
Abstract
Background
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia worldwide. Beyond amyloid-β and tau pathology, accumulating evidence indicates that chronic neuroinflammation, oxidative stress, mitochondrial dysfunction, disrupted calcium homeostasis, and diabetes are critically contributing to disease onset and progression, underscoring the need to identify modulatory factors that influence multiple pathogenic pathways.
Objective
This review evaluates the role of vitamin D in modulating cognitive function and its association with the pathophysiology of AD.
Methods
This narrative review examines the connection between vitamin D and cognitive and neurological outcomes by integrating findings from preclinical and clinical studies. Relevant literature was identified through a PubMed search, restricted to English language publications available up to 2025.
Results
Vitamin D, a secosteroid hormone traditionally associated with bone metabolism, has emerged as a potential neuroprotective factor in the aging brain. Vitamin D receptors are widely expressed in neurons and glial cells, and vitamin D readily crosses the blood–brain barrier. Despite increasing evidence linking vitamin D deficiency to AD, its mechanistic roles in neuroinflammation and metabolic dysfunction, as well as its translational relevance, remain incompletely understood. Experimental and clinical studies suggest that vitamin D modulates amyloid metabolism, microglial activation, inflammatory signaling, redox balance, mitochondrial function, synaptic integrity, and regulates insulin resistance.
Conclusions
In this review, we integrate evidence from molecular, cellular, animal, and human studies to examine the role of vitamin D in AD. Finally, we highlight current limitations and knowledge gaps and outline future directions for clinical and translational research.
Introduction
Alzheimer's disease (AD) is a complex, age-related neurodegenerative disorder that affects millions of people worldwide. According to the Centers for Disease Control and Prevention (CDC), an estimated 5.8 million Americans were living with AD in 2020, and this number is projected to nearly double every five years as the population ages. 1 Neuropathologically, AD is characterized by the accumulation of amyloid-β (Aβ) plaques, hyperphosphorylated tau tangles, mitochondrial dysfunction, oxidative stress, cholinergic deficits, and neuroinflammation, all of which interact to drive disease progression. 2 Among these, alterations in Aβ metabolism remain central to the amyloid hypothesis, with Aβ40 and Aβ42 being the most studied isoforms. Notably, Aβ42 has a greater propensity to aggregate and is the predominant toxic species found in neuritic plaques. 3 While extensive research has emphasized the roles of Aβ and tau, these factors alone cannot fully account for the complexity of AD pathogenesis, particularly in the sporadic form, which likely arises from a combination of gene–environment interactions and epigenetic modifications in the aging brain.4,5
In recent years, vitamin D has gained increasing attention as a potential neuroprotective factor in AD. Traditionally recognized for its role in calcium regulation and bone health, vitamin D also influences brain function via the vitamin D receptor (VDR), which is widely expressed throughout the central nervous system (CNS). Epidemiological studies have associated low serum vitamin D levels with an elevated risk of cognitive decline and dementia.6–8 Moreover, preclinical evidence suggests that vitamin D exerts neuroprotective effects by modulating neuroinflammation, enhancing neuronal survival, regulating calcium and redox homeostasis, and mitigating Aβ toxicity. 9
Despite the growing number of studies investigating vitamin D in the context of AD, existing reviews have largely focused on isolated aspects, such as vitamin D deficiency, cognitive outcomes, or epidemiological associations. A comprehensive synthesis integrating neuroinflammatory, metabolic, mitochondrial, Insulin resistance, and calcium-related mechanisms within the broader framework of AD pathology remains limited. Given the multifactorial nature of AD, an integrated perspective may provide a more informative understanding of how vitamin D influences disease-relevant pathways.
Accordingly, the present review aims to summarize and integrate current molecular, cellular, animal, and human evidence regarding the role of vitamin D in AD. By linking immune modulation, redox regulation, mitochondrial function, and neuronal homeostasis, this review aims to provide a balanced, disease-relevant overview that may inform future mechanistic and translational studies. This review aims to synthesize current evidence regarding the neuroprotective functions of vitamin D in AD, with particular focus on its capacity to modulate key pathological pathways. By highlighting both preclinical and clinical findings, we seek to evaluate the therapeutic potential of vitamin D in slowing or altering the course of AD and to outline future research directions necessary to clarify its role in disease prevention and treatment.
Vitamin D and its relevance to dementia and AD
Vitamin D is a fat-soluble secosteroid that can be obtained through dietary intake or synthesized in the skin upon exposure to ultraviolet B (UVB). In humans, the two primary forms cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2) undergo sequential hydroxylation: first in the liver to produce 25-hydroxyvitamin D [25(OH)D, Calcidiol], the main circulating form and biomarker of vitamin D status, and subsequently in the kidneys to generate the hormonally active form, 1,25-dihydroxyvitamin D [1,25(OH)2D, calcitriol], catalyzed by 1α-hydroxylase (CYP27B1). Beyond the kidneys, several extrarenal tissues, including the brain, also express CYP27B1, allowing local synthesis of calcitriol. 10
Vitamin D readily crosses the blood–brain barrier (BBB), and both neurons and glial cells express VDR, enabling genomic and rapid non-genomic signaling. In the CNS, calcitriol modulates calcium homeostasis, supports neurotrophic factor production, regulates immune responses, and reduces oxidative stress—functions critical for neuronal survival. Enzymes involved in vitamin D catabolism, such as CYP24A1, are also expressed in the brain, suggesting tightly regulated intracerebral vitamin D metabolism.6,11
Accumulating evidence links vitamin D deficiency to increased risk of cognitive decline and dementia, particularly AD. Proposed mechanisms include dysregulation of calcium signaling, mitochondrial dysfunction, neuroinflammation, and impaired clearance of Aβ peptides.12,13 Experimental studies indicate that vitamin D enhances Aβ phagocytosis by macrophages and microglia, reduces Aβ-induced oxidative injury and inflammation, and preserves synaptic integrity. 14 In animal models, vitamin D supplementation improves learning and memory, lowers Aβ burden, and attenuates glial activation. 15
Although both vitamin D2 and D3 can raise serum 25(OH)D levels, their relative bioavailability and efficacy remain debated, with some studies suggesting D3 is more potent. 16 Given the high prevalence of vitamin D deficiency and its potential neuroprotective effects, optimizing vitamin D status could represent a promising adjunctive strategy for the prevention or management of AD.
An important unresolved question is whether vitamin D deficiency acts as a cause of AD development or is a secondary effect of disease progression. Older individuals or patients with cognitive impairment may have lower circulating vitamin D levels due to reduced sunlight exposure, poor nutrition, and decreased physical activity. On the other hand, experimental evidence indicates that inadequate vitamin D signaling might worsen neuroinflammation, oxidative stress, and synaptic dysfunction, thereby speeding up neurodegenerative processes.
It is therefore plausible that the relationship between vitamin D and AD is bidirectional, forming a self-reinforcing cycle in which disease-related changes reduce vitamin D availability, while vitamin D insufficiency further amplifies pathological mechanisms. Longitudinal studies assessing vitamin D status before disease onset will be essential to clarify temporal relationships and causality.
Vitamin D and its metabolic mechanisms
The discovery of vitamin D traces back to the late nineteenth century during England's Industrial Revolution, when a widespread outbreak of rickets affected many individuals. In 1918, Professor Edward Mellanby identified the underlying cause of rickets as a deficiency in essential nutrients. 17 This discovery led to the therapeutic use of cod liver oil, which effectively treated children afflicted with the condition. In 1922, Elmer McCollum isolated 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), the active form of vitamin D, which was subsequently named vitamin D, following the earlier identification of vitamins A, B, and C. 18 Vitamin D is a fat-soluble secosteroid that can be obtained through dietary intake or synthesized in the skin upon exposure to UVB.19–21
It plays a pivotal role in numerous biochemical pathways and is associated with a broad spectrum of health conditions, including brain function. In humans, the two primary forms of cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2) undergo sequential hydroxylation: first in the liver to produce calcidiol, and subsequently in the kidneys to generate the hormonally active form calcitriol, catalyzed by CYP27B1. 22 Beyond the kidneys, several extrarenal tissues, including the brain, also express CYP27B1, allowing local synthesis of calcitriol. 8 The metabolic pathways of vitamin D are illustrated in Figure 1.
