4.33% and 22.89 ± 3.85% at 8 months of age (5 months after BMC transfer), indicative of clonal expansion (Figure 1B, C). Conversely, in PS19WT mice, CD45.2 WT blood monocytes and neutrophils did not expand (Figure 1B, C). We noted that IL-1β in the plasma of PS19WT mice tended to be higher relative to WT mice non-conditionally transplanted with WT BM (WTWT) and that IL-1β was not further increased in PS19Tet2−/− mice (Figure 1D). In line with these blood leukocyte dynamics, 5 months after cell transfer the proportion of donor-derived CD45.2 common myeloid progenitors (CMPs), granulocyte macrophage progenitors (GMPs), macrophage dendritic progenitors (MDPs), monocytes, and neutrophils in the femur (Figure 1E) and skull (Figure 1F and Supplemental Figure 1C) BM was larger in PS19Tet2−/− mice relative to PS19WT mice, indicating successful engraftment and expansion of the Tet2−/− cells. We also observed a modest expansion of lymphoid cells in PS19Tet2−/− mice (Supplemental Figure 1D-F). Together, these findings establish and validate a novel murine model of Tet2 CH in tauopathy that does not rely on cytotoxic BM depletion, aligns with human VAFs, allows Tet2−/− clonal expansion, and enables the distinction between endogenous (CD45.1) and donor-derived (CD45.2) leukocytes.
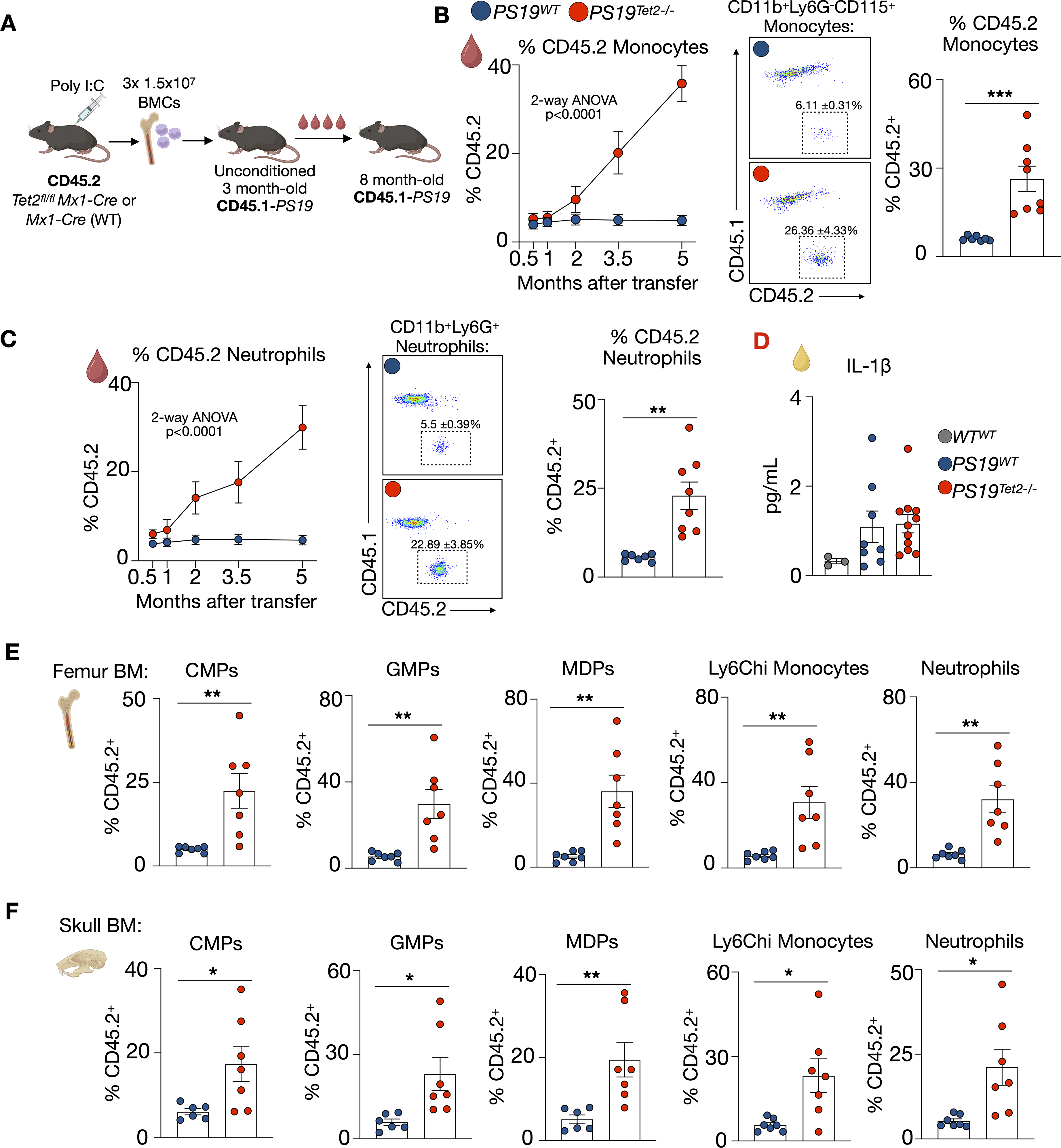
Non-conditioned bone marrow transfer leads to robust Tet2 CH and clone expansion in PS19 mice. (A) Schematic of experimental design. (B) Expansion of blood CD45.2 WT or Tet2−/− monocytes over time (n = 5 PS19WT (4 M/1F), n = 7 PS19Tet2−/− (5 M/2F)); 2-way ANOVA + Tukey's multiple comparisons test (left panel); Representative flow cytometry plots (middle panel); Percentage of CD45.2 monocytes in the blood of PS19WT or PS19Tet2−/− at 8 months of age (n = 7 PS19WT (6 M/1F), n = 8 PS19Tet2−/− (6 M/2F), right panel). (C) Expansion of blood CD45.2 WT or Tet2−/− neutrophils over time (n = 5 PS19WT (4 M/1F), n = 7 PS19Tet2−/− (5 M/2F)); 2-way ANOVA + Tukey's multiple comparisons test (left panel); Representative flow cytometry plots (middle panel); Percentage of CD45.2 neutrophils in the blood of PS19WT or PS19Tet2−/− at 