
Abstract
The autophagy–lysosomal pathway is key for the removal of harmful substances in cells. This article integrates evidence that highlights the role of lysosomal function and the autophagy–lysosomal pathway in maintaining intracellular homeostasis and the effects of their dysfunction on protein secretion and metabolic disorders, leading to the pathogenesis of Alzheimer's disease (AD) and other tau diseases. Dysfunction of the autophagy-lysosomal pathway is believed to be the main factor leading to the accumulation of amyloid-β and tau proteins, which are also pathological features of AD. This article also discusses why autophagy is indispensable in the early to mature stages of neuronal development and how damage to the function of autophagy can cause neurodevelopmental abnormalities and neurodegenerative diseases. We also summarized the potential role of oligodendrocytes. We believe that its relationship with lysosomes can provide a new perspective and research direction for future research on neurodegenerative diseases. Autophagy-lysosomal pathway damage is considered to be a key factor in the pathology and diagnosis of multiple sclerosis, but we believe that the challenge associated with its transformation into clinical treatment is enormous. These findings suggest that enhancing or improving autophagy function may be an effective treatment method to alleviate the condition of AD patients, which can provide new strategies for clinical treatment and intervention of AD in the future.
Introduction
The lysosome is an organelle for catabolism in eukaryotic cells. It contains more than 60 hydrolases, which can degrade a variety of biological macromolecules and damaged organelles that are delivered to lysosomes through endocytosis and autophagy and transport the degradation products inside and outside the cells for reuse. Therefore, since the discovery of lysosomes by Duve et al. in 1955, 1 lysosomes have been regarded as intracellular recycling stations. However, recent studies have revealed that in addition to degradation, lysosomes have broader significance, such as by participating in cell processes such as nutritional perception, signal transduction, gene expression regulation, cell death, plasma membrane repair, antigen processing and presentation, and organelle integrity monitoring. 2 Therefore, lysosomal dysfunction affects cell and body health and is closely related to a variety of diseases, such as lysosomal storage disease (LSD), neurodegenerative diseases, neuroinflammation, autoimmune diseases, and tumors. 3
Lysosomal-related gene mutations can lead to abnormal lysosomal function, leading to the accumulation of decomposition substrates in lysosomes, resulting in cell dysfunction and cell death. 4 Neurons are more sensitive to lysosomal storage. Since neurons are terminally differentiated cells that cannot be effectively supplemented during their death, most lysosomal storage disorders manifest as symptoms of neurodegenerative diseases. 5 For example, severe lysosomal dysfunction and a large number of lysosomal gene mutations have been found in LSD, which is very common in children with neurodegenerative diseases and neurodegenerative diseases such as delayed Alzheimer's disease (AD), Parkinson's disease (PD), frontotemporal dementia, and amyotrophic lateral sclerosis (ALS).6–8 Previous studies have shown that lysosomes are closely related to AD. According to the observation of the accumulation of prelysosomal autophagic vacuoles in the brains of AD patients by electron microscopy and the results of a genome-wide analysis of AD brain samples, the expression of autophagic mechanism components in the AD brain decreases with age,9–12 indicating that the study of this relationship is increasingly detailed. Moreover, increasing evidence emphasizes the key role of the autophagy-lysosomal pathway (ALP) in clearing amyloid-β (Aβ) from the brain.13–15 Therefore, the integrity of lysosomal function is an important determinant of nervous system health.