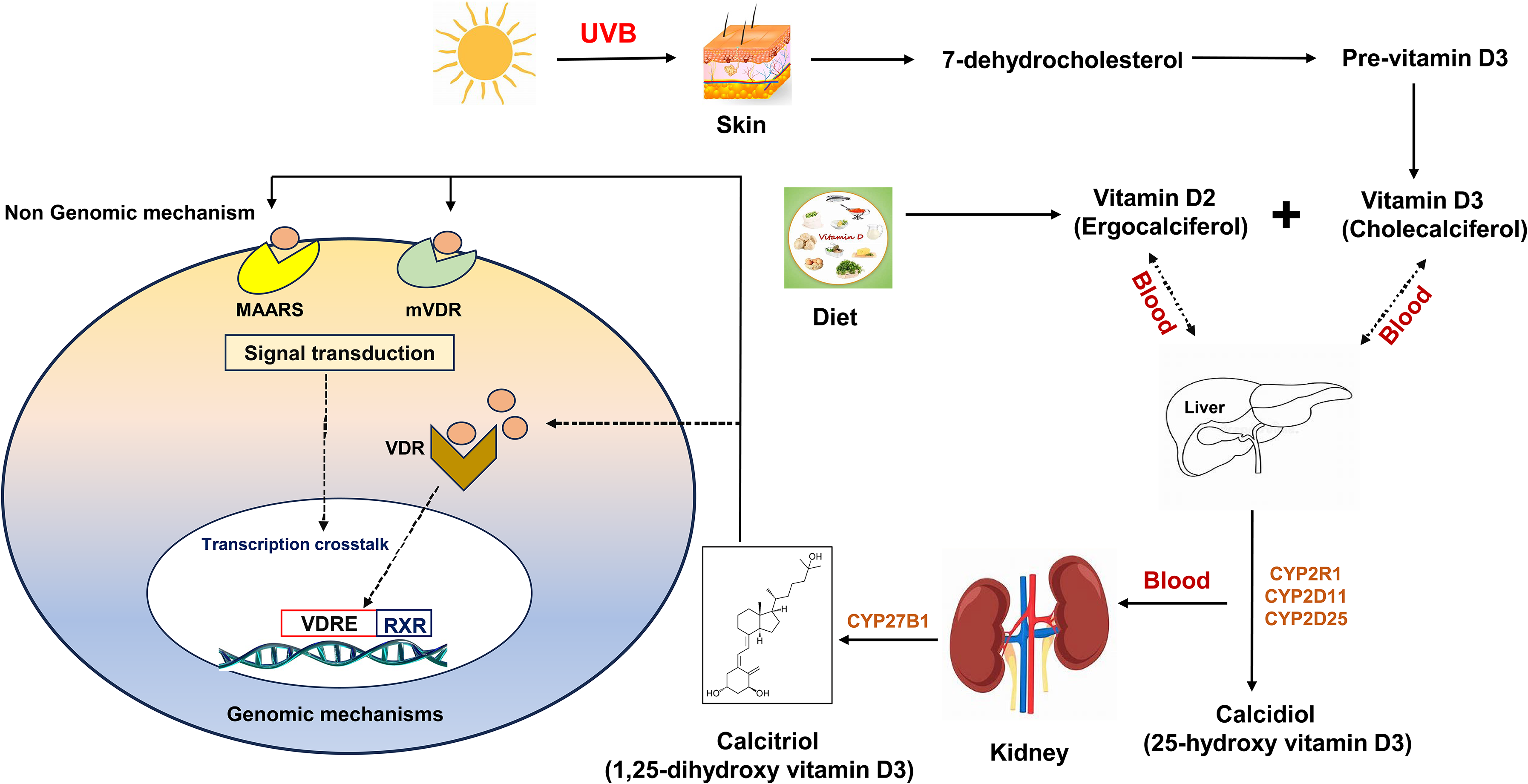
Vitamin D metabolism and mechanism of action.
Calcitriol, the biologically active form of vitamin D, regulates a wide range of physiological processes in the body. The concentration of circulating 1,25(OH)2D3 is tightly controlled by feedback mechanisms influenced by parathyroid hormone, calcium ions (Ca2+), and cytokines. Recent evidence indicates that, beyond renal cells, several other cell types, including keratinocytes in the skin, monocytes and macrophages, osteoblasts in bone, and cells of the prostate and colon, can perform the second hydroxylation step of vitamin D metabolism. 23 Notably, 25(OH)D can cross the BBB, and both neurons and glial cells express the VDR, allowing for genomic and rapid non-genomic signaling. 6 Within the CNS, 25(OH)D can be converted into its active form, 1,25(OH)2D, by Cytochrome P450 Family 27 Subfamily B Member 1 (CYP27B1), or metabolized into its inactive form, calcitroic acid, by CYP24A1.11,24 Additionally, the VDR mediates transcriptional regulation upon binding to the bioactive form, 1,25(OH)2D.25,26
Research has established a strong association between vitamin D deficiency and an increased risk of cognitive decline and AD. 6 Dysregulation of vitamin D metabolism contributes to AD pathology through disturbances in calcium homeostasis, immune regulation, and neuroprotection. 27 The active metabolite, 1,25(OH)2D, synthesized from 25(OH)D in the kidneys and other tissues, is essential for calcium absorption and neuronal function.28,29 Impaired calcium signaling, a hallmark of AD, can promote neuronal dysfunction and amyloid plaque accumulation. 29
Beyond calcium regulation, vitamin D influences immune responses and neuroinflammation, another central feature of AD. 30 VDRs are widely distributed throughout the brain, where active vitamin D regulates neurotrophic factors and supports neuronal survival. 31 Experimental studies show that vitamin D enhances Aβ clearance by stimulating macrophage phagocytosis, while reducing Aβ-mediated toxicity, neuronal death, and inflammation.12,13 Supplementation studies further support these effects: vitamin D has been shown to improve cognition in AD patients, 8 and dietary vitamin D2 from mushrooms reduced Aβ deposition, astroglial activation, and pro-inflammatory cytokine production while enhancing interleukin (IL)-10 expression in mouse models.12,13,15 Moreover, combined therapy with vitamin D and memantine protected cortical neurons against Aβ- and glutamate-induced axonal degeneration. 32
Apart from this, recent studies have identified novel pathways of vitamin D3 activation initiated by CYP11A1 and CYP27A1 that produce biologically active hydroxymetabolites of vitamin D3, lumisterol 3 (L3) and tachysterol 3 (T3).33,34 These metabolites exert diverse functions, including antioxidants, anti-inflammatory, and antiproliferative activities, and act through various nuclear receptors such as VDR, Aryl hydrocarbon receptor, Liver X receptor, and peroxisome proliferator-activated receptor gamma. 34 Additionally, they modulate key signaling pathways, including NRF2 and p53 activation, and inhibition of NF-κB, IL-17, and Wnt/β-catenin signaling, suggesting broader roles in both skin, systemic disease and could be protective in neurological disorders.
Emerging evidence highlights vitamin D's role in maintaining mitochondrial health. By regulating energy metabolism, reducing oxidative stress, and preserving mitochondrial integrity, vitamin D mitigates two major drivers of AD pathogenesis: disrupted calcium signaling and mitochondrial dysfunction. 35 Its ability to modulate oxidative and inflammatory pathways underscores its therapeutic potential. 36 Collectively, these findings suggest that enhancing vitamin D metabolism may provide a novel strategy to prevent or slow AD progression by supporting neuronal survival, reducing neuroinflammation, and preserving mitochondrial function.