8 months of age (n = 7 PS19WT (6 M/1F), n = 8 PS19Tet2−/− (6 M/2F) , right panel). (D) Plasma levels of interleukin-1β (IL-1β) (n = 8 PS19WT (6 M/2F), n = 11 PS19Tet2−/− (7 M/4F)). Left bar shows control IL-1β Plasma levels in WT mice that received WT BM. (E) Percentage of CD45.2 common myeloid progenitors (CMPs), granulocyte-progenitors (GMPs), monocyte-dendritic cell progenitors (MDPs), Ly6Chi monocytes, and neutrophils in in the femur bone marrow (BM) of PS19 mice (n = 7 PS19WT (6 M/1F), n = 7 PS19Tet2−/− (5 M/2F)) (F) Percentage of CD45.2 CMPs, GMPs, MDPs, Ly6Chi monocytes, and neutrophils in the skull BM (n = 7 PS19WT (6 M/1F), n = 7 PS19Tet2−/− (5 M/2F)). Data are mean ± s.e.m.; unpaired t-test *p < 0.05, **p < 0.01, ***p < 0.001.
Tet2 CH does not alter immune cell infiltration, tau burden or atrophy in the brain of PS19 mice
We then evaluated if Tet2 mutant cells influx the brain and if Tet2 CH modifies tau burden and brain atrophy in PS19 mice. First, we performed flow cytometry analysis on the brain of 8-month-old PS19Tet2−/− and PS19WT mice, 5 months after unconditioned cell transfer (Figure 2A) and confirmed efficient CD45.1 and CD45.1 microglia separation by flow cytometry (Supplemental Figure 2A). The number of CD45midCX3CR1+ CD11b+ microglia and non-microglia CD45hi leukocyte was unchanged in PS19Tet2−/− mice relative to PS19WT controls (Figure 2B). Moreover, the proportion of donor-derived CD45.2 microglia and non-microglia CD45hi cells was small (3.49 ± 1.35% and 2.86 ± 1.01%, respectively) and did not differ between PS19Tet2−/− and PS19WT mice (Figure 2B). Given that in PS19Tet2−/− mice at this timepoint, Tet2−/− cells comprise 26.36 ± 4.33% of circulating monocytes (Figure 2B, C), these observations suggest that Tet2−/− cells do not have an increased capacity to influx the brain. Indeed, immunofluorescent imaging identified few CD45.2+ cells in the parenchyma of the hippocampus of PS19Tet2−/− and PS19WT mice (Figure 2C). Together, these data demonstrate that a small fraction of peripheral immune cells influx the brain of PS19 mice but Tet2−/− cells do not have an increased capacity to do so.