The autophagy–lysosome system
In recent years, the autophagy–lysosomal system has received extensive attention, especially its important role in the pathological process of AD. This intricate system represents the confluence of the endolysosomal and autophagic pathways and is now considered a key player in maintaining cellular homeostasis.16,17 The formation and function of this system involve complex interactions between autophagosomes and lysosomes, which synergistically decompose and recycle autophagic cargoes. Therefore, it is referred to as the autophagy–lysosomal system, also known as the “endolysosomal–autophagy system” in some studies.18–20
Autophagosomes
Macroautophagy (hereafter referred to as autophagy) is an important catabolic mechanism, similar to the waste disposal framework, in which damaged organelles and other materials are degraded into small molecules for cell recycling and reuse. 21 This process ensures the continuous renewal of intracellular components, thereby maintaining cell viability. In terms of its mechanism of action, autophagy can be divided into three main types: macroautophagy, microautophagy and molecular chaperone-mediated autophagy. Macroautophagy is the main way in which most long-lived proteins and aggregation-prone toxic proteins are degraded in eukaryotic cells and is the most widely discussed form in the context of AD. Autophagy begins with the activation of the unc-51-like kinase (ULK) complex (composed of ULK1 kinase, ATG13, FIP200 and ATG101).22–24 The complex then binds to class III phosphatidylinositol 3-kinase (PI3K) complex I (including Beclin1, VPS15, VPS34, Atg14 and UVRAG) to form a core PI3K complex. This causes the autophagic vesicle assembly site (PAS) to nucleate and fuse with cytoplasmic separation membranes to form a preautophagosome structure.25,26 Mammalian target of rapamycin (mTOR) is a 289 kDa serine-threonine kinase that belongs to the PI3K-related kinase family. mTOR is the catalytic subunit of two major protein complexes, mTOR complex 1 (mTORC1), that regulate cell metabolism and growth, and mTOR complex 2 (mTORC2), which controls cell proliferation and survival.27,28 mTORC1 is a critical regulator of autophagy 29 . It consists of mTOR, Raptor (mTOR regulatory related protein), mLST8 (SEC13 protein lethal to mammals 8), DEPTOR (mTOR interacting protein containing DEP domain) and PRAS40 (proline-rich Akt substrate is 40 kDa).30,31 Its regulatory mechanism in autophagy is complex. In the process of autophagy induction, mTOR integrates various intracellular signals and plays a major role in regulating autophagy by controlling downstream autophagy-related genes. Autophagy includes three main stages. 32 In the initiation phase, also known as nucleation, mTORC1 inhibits the initiation of autophagy by directly or indirectly inhibiting the ULK1 complex under nutrient-rich conditions. For example, mTORC1 directly phosphorylates ATG13 to inhibit the activity of the ULK1 complex. 33 AMBRA1 is another key autophagy regulator. Interestingly, mTORC1 also directly phosphorylates AMBRA1, preventing it from activating ULK1 and thereby inhibiting autophagy.29,34 However, under cell starvation or oxidative stress conditions, AMBRA1 promotes autophagosome nucleation by promoting the interaction between Beclin1 and VPS34.35,36 In summary, ULK1 is a positive regulator of autophagy, whereas mTORC1 is a negative regulator of autophagy; therefore, the induction of autophagy requires the inhibition of mTORC1 function. AMP-activated protein kinase (AMPK) is a member of the AMPK protein kinase family and acts as an upstream regulator of mTOR during starvation. AMPK activates autophagy through three main mechanisms: First, it directly activates ULK1 to induce autophagy through the AMPK-mTOR pathway.37,38 Second, AMPK activates autophagy by activating its negative regulator TSC2, inhibiting its subunit Raptor and inhibiting mTORC1 activity.39,40 Third, AMPK can also phosphorylate key components of autophagy, such as VPS34 and Beclin1, to promote autophagy.41,42 In addition, under starvation conditions, ATG13 can be dephosphorylated to form the Atg1-Atg13-Atg17 complex, which further promotes the initiation of autophagy. 43
Elongation phase
The preautophagosome structure expands into a double-layered membrane structure, engulfs part of the cytoplasm, and finally closes to form spherical double-layered membrane vesicles called autophagosomes. At this stage, most Atg proteins are dissociated from autophagosomes for recycling, while LC3 (microtubule-associated protein 1A/1B light chain 3) remains associated until degradation. 44 During the formation of the autophagosome membrane, LC3-I and phosphatidylethanolamine (PE) form the LC3-PE complex (autophagy marker) through a ubiquitin-like binding process involving the E1-like enzyme Atg7 and the E2-like enzyme Atg3.45–48 Therefore, LC3 can be used as a classic marker for tracking autophagosomes.
Maturation phase
The maturation of autophagosomes includes fusion with endosomes (formation of zwitterions) and, eventually, fusion with lysosomes to form autolysosomes. In autophagic lysosomes, the contents of phagocytosis are degraded, the autophagy cycle is completed, and the cells are resurrected (Figure 1).