Amyloidosis and the modulatory role of vitamin D in AD
Amyloidopathy is a central feature of AD, driven by dysregulated production, degradation, and clearance of Aβ peptides. 37 The proteolytic cleavage of amyloid-β protein precursor (AβPP) is mediated primarily by β-secretase (BACE-1) and γ-secretase. 38 Non-amyloidogenic processing via α- and γ-secretase produces soluble AβPPα and P3 fragments, whereas amyloidogenic cleavage by BACE-1 and γ-secretase generates Aβ40/42 monomers that aggregate into oligomers and plaques. 39 Soluble Aβ oligomers are now considered the most synaptotoxic species, correlating strongly with excitatory synapse loss, disrupted neurotransmission, and memory deficits. Chronic microglial activation further exacerbates synaptic elimination, while Aβ-driven upregulation of extracellular matrix components such as chondroitin sulfate proteoglycans impedes synaptic remodeling.40,41 In addition, Aβ induces oxidative stress, microglial activation, and neuroinflammation, all of which contribute to progressive neurodegeneration. 42
Vitamin D has emerged as a regulator of AβPP metabolism and Aβ clearance. 1,25(OH)2D enhances macrophage phagocytosis of soluble Aβ via genomic (VDR-mediated transcription) and non-genomic (ion channel–mediated) signaling.43,44 It also reduces Aβ aggregation, promotes its transport across the BBB, and decreases plaque burden. 45 Furthermore, vitamin D–VDR interactions influence AβPP processing through SMAD3 and the TGF-β pathway, 46 underscoring vitamin D's potential to modulate amyloid toxicity and support neuronal and mitochondrial health.
Vitamin D deficiency exacerbates calcium dysregulation
Aberrant neuronal Ca2+ homeostasis is a hallmark of AD and contributes to synaptic dysfunction and memory loss. 47 In transgenic AD models, neurons near plaques display elevated intracellular Ca2+, impairing short-term memory while sparing basic sensory processing. 48 Mechanistic studies implicate Aβ interactions with multiple signaling cascades: binding to prion protein (PrPC)–mGluR5 complexes, activation of calcium-sensing receptors, and stimulation of PLC-dependent InsP3 pathways.49–52 Aβ-mediated activation of mGluR5 also enhances long-term depression, further compromising synaptic plasticity. 53 Familial AD mutations in presenilin-1/2 amplify InsP3 receptor sensitivity, exaggerating ER Ca2+ release and increasing Aβ42 generation.53,54 Genetic downregulation of InsP3 receptors in presenilin mutant mice alleviates aberrant Ca2+ signaling and rescues memory deficits, highlighting the InsP3/Ca2+ axis as a critical contributor to AD progression.55,56
Vitamin D deficiency exacerbates Ca2+ dysregulation. Low serum cholecalciferol and VDR polymorphisms are linked to cognitive decline and AD risk. 57 Vitamin D regulates Ca2+ homeostasis by upregulating plasma membrane Ca2+ ATPase, sodium-calcium exchangers, and Ca2-binding proteins such as calbindin and parvalbumin, all of which are reduced in AD brains.58–61 It also limits Ca2+ influx through L-type calcium channels. 62 Vitamin D deficiency has been shown to elevate intracellular Ca2+ levels and promote the formation of oligomeric Aβ, which in turn drives further Aβ generation and accumulation, creating a self-perpetuating Aβ/Ca2+ feedback loop that accelerates AD pathology. 63 Consistent with this mechanism, inhibition of ryanodine receptor 2 (RYR2) by dantrolene reduces Ca2+ release and attenuates Aβ formation. 63 These findings strengthen the evidence linking vitamin D deficiency to a heightened risk of AD and its clinical manifestations. Figure 2 illustrates the proposed mechanism through which vitamin D deficiency exacerbates Ca2+-mediated AD progression.
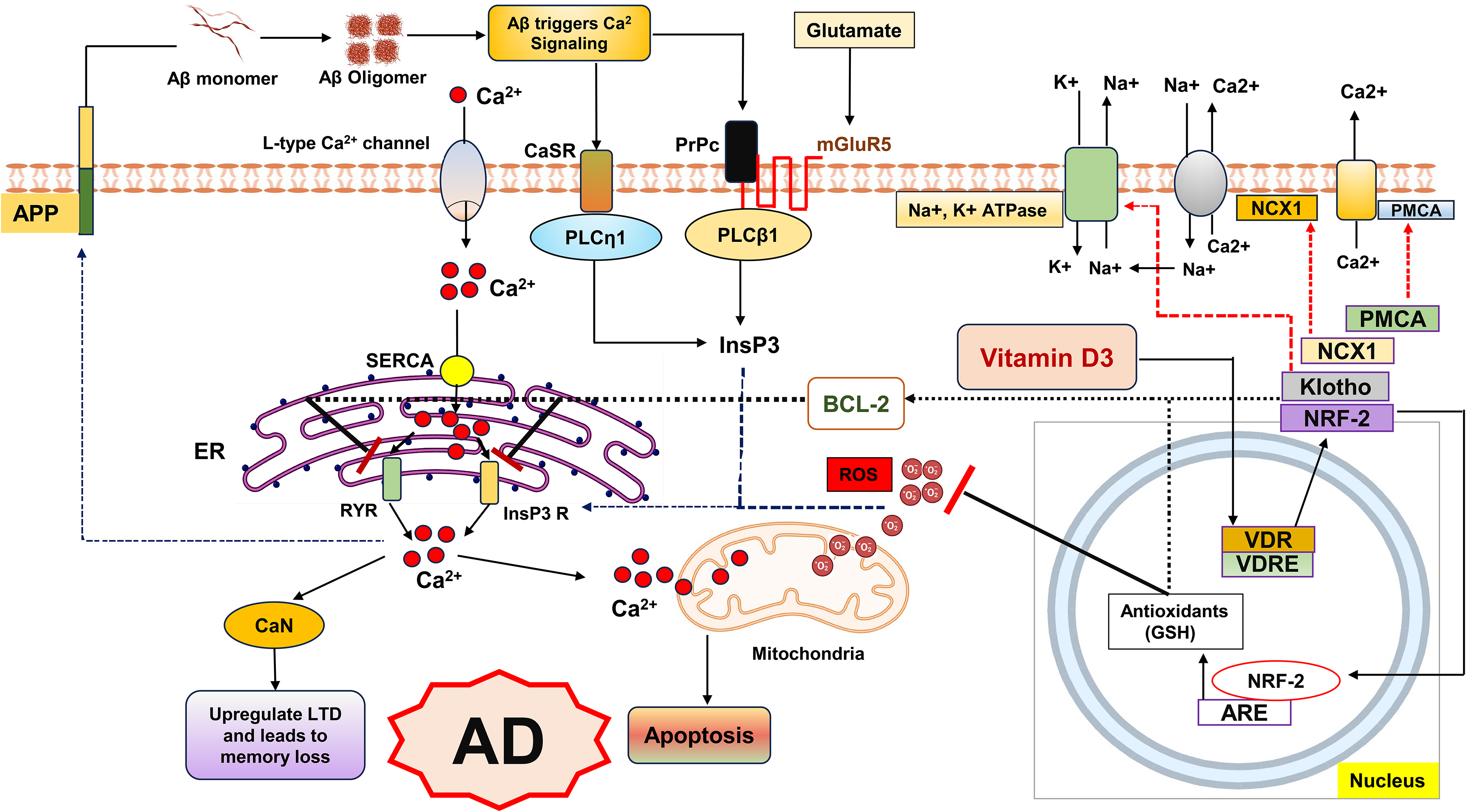
Dysregulated Ca2+ and redox signaling in Alzheimer's disease (AD).
Vitamin D functions in oxidative stress and mitochondrial dysfunction
Maintaining adequate vitamin D levels is essential for mitigating oxidative stress and preserving cellular homeostasis. A deficiency in serum 25(OH)D impairs antioxidant defense, leading to increased intracellular damage and apoptosis. A critical mechanism involves mitochondrial-derived reactive oxygen species (ROS), which inversely correlate with nuclear factor erythroid 2-related factor 2 (Nrf2), a master regulator of antioxidant responses tightly modulated by vitamin D. 64 Disruption of this redox balance contributes to uncontrolled cell death, neurodegeneration, and accelerated aging. 65
In AD, vitamin D deficiency has been associated with reduced levels of both Nrf2 and klotho, two key mediators of neuroprotection. 58 Nrf2 is markedly diminished in AD, and its hippocampal expression in transgenic mice has been linked to cognitive restoration. 61 By maintaining intracellular glutathione (GSH) levels and promoting the expression of anti-apoptotic proteins such as Bcl-2, Nrf2 helps combat oxidative stress and regulate Ca2+ homeostasis through inhibition of IP3R and RyR-mediated Ca2+ release.66–69 Similarly, klotho, which is decreased in the cerebrospinal fluid of AD patients, supports neuronal survival, regulates Aβ metabolism, and its reduction has been associated with cognitive impairment and Aβ1–42 accumulation.70,71 Collectively, these findings suggest that vitamin D, Nrf2, and klotho form an interconnected regulatory axis that protects neurons by maintaining redox balance, Ca2+ stability, and anti-apoptotic signaling.