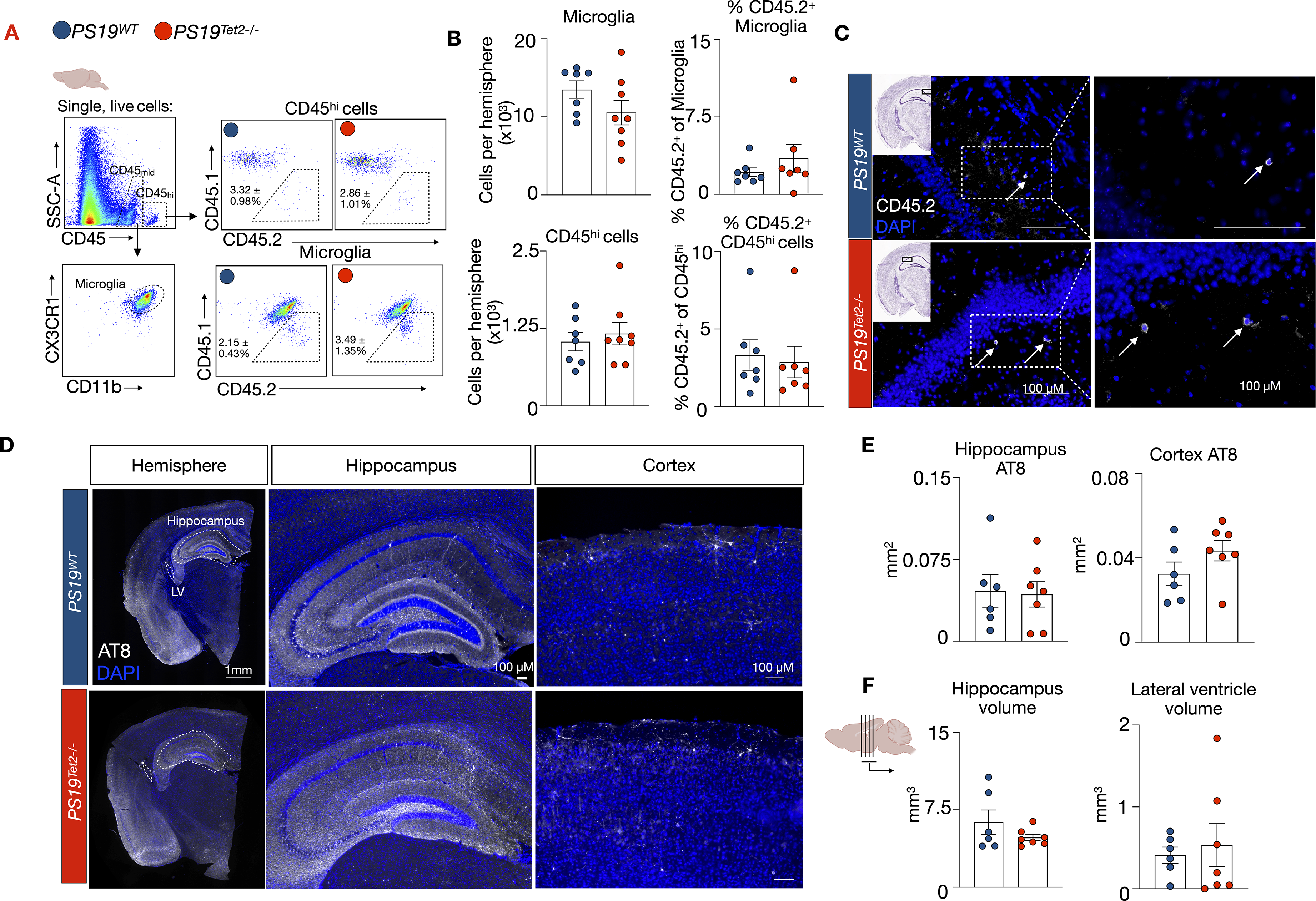
Tet2 CH does not alter tau burden or brain atrophy in PS19 mice. (A) Flow cytometry gating strategy in the brain tissue. (B) Flow cytometry quantification of microglia and CD45hi numbers, and respective percentages of CD45.2 microglia and CD45hi cells in the brain of PS19WT and PS19Tet2−/− mice (n = 7 PS19WT (6 M/1F), n = 7 PS19Tet2−/− (5 M/2F)). (C) Representative CD45.2 immunofluorescence in the hippocampus of PS19WT and PS19Tet2−/− mice. (D) Representative images of p-tau (AT8) staining. (E) Quantification of AT8 staining in the hippocampus and cortex (n = 6 PS19WT (5 M/1F), n = 7 PS19Tet2−/− (5 M/2F)). (F) Quantification of volume measurements of the hippocampus and lateral ventricle (n = 6 PS19WT (5 M/1F), n = 7 PS19Tet2−/− (5 M/2F)). Data are mean ± s.e.m.; unpaired t-test.