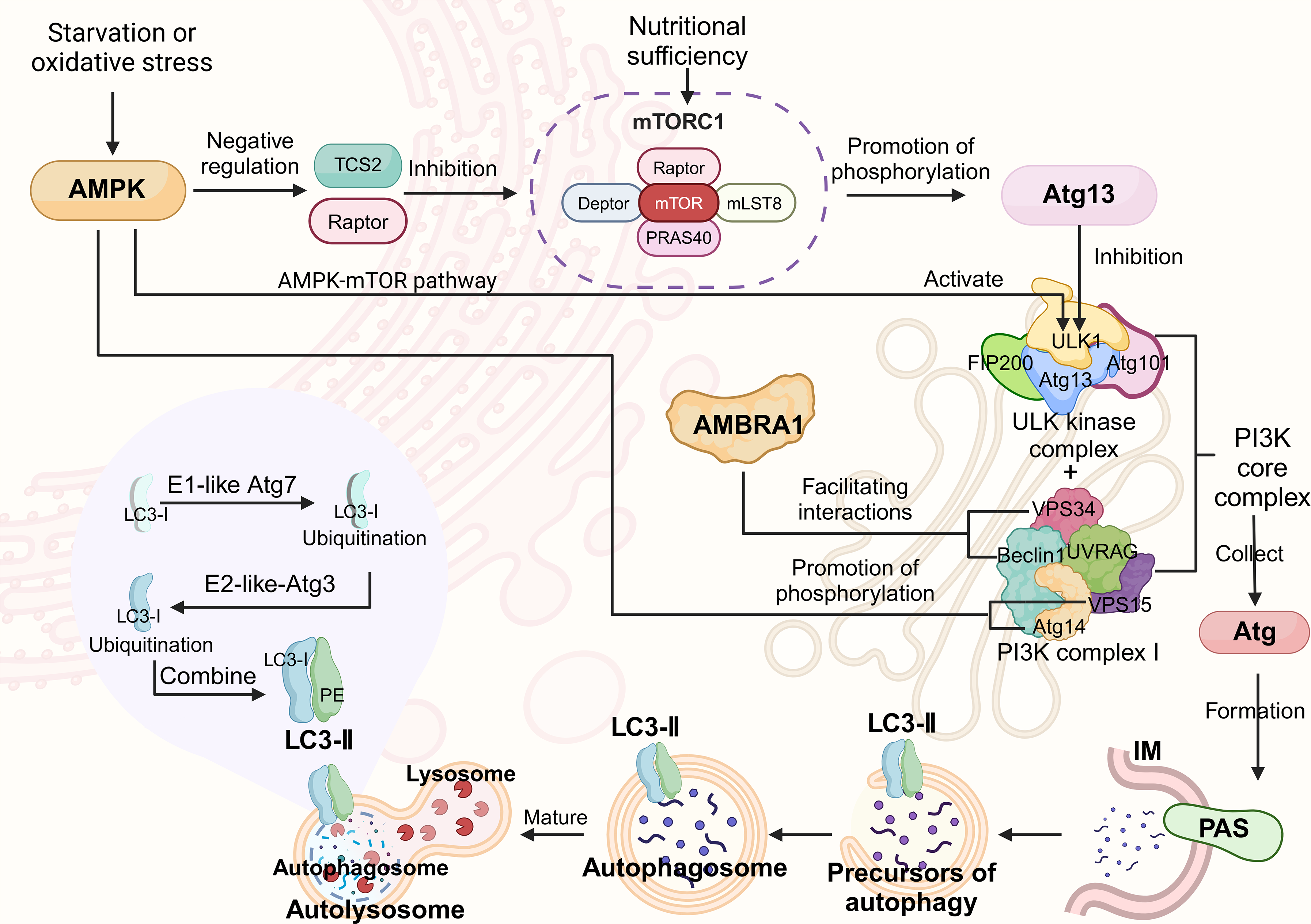
The molecular mechanism of autophagy. Autophagy begins with the activation of the ULK kinase complex, which then binds to type III PI3K complex I to form the core PI3K complex. The complex recruits the Atg protein to nucleate the autophagic vesicle assembly site (PAS), and the free separation membrane fuses to form an autophagosome structure. mTORC1 plays a key role in regulating autophagy. Under nutrient-rich conditions, mTORC1 inhibits the initiation of autophagy by directly or indirectly inhibiting the ULK1 complex; it can also phosphorylate AMBRA1 and prevent it from activating ULK1. In contrast, AMBRA1 promotes utophagosome nucleation under cell starvation or oxidative stress conditions. Under starvation conditions, AMPK acts as an upstream regulator of mTOR. It activates autophagy through three main mechanisms: (1) ULK1 is directly activated through the AMPK-mTOR pathway; (2) it activates the negative regulator of mTORC1 and inhibits its subunits; and (3) key components of autophagy are phosphorylated. In addition, starvation-induced Atg13 dephosphorylation contributes to the formation of specific complexes that promote autophagy. During the stretching phase, the preautophagic structure expands into a double-membrane structure, engulfs cytoplasmic components, and finally closes to form a closed spherical double-membrane vesicle called an autophagosome. Most Atg proteins are subsequently dissociated and recovered from autophagosomes, while LC3-I binds to PE through a two-step ubiquitination process mediated by the enzymes Atg7 (E1-like) and Atg3 (E2-like) to form autophagy markers.
Endosomes
Endosomes are single-membrane-bound organelles in eukaryotic cells that originate from endocytosis. They are important hubs in vesicle transport and are involved in many important cellular activities, often referred to as “sorting centers” or “sorting machines”. 49 In the sequential stage of endocytosis, endosomes are divided into early endosomes, late endosomes and recycled endosomes. 50 Extracellular substances such as growth factors, nutrients, hormones, viruses and toxic proteins are internalized by endocytosis to form vesicles that fuse with early endosomes. Some early endosomes are fused with various organelles for sorting and transport, whereas others integrate with autophagosomes into the autophagy pathway and mature into late endosomes. The intracavitary p H of the late endosome is mainly acidified by the activity of the vacuolar ATPase (V-ATPase) on its membrane. 51 They mature by fusing with homologous endosomes to form larger vesicles; thus, they are also called multivesicular bodies (MVBs). 52 Late endosomes can fuse with autophagosomes to form bipolar bodies and then fuse with lysosomes to form autolysosomes. In addition, late endosomes can directly fuse with lysosomes to form endosomes, resulting in the degradation of their contents.