The vitamin D–Nrf2 axis also interacts with the peroxisome proliferator-activated receptor coactivator 1α (PGC-1α)/SIRT3 pathway, a central regulator of mitochondrial health. Calcitriol modulates this pathway by upregulating SIRT3 expression through Nrf2 and PGC-1α, thereby enhancing mitochondrial antioxidant defenses and stimulating anti-inflammatory cytokine production. 67 This Nrf2/PGC-1α/SIRT3 signaling network safeguards mitochondrial function, protects against oxidative damage, and maintains ROS homeostasis (Figure 3).
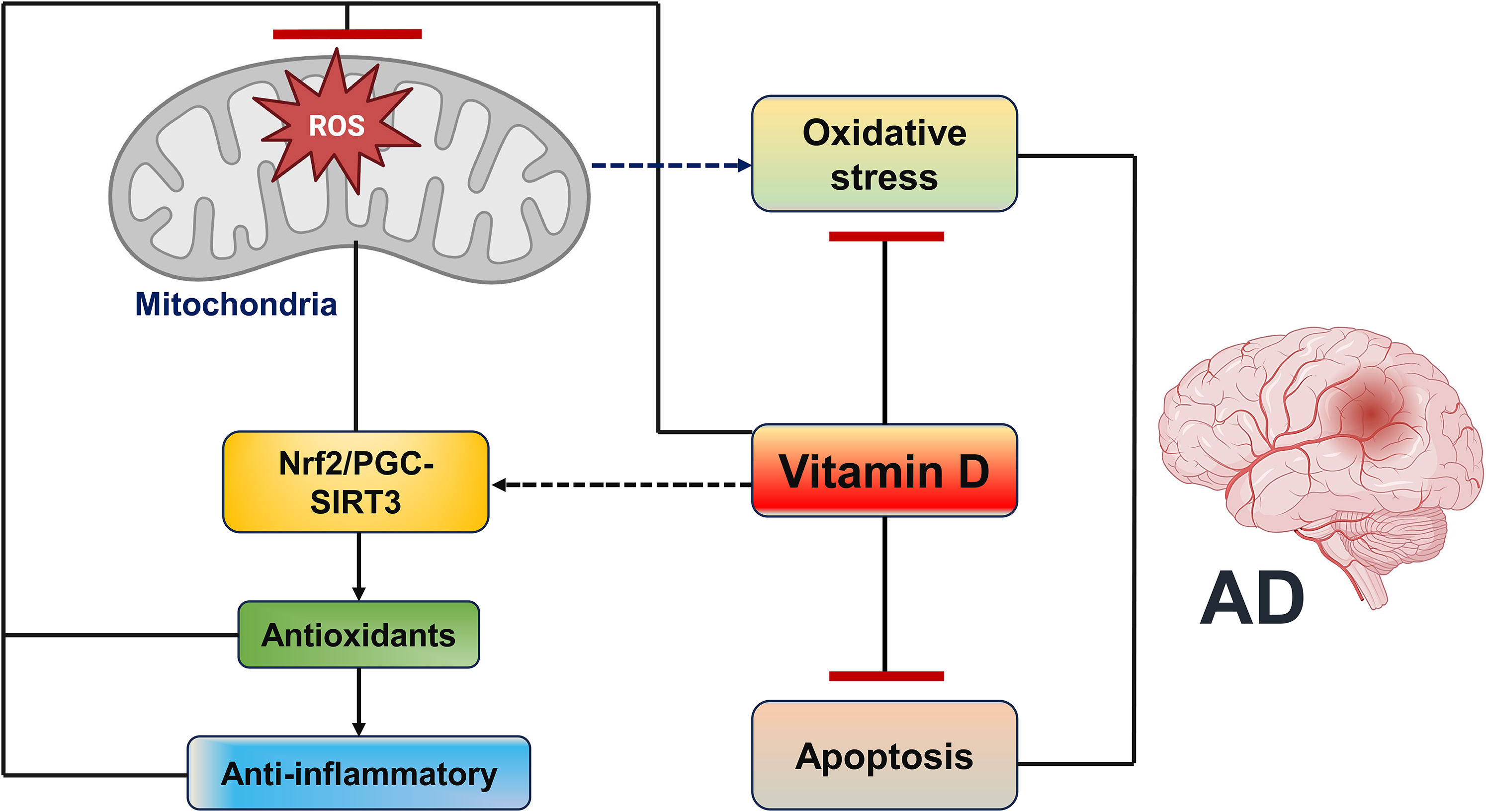
Antioxidant and anti-apoptotic effects of vitamin D.
Mitochondrial dysfunction, including cristae loss, impaired ATP production, and excessive mitophagy, is a hallmark of AD pathology. Vitamin D supplementation improves mitochondrial health by preserving cristae structure, reducing mitophagy, and enhancing mitochondrial biogenesis via the Nrf2/PGC-1α/SIRT3 pathway. 72 In AD models, vitamin D restores mitochondrial function, decreases oxidative stress, and improves cognition. Other antioxidants such as resveratrol, curcumin, CoQ10, and N-acetylcysteine also support mitochondrial integrity and redox balance, underscoring their potential as complementary therapies against AD-related mitochondrial dysfunction. 73
Neuroinflammation and the anti-inflammatory effects of vitamin D
Neuroinflammation in AD arises from disruptions in the interplay between the brain, immune system, and gut microbiota, resulting in sustained immune activation. Both Aβ and tau play pivotal roles in driving this process by activating microglia and astrocytes, the principal immune cells of the CNS. Soluble Aβ and aggregated plaques act as potent triggers of microglial activation, leading to the release of pro-inflammatory cytokines (IL-1β, TNF-α), ROS, and reactive nitrogen species, which contribute to neuronal injury, tau hyperphosphorylation, and aggregation. 74 Hyperphosphorylated tau itself can further stimulate microglia even before neurofibrillary tangle formation, perpetuating a vicious cycle of neuroinflammation and tau pathology. Similarly, astrocytes activated by Aβ and tau release cytotoxic mediators that exacerbate neuronal damage. Although both microglia and astrocytes exhibit protective roles through phagocytosis and degradation of pathological proteins, their prolonged activation results in a chronic inflammatory state that diminishes clearance capacity and accelerates neurodegeneration.42,75,76 Thus, the dual roles of glial cells, initially protective but ultimately neurotoxic under sustained stimulation, highlight their central involvement in Aβ- and tau-driven AD pathology.
Microglial interactions with Aβ oligomers via Toll-like receptors (TLRs) activate intracellular signaling cascades, including MyD88 and NF-κB, which drive NLRP3 inflammasome activation and the release of IL-1β and IL-18. 77 Notably, NLRP3 activation suppresses microglial phagocytic function, thereby promoting Aβ accumulation. Genetic or pharmacological inhibition of NLRP3 enhances Aβ clearance, reduces neuroinflammation, and improves cognitive outcomes, positioning NLRP3 as a promising therapeutic target in AD. 78 Activated microglia further secrete pro-inflammatory cytokines (IL-1β, IL-6, TNF-α, IFN-γ), which enhance β-site APP-cleaving enzyme 1 (BACE-1) activity and increase Aβ production. 76 Aβ accumulation in turn elevates ROS levels, amplifying oxidative stress and neuronal cell death. Moreover, microglia release mediators that activate astrocytes, leading to elevated CCL2 expression and disruption of the BBB via downregulation of tight junction proteins. 79 This increased BBB permeability facilitates the infiltration of peripheral immune cells and inflammatory factors into the CNS.79–81 Activated astrocytes also contribute to neurofibrillary tangle formation. For instance, Aβ-induced NF-κB activation in astrocytes upregulates complement component 3 (C3), which subsequently activates C3a receptors on neurons and microglia, driving further neurotoxicity. 81
Over the past decade, vitamin D has emerged as a promising modulator of neuroinflammation in AD. Its active metabolite, 1,25(OH)2D3, and precursor, 25(OH)D3, exert both anti-inflammatory and antioxidant effects. 82 1,25(OH)2D3 suppresses pro-inflammatory mediators such as IL-6, TNF-α, and nitric oxide (NO), predominantly produced by activated microglia, 83 while promoting the expression of anti-inflammatory cytokines, including IL-10, IL-4, and TGF-β. 84 Furthermore, 1,25(OH)2D3 modulates immune signaling by upregulating TLR-10 and downregulating TLR-2/4, thereby polarizing microglia toward an anti-inflammatory phenotype with reduced IL-1β, TNF-α, and iNOS expression. 84 Vitamin D3 also increases VDR expression in the prefrontal cortex and upregulates CYP27B1 in microglia, enhancing responsiveness to cholecalciferol and reducing IL-6, IL-12, and TNF-α production. In addition, vitamin D3 attenuates NF-κB signaling via MKP-1 activation. 81 Importantly, VDR interacts with NLRP3 inflammasomes to inhibit their oligomerization and activation. 85 Cholecalciferol suppresses NLRP3 signaling by reducing ASC, caspase-1, and TXNIP levels in the hippocampus, while 1,25(OH)2D3 inhibits the p38/NF-κB/NLRP3 axis.81,84
Besides NLRP3, NLRP1 inflammasomes are also implicated in AD-related neuroinflammation. Both NLRP1 and NLRP3 are co-activated in monocytes of AD patients. 86 Calcitriol has been shown to suppress NLRP1 activity, thereby reducing IL-1β levels via activation of the Nrf2–heme oxygenase-1 (HO-1) antioxidant pathway. Since Nrf2 regulates key antioxidant enzymes and mitigates oxidative stress, a known driver of microglial activation, vitamin D-mediated Nrf2 activation may provide additional neuroprotective effects in AD (Figure 4).