Next, we sought if Tet2 CH modified tau accumulation and brain atrophy in PS19 mice. At 8 months of age, 5 months after cell transfer, we probed and imaged tau (AT8) on brain sections (Figure 2D) and found equivalent levels in the hippocampus and cortex of PS19Tet2−/− and PS19WT mice (Figure 2E). Further, the volume of the hippocampus and lateral ventricle were similar in PS19Tet2−/− and PS19WT mice (Figure 2F), suggesting that atrophy and neuronal tissue loss was not affected. Together, these findings demonstrate that the presence of Tet2 CH does not alter tau accumulation or neurodegeneration in PS19 mice.
Increased macrophage inflammasome activation and IL-1β generation is a causal mechanism by which Tet2 CH aggravates inflammatory pathologies like atherosclerosis.6,8,31 It remained unclear, however, whether Tet2 deficiency influences inflammasome activation and IL-1β production by primary microglia. It is conceivable that tau accumulation and neurodegeneration is unaltered in PS19Tet2−/− mice because, unlike peripheral macrophages, Tet2 deficient microglia do not display altered inflammatory programing. To test this, we generated astrocyte and microglia cocultures, mirroring the multicellular in vivo environment, from the brains of P3 WT and Tet2+/− pups. We used Tet2+/− mice to avoid confounding inflammatory issues associated with Poly(I:C) induction of Cre in the in vitro setting. We stimulated inflammation in the cultures by exposing them to lipopolysaccharide (LPS) (Supplemental Figure 2B). LPS exposure resulted in a higher expression of Il1b, Nlrp3, and Aim2, in Tet2+/− glia relative to WT glia (Supplemental Figure 2C). Moreover, IL-1β concentration in the supernatant of the Tet2+/− glia cultures was higher than the WT cultures (Supplemental Figure 2D). These data affirm that Tet2 deficiency in glia augments the NLRP3 and AIM2 inflammasomes and IL-1β production, thus the lack of neuropathology changes in PS19Tet2−/− mice cannot be explained by dampened inflammatory programing of Tet2 mutant cells in the brain microenvironment.
Establishment of Jak2V617F CH in PS19 mice by unconditioned hematopoietic cell transfer
Each CH mutation elicits unique cellular phenotypes and biological mechanisms which confer different disease risks. For example, in humans, TET2 CH elevates the risk of atherosclerotic cardiovascular disease 2-fold, while JAK2V617F elevates risk over 12-fold. 6 Thus, it is possible that each CH mutation has different effects on the brain and risk for neurodegeneration. Therefore, we established a model of Jak2V617F CH in unconditioned PS19 mice. Using a similar strategy as our Tet2 CH model, Jak2V617Ffl/flMx1-Cre (CD45.2) mice were i.p. injected with Poly(I:C) to induce the V617F CH mutation in Jak2. Alongside, Mx1-Cre mice (CD45.2) were also injected with Poly(I:C) and served as WT controls. Seven days later, the bones of the Jak2V617Ffl/flMx1-Cre and Mx1-Cre mice were collected and 1.5 × 107 BMCs were i.v. injected daily for three days into unconditioned, naïve, 4-month-old CD45.1-PS19 mice. This strategy generated recipient CD45.1-PS19 mice with a proportion of blood leukocytes deriving from CD45.2 Jak2V617F or WT donors which we refer to as PS19Jak2V617F and PS19WT, respectively. Over time, the fraction of CD45.2 Jak2V617F blood monocytes and neutrophils in PS19Jak2V617F mice steadily increased, reaching 28.91 ± 4.656% and 42.6 ± 5.914% respectively at 8 months of age (5 months after transfer), indicative of clonal expansion (Figure 3B, C). Conversely, WT CD45.2 blood monocytes and neutrophils in PS19WT mice did not expand (Figure 3B, C). In line with this cellular expansion, plasma IL-1β was elevated in PS19Jak2V617F mice (Figure 3D). Further, 4 months after cell transfer, the proportion of CD45.2 CMPs, GMPs, MDPs, monocytes, and neutrophils in the BM of the femur (Figure 3E) and skull (Figure 3F) was substantially larger in PS19Jak2V617F mice relative to PS19WT mice, indicating successful engraftment and expansion of Jak2V617F cells in the BM. We also observed a modest expansion of Jak2V617F lymphoid cells (Supplemental Figure 3). Together, these findings establish and validate a novel murine model of Jak2V617F CH in tauopathy that relies on unconditioned BMC transfer and recapitulates CH clonal dynamics.