The main function of endosomes is to sort and transport extracellular substances that are internalized by endocytosis. Afterward, the endosomal system allocates these goods to different cell destinations; waste or harmful substances are directed to lysosomes or autophagy systems for degradation. Since both endocytosis and autophagy depend on lysosomes to degrade extracellular and intracellular “waste” and because some endosomes are eventually incorporated into autophagosomes, autophagy and the endosomal system are functionally intertwined. Their convergence constitutes an endosomal-autophagic lysosomal system. In neurons, the system is essential for clearing abnormal intracellular and extracellular proteins, and its efficiency decreases with age. Dysfunction of the lysosomal-autophagic lysosomal system occurs in the early pathological stage of AD and is considered to be a potential contributor to its pathogenesis.53,54
Neurodegenerative diseases and the pathogenesis of AD
Neurodegenerative diseases are a class of diseases characterized by progressive sexual dysfunction and loss of neuronal structure and function, leading to neurological impairment. These diseases include AD, PD, Huntington's disease, and ALS. Except for a few familial diseases with clear pathogenic genes, the pathogenesis of most neurodegenerative diseases is still elusive, hindering the development of effective treatments. 55
AD is the most common neurodegenerative disease characterized by progressive cognitive decline and behavioral disorders. Core symptoms include but are not limited to memory loss, language and movement disorders, and personality and behavior changes. These symptoms are caused mainly by extensive neuronal death and synaptic loss, although the underlying mechanisms are not fully understood. The accumulation of Aβ plaques and neurofibrillary tangles composed of hyperphosphorylated tau proteins in the brain are hallmark pathological features of AD and are considered to be the main drivers of neuronal death.56,57 Aβ is produced by the sequential cleavage of amyloid-β protein precursor (AβPP) by β-secretase and γ-secretase. Lysosomes are considered potential key sites for this proteolytic process,58,59 and β-secretases exhibit peak activity near a pH of 4.5, which is consistent with the acidic pH of lysosomes. 60 The functional integrity of γ-secretase depends on its complex with the AD-related protein presenilin 1 (PS1) and several other proteins. 61 Mutations in the PS1 gene are the main cause of familial AD, and these mutations also affect autophagic flux and lysosomal enzyme activity. 62 The role of lysosomal enzymes (e.g., cathepsin B and D) in the production and degradation of Aβ is still a research hotspot. Initial studies have shown that the expression and activity of cathepsin D are elevated in AD patients and are present in amyloid plaques,63–66 suggesting a potential correlation between cathepsin D and the pathogenesis of AD. Some studies have reported an association between nucleotide polymorphisms of the cathepsin D gene and the pathogenesis of AD. 67 Cathepsin D is thought to cleave AβPP similarly to β- and γ-secretases 68 ; however, other studies have reported no abnormalities in AβPP processing in cathepsin D-deficient hippocampal neurons. 69 In addition to Aβ, tau pathology may also be related to cathepsin D. Studies have shown that in AD models,65,70 cathepsin D inhibitors can inhibit the hyperphosphorylation of tau. Similarly, although cathepsin B was initially thought to be effective at degrading Aβ,71,72 subsequent studies have shown that inhibition of cathepsin B can reduce Aβ accumulation and improve memory in AD mouse models. 73
There are significant contradictions in the literature on the specific role of cathepsins (especially cathepsin B and D) in the pathogenesis of AD. This inconsistency profoundly reflects the high heterogeneity of the ALP system in complex microenvironments. We speculate that these differences are due mainly to the following two core dimensions:
First, changes in lysosomal membrane permeability and subcellular localization are key to explaining the different roles of cathepsin B (CTSB) in AD. Under normal physiological conditions, CTSB is stored in the lysosomes of healthy cells and participates in proteolysis to maintain cell homeostasis. 74 However, in the AD model, persistent lesion stimulation leads to lysosomal membrane permeability, which causes CTSB to leak from lysosomes to the neutral pH of the cytoplasm, thereby inducing neuronal death. 75 Other studies have shown that targeted knockout of CTSB can not only reduce Aβ deposition but also significantly improve the behavioral defects of sporadic AD models and other neurological disease models.76,77 These findings indicate that the neurotoxicity of CTSB is not due to an increase in its total expression but to a change in its subcellular localization and activation in an abnormal pH environment.