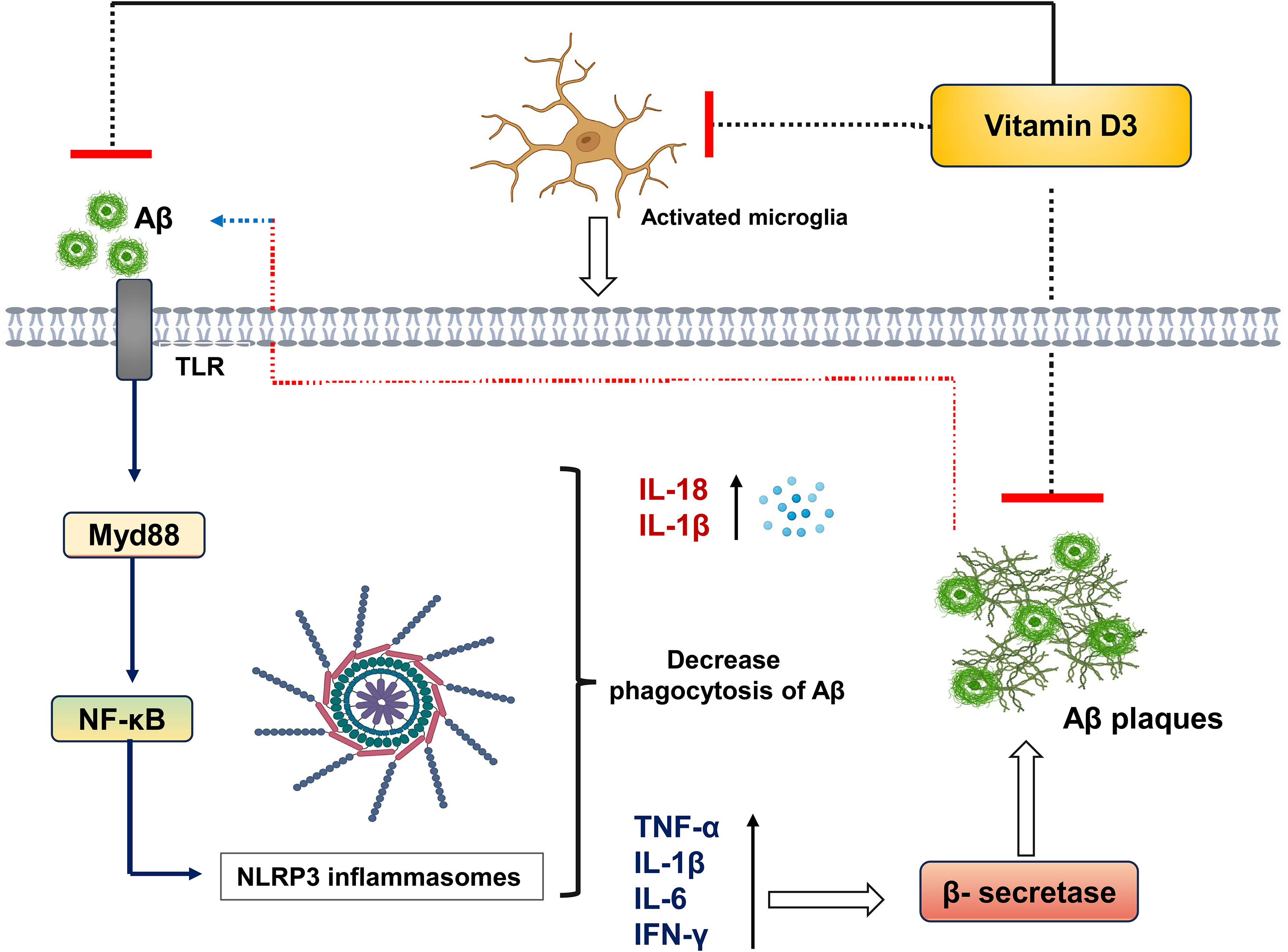
Neuroinflammatory mechanisms in AD. Aβ fibrils activate microglia through TLR–MyD88–NF-κB signaling, inducing NLRP3 inflammasome activation and pro-inflammatory cytokine release. This enhances β-secretase activity, increasing Aβ production, while vitamin D3 suppresses Aβ accumulation and microglial activation.
Vitamin D in diabetes-mediated AD
AD is a progressive pathological condition marked by the gradual decline in memory and cognitive abilities required for daily functioning. A strong association exists between AD and metabolic disorders, specifically type 2 diabetes mellitus. 87 Individuals with diabetes have a significant risk of developing AD, and many patients exhibit impaired glucose metabolism. 88 This has led to the concept of AD as type 3 diabetes, characterized by brain insulin resistance and defective insulin signaling. 89 Insulin plays a central role in neuronal survival, synaptic plasticity, learning, and memory. Disruption of insulin signaling cascades enhances Aβ accumulation and causes tau hyperphosphorylation through the glycogen synthase kinase-3β (GSK-3β), a major kinase involved in tau pathology. 90
Furthermore, insulin resistance and diabetes elevate oxidative stress through ROS production and impair antioxidant defenses, which accelerates neuronal damage and the progression of AD. 91 Oxidative stress plays a key role in neurodegeneration, linking metabolic dysfunction with AD pathology. Vitamin D, mostly known for regulating phosphate and calcium levels, has also proven to be a strong neuroprotective agent. Notably, vitamin D influences critical signaling pathways, including the inhibition of GSK-3β activity, which helps to decrease tau hyperphosphorylation and Aβ toxicity. 90 Moreover, vitamin D demonstrates antioxidant effects by reducing ROS formation, partly through lowering the expression of NADPH oxidase. 92 Vitamin D supplementation has also been reported to reduce the risk of progression of type 2 diabetes in individuals with prediabetes, as shown in the D2d study. 93
Overall, these findings demonstrated that vitamin D may exert protective effects against AD by improving insulin signaling, modulating GSK-3β–mediated pathways, and reducing oxidative stress, highlighting its potential as a therapeutic target in neurodegenerative and metabolic disorders.