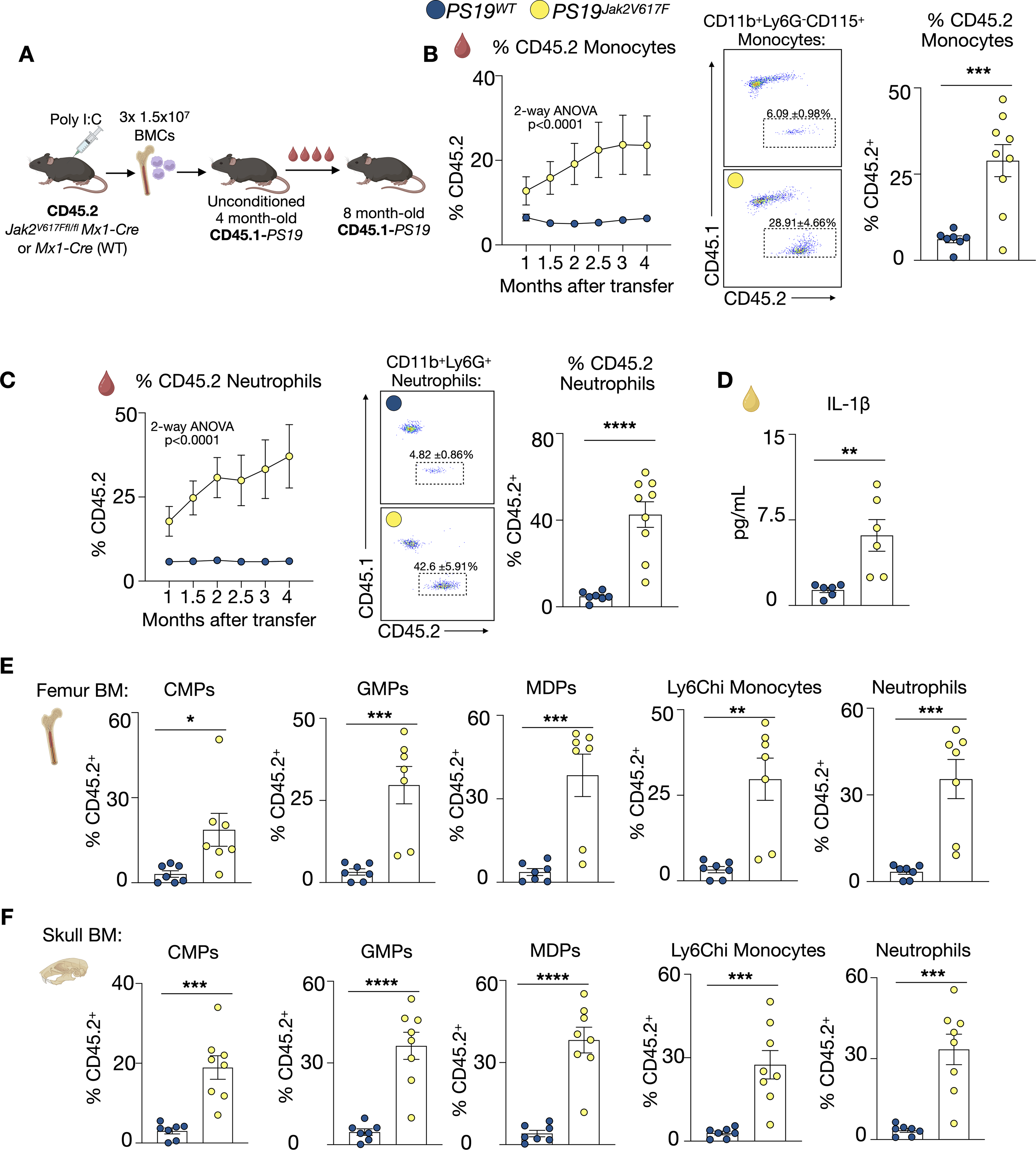
Non-conditioned bone marrow transfer leads to robust Jak2V617F CH and clone expansion in PS19 mice. (A) Schematic of experimental design. (B) Expansion of blood CD45.2 WT or Jak2V617F monocytes and neutrophils over time (n = 6 PS19WT (5 M/1F), n = 7 PS19Jak2V617F (5 M/2F)); 2-way ANOVA + Tukey's multiple comparisons test (left panel); Representative flow cytometry plots (middle panel); Percentage of CD45.2 monocytes in the blood at 8 months of age (n = 7 PS19WT (5 M/2F), n = 9 PS19Jak2V617F (6 M/3F), right panel). (C) Expansion of blood CD45.2 WT or Jak2V617F neutrophils over time (n = 6 PS19WT (5 M/1F), n = 7 PS19Jak2V617F (5 M/2F)); 2-way ANOVA + Tukey's multiple comparisons test (left panel); Representative flow cytometry plots (middle panel); Percentage of CD45.2 neutrophils in the blood of PS19WT or PS19Jak2V617F at 8 months of age (n = 7 PS19WT (5 M/2F), n = 9 PS19Jak2V617F (6 M/3F) , right panel). (D) Plasma levels of interleukin-1β (IL-1β) (n = 6 PS19WT (5 M/1F), n = 6 PS19Jak2V617F (3 M/3F)). (E) Percentage of CD45.2 CMPs, GMPs, MDPs, Ly6Chi monocytes, and neutrophils in in the femur BM of PS19 mice (n = 7 PS19WT (5 M/2F), n = 7 PS19Jak2V617F (4 M/3F)) (F) Percentage of CD45.2 CMPs, GMPs, MDPs, Ly6Chi monocytes, and neutrophils in the skull BM (n = 7 PS19WT (5 M/2F), n = 8 PS19Jak2V617F (5 M/3F)). Data are mean ± s.e.m.; unpaired t-test *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Jak2 CH does not alter tau burden nor brain atrophy in PS19 mice
We then evaluated if Jak2 mutant cells influx the brain of PS19 mice and if Jak2 CH modifies tau burden and brain atrophy. First, we conducted flow cytometry on the brains of 8-month-old PS19Jak2V617F and PS19WT mice, 4 months after BMC transfer (Figure 4A). The number of CD45midCX3CR1+ CD11b+ microglia was unchanged in PS19Jak2V617F mice relative to PS19WT controls while there were slightly more non-microglia CD45hi cells in the brains of PS19Jak2V617F animals (Figure 4B). The proportion of donor derived CD45.2 microglia was small (1.123 ± 0.275%) and did not differ between PS19 Jak2V617F and PS19WT mice (Figure 4B). Meanwhile, the proportion of non-microglia CD45.2 