Second, clinical evidence has shown that the expression of cathepsin D (CTSD) is significantly upregulated in the cortex and plasma of AD patients and can be used as a biomarker for neurofibrillary degeneration.78,79 However, this upregulation is a compensatory stress response to Aβ in the early stage of AD, which aims to reduce Aβ accumulation. In fact, maintaining or restoring the normal physiological function of CTSD has a clear neuroprotective effect. 80 Recent studies have confirmed that targeted demethylation by epigenetic genome editing technology to restore the expression of CTSD can effectively rescue the pathological phenotype of AD mice. 81 These breakthroughs in animal models have laid a solid experimental foundation for the development of targeted treatments for human patients. Therefore, in terms of the clinical transformation pathway, precise intervention for CTSD promoter methylation in human patients also has great potential. Studies have confirmed that the use of inactivated Cas9 (dCas9) to fuse the catalytic domain of DNA demethylases (such as TET1) can achieve highly specific DNA methylation regulation in mammalian genomes. 82 This process first guides the dCas9-TET1 fusion protein to be accurately anchored to the hypermethylated promoter region of the CTSD gene by designing a specific guide RNA (sgRNA). This targeted editing can effectively reverse the epigenetic inhibition of the promoter region, thereby reactivating the expression of endogenous CTSD at the transcriptional level. Such strategies have shown excellent potential in the phenotypic rescue of neurological diseases. For example, in a fragile X syndrome study, targeted methylation editing of the FMR1 gene successfully reactivated the silenced gene and rescued the pathological phenotype of neurons. 83 Considering the safety of clinical application, this technology has unique advantages. Experiments have shown that the transient expression of epigenetic editing tools is sufficient to induce a stable and heritable modification state of the target, which means that the therapeutic effect can be maintained without the long-term presence of exogenous proteins. 84 In terms of delivery methods, future clinical transformation can rely on optimized adeno-associated virus vectors or lipid nanoparticles to achieve long-term regulation of CTSD levels in the central nervous system through controlled transient expression. This strategy can not only minimize the risk of off-target effects and immunogenicity caused by long-term expression of exogenous proteins but also provide a promising epigenetic treatment paradigm for the precise intervention of AD and other neurodegenerative diseases.
Restoring the physiological level of CTSD is essential for reversing the pathological process of AD. In vivo, CTSD is a key regulator of Aβ protein homeostasis in the brain, and a large amount of Aβ is transported to lysosomes for clearance.85,86 A decrease in CTSD expression in vivo leads to a significant increase in Aβ in the brain. Moreover, Aβ in lysosomes cannot be cleared in time and accumulates to a high level because of the decrease in CTSD expression. When this accumulation reaches the critical point, it leads to impaired lysosomal function, which in turn causes changes in the lysosomal environment, eventually leading to the collapse of the Aβ clearance mechanism and accelerating the accumulation of the Aβ protein.
In summary, simply dividing cathepsins into beneficial or harmful is not comprehensive. The conflicting findings in the literature suggest that future research and drug development should not be limited to whole-cell inhibition or activation of cathepsins but should focus on exploring the repair of lysosomal membrane integrity, the recovery of lysosomal function, and the promotion of clearance of damaged lysosomes.
In addition, we compared the key pathogenic proteins and related ALP risk genes in various neurodegenerative diseases (Table 1), providing a systematic view of the key role of the ALP in neurodegenerative diseases.
Comparison of ALP characteristics and pathological effects in neurodegenerative diseases.