VDR in AD: Implications of VDR polymorphism and silencing
Genome-wide, transcriptomic, and proteomic studies have identified multiple genes associated with AD risk, including those involved in neuroinflammation (IL-1, IL-6), oxidative stress (iNOS), and other factors such as VDR, cathepsin, ubiquilin, and acetylcholinesterase. Among these, VDR is of particular interest due to its direct link with vitamin D signaling.94,95 Recent studies have examined the role of VDR polymorphisms such as FokI, BsmI, ApaI, TaqI, Tru9I, and Cdx2 in late-onset AD, and consistently report reduced VDR mRNA levels in the hippocampus of AD patients. 57 Vitamin D and its receptor (VDR) play a critical role in regulating energy metabolism by modulating gene expression of mtDNA-encoded oxidative phosphorylation subunits. It has been found that VDR co-localizes with mitochondria and mtDNA, binds multiple D-loop sites (‘MMHKCA’), and interacts with mitochondrial transcription factor A. This evidence highlights VDR as a significant determinant of metabolic homeostasis and provides important insights into the mechanisms through which vitamin D influences energy balance and cellular function. 96
Reduced expression of VDR and 1,25-MARRS receptors, together with impaired vitamin D metabolism, diminishes the neuroprotective actions of vitamin D, thereby increasing neuronal vulnerability to degeneration and AD pathology. 97 The association between VDR polymorphisms, megalin, and AD progression suggests that disruptions in VDR and 1,25-MARRS signaling pathways contribute to neurotoxicity and neurodegeneration. Collectively, these findings underscore the central role of vitamin D-related signaling in AD.95,98
The VDR knockout (VDR−/−) mouse model, generated by deleting the DNA-binding domain of the VDR gene, has been widely used to investigate impaired vitamin D-VDR signaling in the CNS. 99 First developed at Tokyo University, these mice display shortened lifespan and premature aging, effectively modeling VDR deficiency and its association with accelerated neurodegeneration. VDR−/− mice exhibit cognitive impairments, behavioral abnormalities, and disrupted calcium homeostasis, thereby providing important insights into the neurological impact of impaired VDR signaling.99,100
Evidence indicates that Aβ toxicity downregulates VDR and nerve growth factor (NGF) levels in the brain. This reduction may compromise vitamin D utilization and lower NGF expression, an essential neurotrophin for neuronal survival, synaptic plasticity, and cognitive function. 101 It has been hypothesized that Aβ promotes VDR degradation, although the precise mechanism remains unclear. 101 In AD patients, low serum 25(OH)D levels combined with reduced VDR expression may contribute to pathological cascades, suggesting that adequate 25(OH)D supplementation alone may not restore normal function if VDR signaling is impaired. Furthermore, VDR suppression has been shown to enhance L-type voltage-sensitive calcium channel (LVSCC) activity. Cortical neurons treated with VDR siRNA displayed elevated LVSCC-A1C mRNA and protein levels at 12 and 24 h compared with controls.102–104
VDR-mediated gene regulation involves ligand binding, heterodimerization with the retinoid X receptor (RXR), and subsequent binding of the VDR–RXR complex to vitamin D response elements (VDREs). Genetic polymorphisms within the VDR gene can disrupt these processes, impairing the activation of downstream target genes. 105 Several single-nucleotide polymorphisms, including those identified through restriction enzymes (Tru9I, BsmI, TaqI, ApaI, EcoRV, FokI) and sequencing-based approaches (Cdx2), have been linked to AD susceptibility (Figure 5).106,107 For instance, the ApaI polymorphism but not TaqI was associated with late-onset AD in a Turkish cohort, suggesting that alterations in ligand-binding domains may increase AD risk. However, results remain inconsistent, as other studies have implicated both ApaI and TaqI variants in modifying AD susceptibility. Additionally, the Cdx2 polymorphism has been shown to reduce VDR promoter activity, potentially exacerbating disease risk. Thus, VDR polymorphisms may reduce ligand affinity, impair neurotrophin expression, and compromise the neuroprotective effects of vitamin D.108–110 When combined with other genetic and environmental factors, these alterations may accelerate neuronal aging and neurodegeneration.
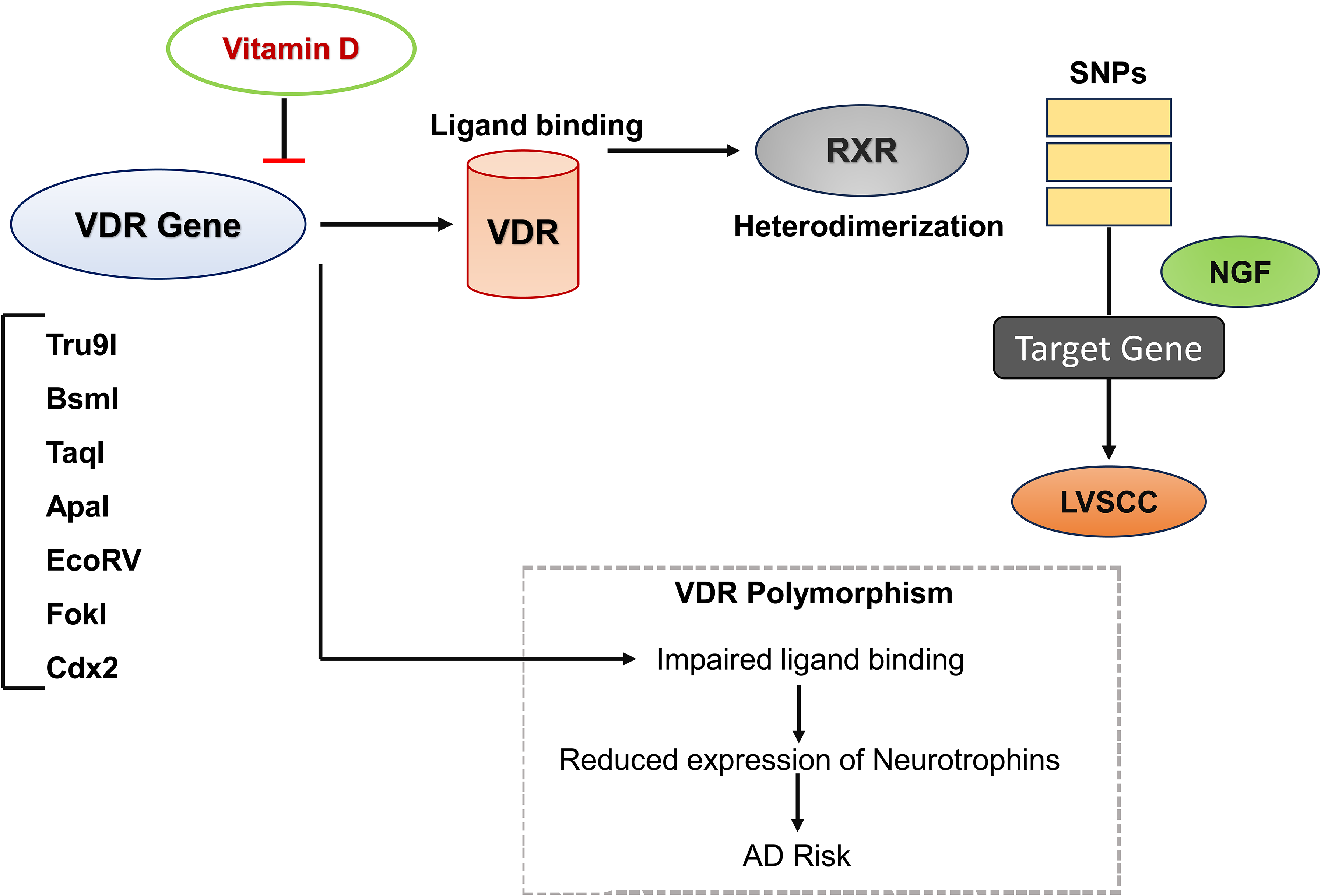
Vitamin D–VDR signaling and disruption by VDR polymorphisms in AD.
Moreover, it has been reported that vitamin D is crucial during pregnancy to support fetal bone development and modulate maternal immune tolerance. Maternal 25(OH)D crosses the placenta and is the primary determinant of neonatal vitamin D status, with deficiency associated with adverse effects such as pre-eclampsia, gestational diabetes, and low birth weight. 111 Emerging reports suggest that maternal VDR polymorphism could influence both maternal and neonatal vitamin D levels, although evidence remains heterogeneous and requires further validation.