CD45hi cells was higher in PS19Jak2V617F mice (Figure 4B). Immunofluorescent imaging identified a few rare CD45.2+ cells in the hippocampus of PS19Jak2V617F and PS19WT mice (Figure 4C). These data suggest and that Jak2V617F cells have a modestly increased capacity to influx the PS19 brain but comprised a small proportion of brain immune cells when compared to their proportion among blood monocytes (28.91 ± 4.66%, Figure 3B, C).
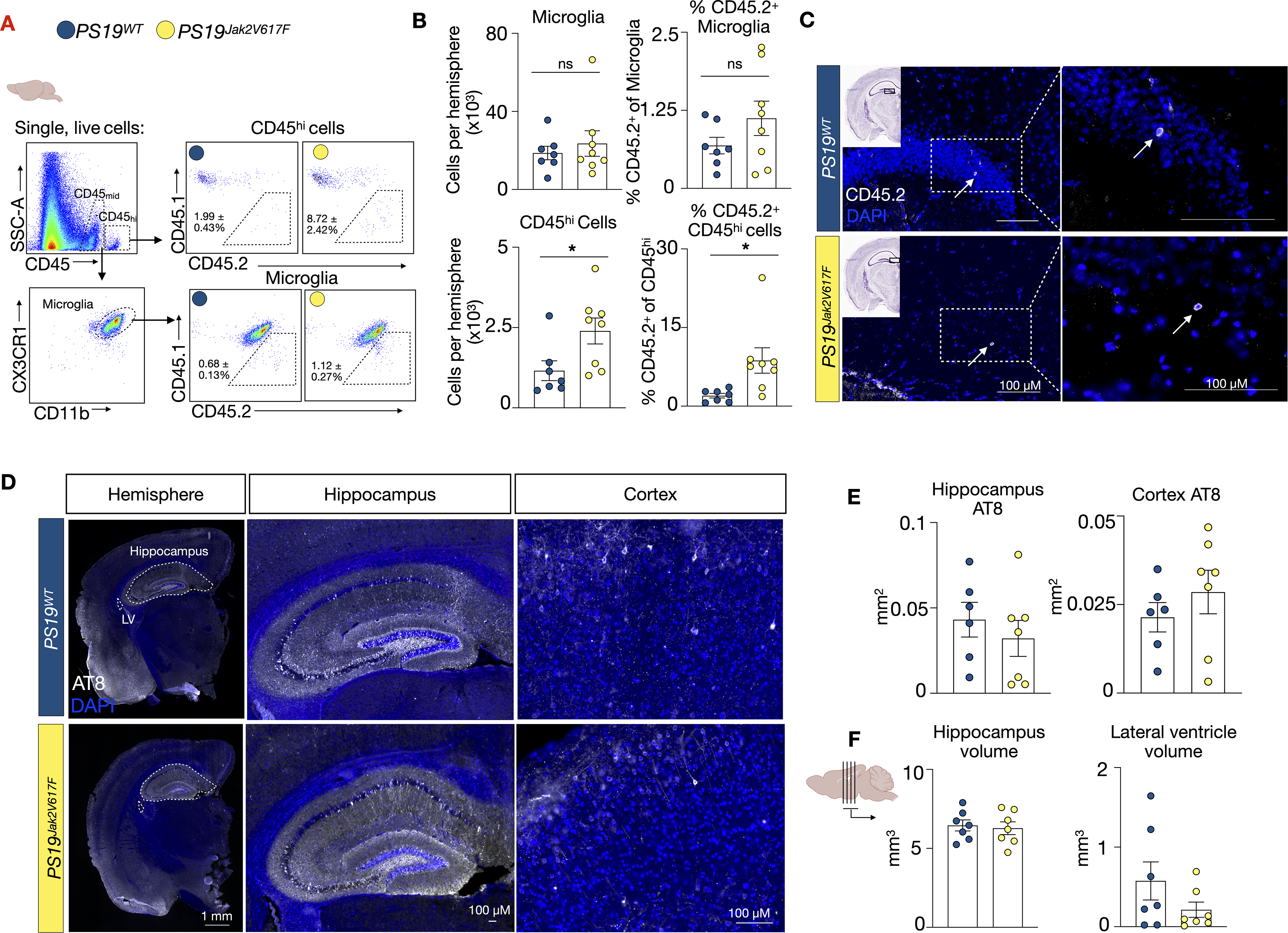
Jak2V617F CH does not alter tau burden or brain atrophy in PS19 mice. (A) Flow cytometry gating strategy in the brain tissue. (B) Flow cytometry quantification of microglia and CD45hi numbers and respective percentages of CD45.2 microglia and CD45hi cells in the brain of PS19WT and PS19Jak2V617F mice (n = 7 PS19WT (5 M/2F), n = 8 PS19Jak2V617F (5 M/3F)). (C) Representative CD45.2 immunofluorescence in the hippocampus of PS19WT and PS19Jak2V617F mice. (D) Representative images of p-tau (AT8) staining. (E) Quantification of AT8 staining in the hippocampus and cortex (n = 6 PS19WT (4 M/2F), n = 7 PS19Jak2V617F (4 M/3F)). (F). Quantification of volume measurements of the hippocampus and lateral ventricle (n = 7 PS19WT (5 M/2F), n = 7 PS19Jak2V617F (4 M/3F)). Data are mean ± s.e.m.; unpaired t-test.
Next, we sought if Jak2 CH modified tau accumulation and brain atrophy in PS19 mice. We imaged tau (AT8) and found equivalent levels in the hippocampus and cortex of PS19Jak2V617F and PS19WT mice (Figure 4E). Further, hippocampal and lateral ventricle volumes were similar in PS19Jak2V617F and PS19WT mice (Figure 4F), suggesting that atrophy and neuronal tissue loss was not affected. Together, these findings demonstrate that Jak2V617F CH does not influence tau accumulation or neurodegeneration in PS19 mice.
Discussion
Studies have reported conflicting data on the association between CH and neurodegenerative diseases.16,17,19–21 Here, we established new murine models of CH in tauopathy that allowed us to distinguish mutant and endogenous non-mutant immune cells via CD45.2 and CD45.1, respectively. Our model uses unconditioned hematopoietic cell transfer in mice with tauopathy, thereby maintaining brain-peripheral