FUS: Fused in sarcoma; SNCA: α-Synuclein; LRRK2: Leucine-rich repeat kinase 2; PINK1: PTEN-induced kinase 1; VPS35: Vacuolar protein sorting 35; FBXO7: F-box protein only 7; PARK7(DJ-1): Parkinson's disease 7; GBA: Glucocerebrosidase; ATO13A2: ATPase Cation Transporting 13A2; TMEM175: Transmembrane protein 175; UCHL1: Ubiquitin carboxyl-terminal hydrolase L1; TDP-43: TAR DNA-binding protein 43; SOD1: Superoxide dismutase 1; C9orf72: Chromosome 9 open reading frame 72; TMEM106: Transmembrane protein 106; CHMP2B: Charged multivesicular body protein 2B
Role of autophagy in AD pathogenesis
Autophagy is a conserved process that is essential for maintaining cell homeostasis. Removing damaged organelles and misfolded proteins in the brain helps prevent neuronal apoptosis and maintains normal brain function. Therefore, autophagy is often referred to as the “purification group” of cells. 111 A key step in autophagy is the fusion of autophagosomes with lysosomes to obtain degradation enzymes. Under the guidance of the Rab-GTP enzyme, tethering factor and SNARE protein, autophagosomes undergo multiple fusion events in the lysosomal system and eventually form degraded autolysosomes. 112 As previously mentioned, impaired autophagic lysosomal function is associated with major neurodegenerative diseases, including AD. he accumulation of Aβ in the brain and the hyperphosphorylation of the tau protein are the hallmark pathological features of AD. The production of Aβ in the brain is affected by many factors, among which apolipoprotein E (APOE) is among the important factors affecting the production of Aβ. APOE is the main component of circulating low-density lipoprotein and very low-density lipoprotein in the blood and is highly expressed by astrocytes in the brain. 113 Studies have shown that APOE can stimulate the production of Aβ in the brain, hinder the clearance of Aβ42 by lysosomes, and destroy the stability of the lysosomal membrane similar to that of Aβ42.114–116 At present, research and treatment strategies for AD focus mainly on the removal of Aβ in the brain, in which ALP plays a major role. Under normal autophagy, ALP degrades “waste”, such as misfolded proteins and damaged organelles, into small molecules for reuse. 21 Therefore, maintaining or restoring ALP function is essential for improving the clinicopathological status of patients with AD. ALP stability is regulated by many factors, the most significant of which is transcription factor EB (TFEB). TFEB is a basic helix-loop-helix leucine zipper transcription factor that acts as a major regulator of lysosomal biosynthesis by binding to lysosomal coordinated expression and regulation (CLEAR) network elements. 117 From a deeper perspective of the pathogenesis of AD, the loss of TFEB function is often not simply a decrease in expression but an abnormality in its posttranslational modification that hinders its entry into the nucleus. Recent studies have shown that acetylation of TFEB can promote TFEB nuclear translocation and lysosomal biosynthesis and alleviate AD-related pathological and cognitive defects in APP/PS1 mice. 118 This TFEB-driven repair mechanism also faces severe challenges related to genetic factors in the pathological environment of AD. Interestingly, some studies have shown that APOE can also bind to sequences in the CLEAR network. The binding of TFEB to CLEAR was significantly reduced in cells with high expression of APOE, suggesting that APOE may compete with TFEB for binding to CLEAR. 119
This competition shields ALP repair instructions at the transcriptional level. As the most important genetic risk factor for AD, APOE (especially the APOE4 allele) destroys the ALP pathway in a multidimensional manner.120,121 The core mechanism through which APOE4 accelerates the pathological progression of AD is that APOE4 can cause lysosomal failure. First, in neurons, APOE4 induces comprehensive remodeling of the lysosomal proteome, which directly leads to lysosomal dysfunction in neurons and hinders the degradation of Aβ. 122 Second, in microglia, APOE4 has a specific negative regulatory effect, which severely decreases ALP levels, greatly weakening the ability of microglia to phagocytose and clear Aβ plaques and collapsing the immune defense barrier of the central nervous system. 123 More lethally, in astrocytes, APOE4 causes a chain pathological response involving multiple organelles. Studies have shown that APOE4 can lead to abnormal pathological accumulation of cholesterol in lysosomes. This lipid metabolism disorder further disrupted mitochondrial homeostasis, severely weakened the oxidative phosphorylation ability of astrocytes, and ultimately led to the depletion of energy supply in the brain microenvironment. 124
In summary, the APOE4 genotype has a devastating effect because it does not simply block a certain signaling pathway but completely disrupts the balance between macromolecular degradation and energy metabolism through the global destruction of the ALP pathway in the brain. This multidimensional ALP disruption and TFEB nuclear translocation disorder together constitute the key molecular causes of early AD neurodegeneration.
For this reason, TFEB, as a master switch that can restart ALP homeostasis globally, has become a core hub for verifying various interventions in recent years. Many studies have confirmed the core position of TFEB from different intervention dimensions: whether it promotes TFEB/TFE3-mediated autophagy through small-molecule drugs or through physical and physiological interventions,125–127 it can significantly ameliorate Aβ pathology and reverse cognitive impairment by specifically activating the TFEB pathway. This evidence not only confirms the pathological importance of TFEB but also provides a wide range of intervention options for clinical transformation. Although ALP activity is complex and its abnormalities are driven by multiple factors, the TFEB-APOE axis and its ability to mediate autophagic flux recovery are undoubtedly key to ensuring the body's metabolic balance and interfering with Aβ and tau protein pathology.