Preclinical evidence of vitamin D in modulating AD pathology
Preclinical studies suggest that vitamin D plays a key role in mitigating Aβ accumulation, a central feature of AD. Single-dose administration of vitamin D3 (1 µg) in mouse models enhanced Aβ clearance within 24 h.112,113 Similarly, Durk and colleagues reported that short-term vitamin D3 treatment increased P-glycoprotein (P-gp) expression and reduced soluble Aβ production, while long-term administration (2.5 µg/kg for 8 weeks) decreased both soluble and insoluble Aβ plaques in the hippocampus and improved memory. These effects were reversed by the P-gp inhibitor elacridar, highlighting the importance of P-gp-mediated Aβ transport. Conversely, vitamin D deficiency reduced cerebral P-gp expression, further implicating vitamin D in Aβ clearance. 98 Additional studies demonstrated that calcitriol treatment (in vitro and in vivo) upregulated low-density lipoprotein receptor-related protein 1 (LRP1), enhancing Aβ1–40 efflux across the BBB, indicating that vitamin D promotes Aβ removal.114,115
Vitamin D also modulates neuroinflammation. Briones et al. showed that vitamin D3 administration reduced IL-1β, increased IL-10, and improved learning in aged rats, while calcitriol incubation in cortical neurons decreased iNOS expression and Aβ-mediated cytotoxicity.98,116 Long-term vitamin D3 treatment in 5xFAD mice modulated genes related to learning and memory and reduced Aβ plaques. 117 Moreover, vitamin D3 attenuated oxidative stress and inflammation by regulating β-secretase and AβPP activity, enhancing SIRT1 signaling, and upregulating Nrf2 and HO-1 while reducing TNF-α, IL-1β, and NF-κB.118,119 In Aβ-challenged Wistar rats, vitamin D3 improved cognitive function by boosting antioxidant defenses and reducing lipid peroxidation, while in streptozotocin-induced AD models, it attenuated NF-κB p65 and TNF-α expression and increased antioxidant enzymes, including catalase, superoxide dismutase, and glutathione. 120 Vitamin D deficiency exacerbates Aβ accumulation, impairs microglial clearance, and accelerates memory decline, whereas supplementation reduces Aβ and improves cognition, partly through MAO-B downregulation in 5xFAD mice. 119
Mechanistically, calcitriol activates the PI3 K/Akt pathway, inhibits GSK-3β, and protects SH-SY5Y cells from Aβ-induced ROS production, apoptosis, and tau hyperphosphorylation. Combining vitamin D3 supplementation with physical activity further reduced IL-6, malondialdehyde, Aβ, and tau while enhancing IL-10 and GSH in LPS-induced AD rat models. 119 Vitamin D2 has also demonstrated neuroprotective effects. In wild-type and transgenic mice, dietary vitamin D2 improved memory, reduced Aβ deposition, prevented astroglial activation, and increased hippocampal IL-10. In BV2 microglial cells, vitamin D2 inhibited Aβ25–35-induced iNOS and COX-2 expression, suppressed NF-κB phosphorylation, reduced ROS, and decreased pro-inflammatory cytokines IL-1β, IL-6, and TNF-α.13,15 Furthermore, co-treatment with vitamin D and memantine prevented Aβ- and glutamate-induced axonal degeneration in cortical neurons. 121
Interestingly, in APP/PS1 mice, serum vitamin D remained low despite sufficient dietary intake, suggesting deficiency may result from AD rather than cause it. In these models, supplementation increased Aβ deposition via activation of the non-genomic VDR/p53 pathway rather than restoring the genomic VDR/RXR pathway, highlighting the complex and context-dependent effects of vitamin D in AD. 122 As observed, several preclinical studies employ 1,25(OH)2D, the active hormonal form, which demonstrates strong neuroprotective effects but has limited clinical relevance due to safety concerns, such as hypercalcemia. 22 Conversely,25(OH)D, the major circulating form, better reflects physiological vitamin D status and is more suitable for translational relevance in preclinical models. Table 1 summarizes the preclinical evidence supporting the protective effects of vitamin D in AD.
Protective role of vitamin D in AD: preclinical studies.

1,25(OH)2D3: 1,25-dihydroxyvitamin D3; AChE: acetylcholinesterase; AD: Alzheimer's disease; Aβ: amyloid-β; BBB: blood-brain barrier; BDNF: brain-derived neurotrophic factor; CAT: catalase; COX-2: cyclooxygenase-2; DIV: days in vitro; GSH: glutathione; ICV: intracerebroventricular; IL: interleukin; iNOS: inducible nitric oxide synthase; NF-κB: Nuclear factor kappa B; NGF: nerve growth factor; ROS: reactive oxygen species; SOD: superoxide dismutase; TNF: tumor necrosis factor; VDR: vitamin D receptor.
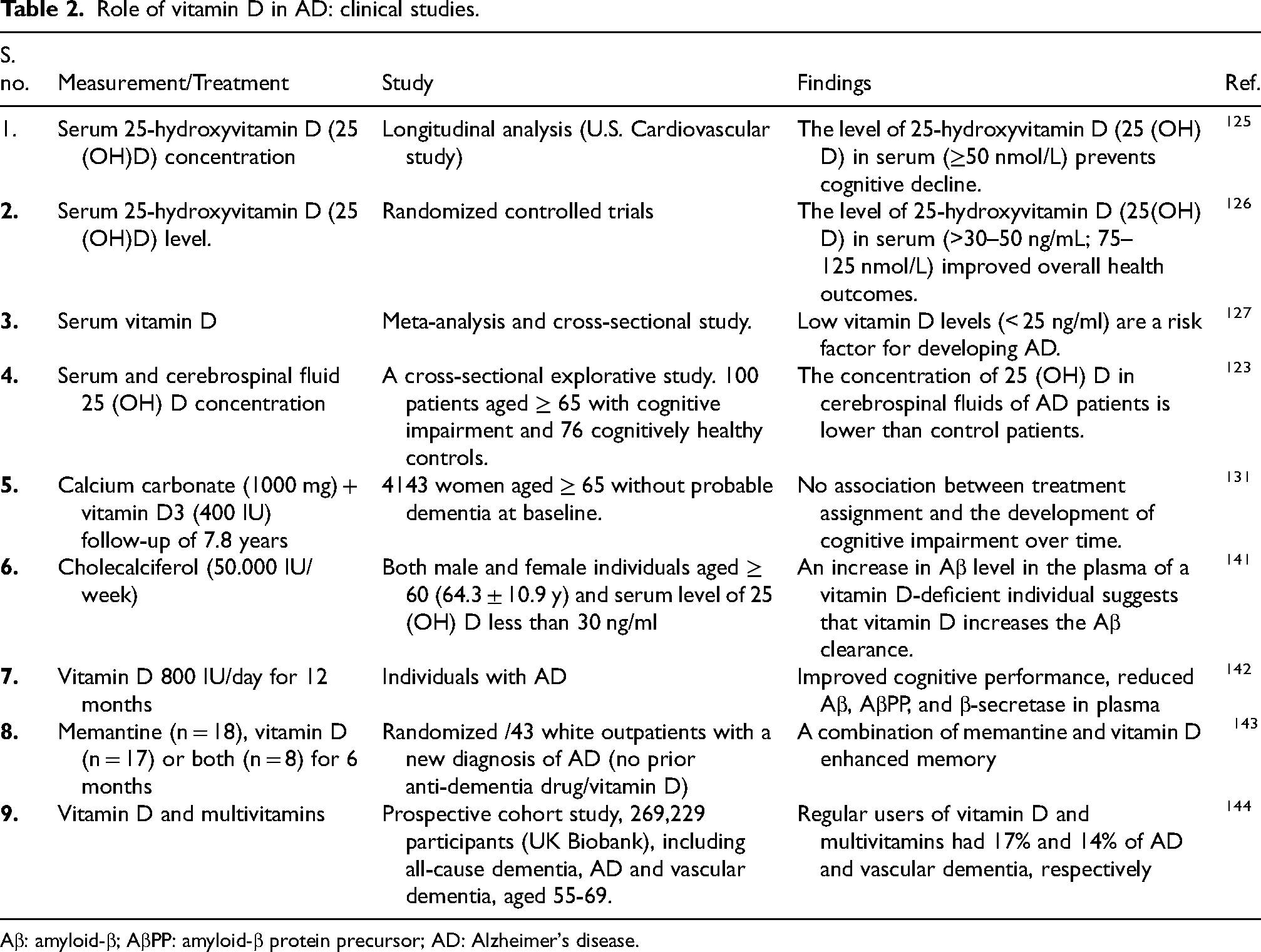
Impact of vitamin D on AD: Insights from clinical studies
Clinical research suggests an inverse relationship between vitamin D status and AD risk. A systematic review and meta-analysis revealed that AD patients typically exhibit lower serum 25(OH)D concentrations compared to healthy controls. 32 Notably, a recent cross-sectional study reported that individuals with positive AD biomarkers had reduced cerebrospinal fluid 25(OH)D levels, even when serum vitamin D was adequate. 123
Large-scale cohort analyses support these findings. Among 12,388 dementia-free adults in the National Alzheimer's Coordinating Center dataset, those who took vitamin D supplements had a 40% lower risk of developing dementia (HR = 0.60, 95% CI: 0.55–0.65) and remained dementia-free longer than non-users. This protective effect varied by sex, cognitive status, and APOE ε4 genotype, indicating that vitamin D may prevent dementia in a population-specific manner. 124 Similarly, the U.S. Cardiovascular Health Study followed 1658 older adults without dementia for 5.6 years and found that severe vitamin D deficiency (<25 nmol/L) more than doubled the risk of all-cause dementia and AD, while moderate deficiency (25–<50 nmol/L) also significantly increased risk compared with sufficient levels (≥50 nmol/L). Risk escalated sharply below 50 nmol/L, highlighting the importance of maintaining adequate vitamin D for cognitive health. 125 Moreover, in humans, optimal serum 25(OH)D levels higher than ∼30 ng/mL (75 nmol/L), and up to 40–50 ng/mL, are linked with improved overall health outcomes. 126
A meta-analysis incorporating five prospective studies and one cross-sectional study up to 2022 found that individuals with low vitamin D (<25 ng/mL) had a significantly higher risk of AD compared with those with sufficient levels (≥25 ng/mL; pooled HR = 1.59, 95% CI: 1.09–2.33; I2 = 77%). 127 These findings suggest that vitamin D deficiency may be a modifiable risk factor for AD, although causality remains to be fully established. However, the observed association could be influenced by follow-up duration, as highlighted by William B. Grant, who demonstrated that longer follow-up periods can attenuate hazard ratios due to regression dilution caused by temporal variability in serum 25(OH)D concentrations. 128 Therefore, studies with shorter follow-up durations tend to suggest stronger associations, indicating that the pooled estimate may underestimate the real effect size.