immune cell dynamics and reliably establishing Tet2−/− and Jak2V617F CH in PS19 mice with mutant blood cell frequencies that closely align with human clonality. We find that, relative to their WT counterparts, Tet2 mutant myeloid cells display similar and limited capacity to influx the brain of PS19 mice, even at advanced stages of disease. Glia with Tet2 deficiency do, however, retain the ability for exaggerated inflammation and inflammasome activity. Jak2V617F myeloid cells appear to have a modestly increased ability to be retained in the PS19 brain. Despite their low abundance in the brain, it is conceivable that peripheral CH mutant cells could modify tauopathy through long distance signaling from the blood via their immune products like IL-1β. Indeed, we find elevated plasma IL-1β in PS19Jak2V617F mice. Our data, however, demonstrate that in PS19 mice with robust Tet2 and Jak2V617F CH, tau accumulation and neurodegeneration are unaffected. These observations support the conclusion that CH does not alter the development brain pathology in a murine model of tauopathy.
It is surprising that Jak2 and Tet2 CH does not influence murine tauopathy positively or negatively. As such strong drivers of inflammation and inflammatory programming, CH might conceivably aggravate neuronal loss or promote clearance of debris. However, these hypotheses necessitate cell entry into the brain in robust numbers. Our data suggest that in murine tauopathy with CH dynamics mimicking human expansion and VAFs, mutant cells are constrained to the periphery. Complementary analyses in alternate murine models, including alternative BM transplantation systems, along with well-characterized human cohorts that integrate CH genotyping with detailed clinical phenotyping and brain pathological scoring across neurodegenerative subtypes will be essential to disentangle mutation-specific effects and their relevance to human disease. Continued exploration in mouse models and human populations will be key to inform risk stratification strategies and support the integration of somatic mutation profiling into personalized approached for diagnosis, prognosis, and therapy in neurodegenerative disease.