Autophagy and Aβ
In the pathological process of AD, AβPP is abnormally cleaved by preβ-site proteolytic enzyme 1 (BACE1) and γ-secretase to produce the Aβ peptide. These peptides are grouped together, with Aβ oligomers exhibiting the strongest neurotoxicity. Many MVBs and autophagosomes containing Aβ oligomers have been observed in the synaptic terminals of AD mouse models and neurons of AD patients. 128 Similarly, in neuronal cell lines overexpressing AβPP, abnormally enlarged endosomes and autophagic vesicles loaded with Aβ can be observed.20,129
Studies have shown that novel stimuli can increase autophagy, thereby promoting synaptic plasticity in hippocampal neurons. In addition, restoring impaired autophagy in the hippocampus can rescue memory deficits to a certain extent and promote the formation of new memories. 130 Autophagy manifests in two main forms: selective autophagy, which involves the recognition of specific cargoes that are degraded (e.g., mitochondria, lipids, and protein aggregates), and nonselective autophagy, which involves the massive transport of damaged organelles or misfolded proteins to lysosomes. 131 Autophagy maintains cell homeostasis by degrading damaged or excess intracellular components. 132 However, in AD, the speed of autophagic clearance lags behind the speed of Aβ production, leading to the accumulation of neurotoxic aggregates and Aβ. 133 Therefore, during the progression of AD, dysfunction of the endosomal-autophagic lysosomal system may create conditions conducive to Aβ aggregation, which may exacerbate this process. Beclin1 and phosphatidylinositol 3-phosphate (PI3P) levels are significantly decreased in AD mouse models.38,134 Beclin1 knockout mice showed impaired lysosomal acidification. Compared with those of AD mice, the levels of AβPP and autophagy markers in the brains of offspring of these mice were greater, indicating that Beclin1 is involved not only in the initiation of autophagy but also in autophagosome‒lysosome fusion. 135 Another study revealed that knocking down Beclin1 in mice not only impaired the formation of autophagosomes but also disrupted the fusion of endosomal–lysosomal pathways.11,136 Some studies have also suggested that a decrease in the level of PI3P in the pathological state of AD hinders the sorting and transport of AβPP in the endosome, impairs the fusion of MVBs and autophagic lysosomes, and thus promotes the deposition of senile plaques.134,137 AD can cause dysfunction of the lysosomal–autophagic lysosomal system and promote Aβ aggregation. In turn, defects in autophagy and the endosomal system can promote the production of Aβ, thereby forming a vicious cycle and accelerating the pathological process of AD.138,139
Autophagy and tau protein
Studies have shown that the toxic effects of Aβ are mediated by the tau protein. The degree of cognitive decline in AD patients is positively correlated with the burden of neurofibrillary tangles in neurons, highlighting the importance of tau pathology in AD. The tau proteins that cause neurofibrillary tangles in the AD brain include abnormally hyperphosphorylated tau protein (P-tau), tau protein oligomers, and paired helical filaments formed by abnormally modified tau protein aggregates. In cells, a portion of soluble tau and P-tau is cleared by autophagy. 140 However, in a pathological model, morphological abnormalities were observed in endosomes, amphisomes, and autophagosomes, which are rich in P-tau. 141 These findings indicate that the damaged lysosome–autophagy–lysosome system hinders the clearance of abnormal tau and promotes its accumulation.
Increased mTOR activity promotes tau protein phosphorylation.142,143 Studies have shown that mTOR interacts with glycogen synthase kinase 3 (GSK3). Knockout of GSK3 in the mouse cerebral cortex inhibits mTOR activity, 144 whereas GSK3 itself affects lysosomal function by inhibiting lysosomal acidification and promoting tau phosphorylation. 145 In addition, autophagy is regulated not only by mTOR but also by the endosomal system, which may be a potential target for TOR regulation. 146 The hyperphosphorylation of the tau protein in AD may be caused by decreased activity or phosphorylation of certain protein phosphatases, such as protein phosphatase 2A (PP2A).147,148 These phosphatases are involved in signal transduction regulated by endosomes, which serve as a signaling platform to receive, diversify and isolate signaling molecules to their respective downstream pathways. 149 Recent studies have shown that these enzymes are also involved in the regulation of the autophagy MAPK/ERK signaling pathway. 150 These findings further emphasize the close relationship between tau phosphorylation and the lysosomal-autophagic lysosomal system.