25(OH)D (≤25 nmol/L) in older adults has also been associated with increased agitation, aggressiveness, and disinhibition, as demonstrated in the CLIP (Cognition and Lipophilic Vitamins) cohort study. 129 These findings suggest that vitamin D insufficiency may contribute to behavioral issues in older populations and emphasize the potential role of vitamin D supplementation as a preventive approach for such disorders.
Vitamin D supplementation may also benefit individuals already affected by AD. In vitamin D-deficient adults aged ≥60, weekly high-dose vitamin D3 (50,000 IU) for up to 8 weeks increased plasma Aβ levels, suggesting improved cerebral clearance. 130 Furthermore, a double-blind randomized trial demonstrated that daily supplementation with 800 IU vitamin D for 12 months improved cognitive function and decreased plasma Aβ, AβPP, β-secretase 1 (BACE-1), and corresponding mRNA expression in AD patients. 131 Synergistic effects have also been observed when vitamin D is combined with conventional therapies; for example, a 6-month co-treatment with memantine and vitamin D enhanced cognitive performance in AD patients. 132
However, clinical evidence related to the cognitive effects of vitamin remains mixed. While some studies report cognitive benefits, others show no significant effect. 122 These inconsistencies could be attributed to variations in study design, population characteristics, and biomarkers used to evaluate vitamin D status. In particular, the use of 1,25(OH)2D as an indicator of vitamin D status can be misleading, as it is tightly regulated and does not influence systemic vitamin D level. 133 These factors underscore the need for careful interpretation, further clinical trials to calculate optimal dosing, and identification of patients’ subgroups most likely to benefit from supplementation. Key clinical studies evaluating vitamin D in AD are summarized in Table 2.
Role of vitamin D in AD: clinical studies.
Aβ: amyloid-β; AβPP: amyloid-β protein precursor; AD: Alzheimer's disease.
The heterogeneous outcomes reported in vitamin D intervention trials warrant careful consideration. Differences in baseline vitamin D status, supplementation dose, duration of treatment, disease stage at enrolment, and genetic variability in VDR signaling may all influence therapeutic responsiveness. Notably, individuals with sufficient baseline vitamin D levels may derive limited benefit from supplementation, whereas those with marked deficiency or early-stage disease could represent more responsive subgroups. In contrast, methodological limitations in designing the trial may be associated with inconsistent findings, as reported in the study, which argued that various clinical trials fail to account for achieved serum 25(OH)D concentrations, instead relying on fixed dosing strategies. This approach may obscure the exact dose-response relationship and lead to underestimations of vitamin D's therapeutic efficacy. 134
These observations underscore the importance of stratified and personalized approaches in future clinical trials. Incorporating biomarker-based selection criteria and longitudinal cognitive assessments may improve the ability to detect disease-modifying or preventive effects of vitamin D in AD populations.
At present, vitamin D should be regarded as a complementary factor influencing disease-relevant pathways rather than a primary therapeutic intervention for AD. In addition, other modifiable factors, such as physical activity, dietary patterns, cognitive and social engagement, and management of comorbid conditions, could have an important role in disease prevention. 135
Conclusion and future directions
AD is a multifactorial neurodegenerative disorder in which oxidative stress, mitochondrial dysfunction, and neuroinflammation intersect with classical amyloid and tau pathology. Growing evidence highlights vitamin D as a potential neuroprotective factor that extends beyond its established role in bone health. By crossing the BBB, vitamin D regulates neurotrophic factors, maintains redox balance, and modulates neuroinflammatory responses. Preclinical and clinical studies suggest that adequate vitamin D may reduce Aβ accumulation, attenuate oxidative stress, and protect against tau-mediated neurotoxicity.
However, findings remain inconsistent. While supplementation has been shown to improve cognition, reduce amyloid burden, and synergize with existing therapies, prolonged or high-dose vitamin D3 is not well established, and may vary based on individual baseline vitamin D status and clinical context. These discrepancies underscore the need to clarify dose-response relationships, the comparative effects of vitamin D2 versus D3, and the influence of genetic variants in VDR signaling.
It is important to note that vitamin D should not be viewed as a standalone therapeutic intervention for AD. Rather, current evidence supports its potential role as an adjunctive or preventive strategy that may modulate disease-relevant pathways, particularly neuroinflammation and metabolic dysfunction. Careful consideration of dosage, timing, and individual susceptibility will be necessary to translate experimental findings into clinical benefit.
It is important to critically compare findings from observational studies and randomized controlled trials (RCTs) when evaluating the effects of vitamin D. Observational studies consistently reported that higher serum 25(OH)D levels are associated with reduced risk of various non-skeletal diseases, whereas RCTs often report null or inconsistent outcomes. 136
These variations largely arise from methodological differences. While well-designed prospective cohort studies could account for various risk-modifying factors and decrease the likelihood of reverse causation, Observational studies may be influenced by confounding factors. At the same time, many RCTs are not optimally designed for nutrient research. In particular, vitamin D trials often include participants with adequate baseline levels, use insufficient dosing, permit external supplementation, and fail to consider achieved serum 25(OH)D concentrations.
Consequently, future research should prioritize improved RCT designs that account for baseline vitamin D status, target optimal serum concentrations, and apply adaptive dosing strategies. Integrating evidence from observational, mechanistic, and genetic studies will further provide a more comprehensive and reliable understanding of vitamin D's role in human health.
Collectively, current evidence suggests that vitamin D may represent a promising adjunct in AD prevention and treatment, but its therapeutic potential likely depends on baseline vitamin D status, timing and duration of supplementation, and individual genetic and metabolic factors. Future clinical trials should prioritize precision-based approaches to optimize efficacy while minimizing risk.
Footnotes
Acknowledgements
We would like to thank the National Science and Technology Council and the Higher Education Sprout Project of the Ministry of Education (MOE) in Taiwan sponsored this study.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from National Science and Technology Council (NSTC 112-2314-B-038 −091 -MY3) to S.Y. Chou, Sunny Brain Tumor and Brain Disease Research and Development Fund to S.Y. Chou, and (NSTC International Internship Pilot Program (IIPP)) to M. Kashif.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.