Further human studies evaluating CH and AD are warranted as data linking CH to neurodegenerative disease remains complex. Yet, a consensus is emerging on the need to evaluate gene-specific effects and the involvement of neuroinflammatory programs.16,19,20 Brain pathobiology as well as disease stage and severity should be considered. It is possible that CH does not influence the instigation of AD but may impact its neuroinflammatory pathology and rate of cognitive decline at later stages of disease. Indeed, neurodegenerative conditions encompass distinct pathological mechanisms, ranging from neurotrophic, neurodegenerative, neuroinflammatory, and Aβ versus tau driven pathology, raising the possibility that specific CH mutations may differentially influence disease onset or progression depending on the underlying neurobiology. The contribution of CH mutant lymphoid cells should also be addressed which may depend on transplantation model used (unconditioned vs. irradiation). Moreover, robust evaluation on the capacity of mutant myeloid cells to enter the neurovascular unit or the brain parenchyma and its surrounding meninges, pia, and dura in humans will offer new mechanistic insights.
Additional causal investigations between CH and ADRDs using mouse models are also warranted. High clone VAFs along with a secondary inflammatory challenge may enhance the effects of CH on ADRD, 20 therefore in experimental and epidemiological investigations, stratification based on clone size and comorbidities (like CVD) will be needed. Other germline genetic susceptibilities, like APOE genotype and ancestry, may not only impact how CH alters AD but also how AD may change CH incidence or emergence.17,21,32 In such investigations, careful consideration of experimental design is needed. The PS19 CD45.1/2 dual-allelic unconditioned transfer system established here not only permits precise tracking of donor-derived cells but also provides a valuable platform for future studies investigating immune cell dynamics, including turnover, competition, and brain infiltration in neurodegenerative disease. Our system maintains natural central-peripheral immune cell dynamics, BBB integrity, and brain cellular health. Beyond modeling CH, this system offers an avenue for tracking cell fate and function within complex tissue environments, particularly in neurodegenerative models where peripheral-to-central immune crosstalk is increasingly implicated. 33
Limitation of the study
Murine models offer invaluable tools to study human disease. Although imperfect, they are robust tools for mechanistic evaluations if human disease and pathobiology is recapitulated as closely as possible. However, immune dynamics differ between PS19 mice and human tauopathy. While we do not observe CH cell influx into the murine tauopathy brain using our unconditioned transplant system, in humans with advanced disease, the BBB may be more permissible to cell infiltration. Further, different transplantation systems may have divergent effects on the impact of CH mutation in the lymphoid lineage. CH may also have differing effects in Aβ driven AD where CH might modify disease outcomes. 20 Our studies do not model secondary inflammatory challenges or comorbid disease. For example, people with AD often have comorbid metabolic and cardiovascular complications which can influence inflammatory parameters. We study two common forms of CH but mutation-dependent responses are likely and further mechanistic investigations in mice should assess the contribution of Dnmt3a, Asxl1, p53, and other CH mutation on tauopathy. Finally, we use an Mx1-Cre inducible system to initiate Tet2 deletion or Jak2V617F in donor mice. Future studies could rely on constitutive mouse models as hematopoietic donors.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261458734 - Supplemental material for Tet2 and Jak2 clonal hematopoiesis do not modify murine tauopathy
Supplemental material, sj-docx-1-alz-10.1177_13872877261458734 for Tet2 and Jak2 clonal hematopoiesis do not modify murine tauopathy by Lena Gaebel, Teresa Gerhardt, Annie Khamhoung, Pacific Huynh, Merlin Heiser, Niki F. Brisnovali, Walter Jacob, Abi G. Yates, Aaron Douglas, Trevor Fidler, Leigh Goedeke and Cameron S. McAlpine in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Ethical considerations
All experimental procedures were approved and conducted in accordance with guidelines set by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai.
Consent to participate
Not applicable.
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the a Walter Benjamin fellowship by the German Research Foundation DFG GE 3588/1-1 (to T.G.); MD fellowship from the Boehringer Ingelheim Fonds (to M.H.); an American Heart Association postdoctoral fellowship 24POST1196847 (to P.H.); NIH R01HL178835, R01HL158534, R01AG082185, the American Heart Association 25TPA147787, the Cure Alzheimer's Fund, and the Alzheimer's Association 25AARG-1378979 (to C.S.M.).
Alzheimer's Association, Cure Alzheimer's Fund, National Institute on Aging, National Heart, Lung, and Blood Institute, (grant number 25AARG-1378979 , R01AG082185, R01HL158534, R01HL178835).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data and methods are available and described within. Materials are available upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.