Impact of autophagy on neurons
Neurons are the basic unit of the brain. Normal cognitive function maintenance depends on proper neuronal development and a healthy and stable brain microenvironment. 151 The pathological process of AD is always accompanied by neuronal damage and destruction of the brain microenvironment, which is often related to microglial dysfunction.152,153 ALP is the main intracellular pathway involved in the removal of misfolded proteins and indigestible cell waste 154 and plays a vital role in maintaining neuronal activity and homeostasis of the brain microenvironment. Some lysosomal proteins, such as progranulin, 155 contribute to the repair mechanism of neurons. Lysosomes are essential for maintaining neuronal function and survival. 156 Given that neurons are highly polarized, functionally special cells with many metabolic needs, they are intrinsically dependent on autophagy. 157
Structurally, neurons are composed of three parts: cell bodies (cell bodies), axons and dendrites. Neuronal autophagy varies by location: nonselective autophagy in distal axons, mitophagy and endoplasmic reticulum autophagy in the cell body, and dendritic autophagy. However, macroautophagy is the main form of autophagy that occurs in neurons. 158 Increasing evidence has shown that neuronal autophagy defects can lead to abnormal axon development and neurodegeneration. Because axons are involved in memory function, dysfunctional neuronal autophagy is considered to be a contributing factor to the pathogenesis of AD.159,160 Autophagy regulates axon growth and development. Ban et al. reported that inhibition of autophagy by Atg7 siRNA resulted in axon elongation, whereas activation of autophagy by rapamycin inhibited axon growth. 161 Moreover, the loss of the autophagy-related protein ATG7 was found to increase synaptic transmission, suggesting that autophagy can inhibit axon growth while promoting synaptic development.158,162–164
Another prominent pathological feature of AD is synaptic dysfunction. The synapse is a unique structure of neurons and is the basic unit of neuronal communication. 87 Advances in technology have produced increasing amounts of experimental data, indicating that functional autophagy is necessary for neurotransmission and synaptic plasticity, which are essential for cognitive functions such as learning and memory. 165 Studies have shown that new stimuli induce autophagy by promoting synaptic plasticity in hippocampal neurons. 139 In addition, restoring the level of autophagy in the hippocampus of elderly individuals can reverse memory deficits and promote the formation of new memories. Neuronal autophagy includes two stages, induction and nucleation, which are mediated by the ULK1 complex and class III PI3K complex. The PI3K complex recruits DFCP1 (double FYVE protein 1), the E3-like Atg12-Atg5-Atg16L1 complex and LC3-PE conjugates to promote autophagosome formation. LC3 is acylated to LC3-PE through the Atg12-Atg5-Atg16L1 complex and integrated into autophagic vesicles. The formed autophagosomes fuse with late endosomes and are retrogradely transported to the cell body by dynein. 157
Lysosomes are located mainly in neuronal cell bodies and are rare in axons. 156 This requires the retrograde transport of autophagosomes produced in axons back to the cell body for fusion with lysosomes and subsequent degradation. The inability of autophagosomes to be correctly transported to the cell body for lysosomal fusion may be involved in the pathogenesis of AD. Some adaptor proteins, such as c-Jun N-terminal kinase interacting protein 1 (JIP1), Huntington-associated protein 1 (HAP1) and c-Jun N-terminal kinase interacting protein 3 (JIP3), have been identified as key proteins for the retrograde transport of autophagosomes. 166 Enhancing the transport capacity of axonal autophagosomes may slow the progression of AD. However, the etiology of AD is multifactorial and has not been fully elucidated. Although there is much evidence that the pathogenesis of AD is related to ALP damage, further research on neuronal autophagy may provide strategies for alleviating or even treating AD and other neurodegenerative diseases.
Role of astrocytes in the pathogenesis of AD
Astrocytes have become a hot spot in the study of the pathogenesis of AD. Although the treatment strategy is aimed mainly at Aβ aggregation, there is currently no effective treatment. An increasing number of studies have revealed the profound influence of astrocytes on the pathological trajectory of Aβ and tau aggregation. 167 Genomic studies have shown that most of the more than 40 gene loci associated with late-onset AD are expressed in astrocytes. These findings highlight the important role of astrocytes in the development of neuroinflammation and neurodegeneration in AD.168,169 Therefore, astrocytes play a pathological role in the occurrence and development of AD, suggesting that the relationship between astrocytes and AD may provide a new direction for the treatment and diagnosis of AD.
Astrocytes account for a large proportion of brain cells, approximately 20–40% of which are glial cells, and glial cells account for approximately 40% of all brain cells. 170 Physiologically, astrocytes regulate a series of key processes, including the management of neurotransmitters and calcium balance, the formation and maturation of synapses, the maintenance of the blood‒brain barrier (BBB), the control of extracellular volume and ion homeostasis, and the provision of nutritional support to the brain through neurovascular units. 171 Under pathological conditions, astrocytes undergo diverse changes, which can be morphologically divided into three categories: (1) atrophied astrocytes; (2) pathological remodeling of astrocytes; and (3) reactive astrocyte proliferation. 172 Atrophic astrocytes are characterized by reduced cell volume, reduced processes, and weakened steady-state function. Astrocyte atrophy is seen in many central nervous system diseases, such as AD, ALS, epilepsy, and schizophrenia. 173 The pathological remodeling of astrocytes is characterized by the presence of specific cytoplasmic inclusions called “Rosenthal fibers”, which are usually found in diseases caused by nutritional deficiencies in white matter, such as Alexander's disease. 172 Reactive astrocyte proliferation, marked by astrocyte proliferation with cell hypertrophy and thickened protrusions, is also common in a variety of central nervous system diseases, including AD. Genetic studies have shown that the overall risk of AD is largely related to glial gene expression. Notably, some AD risk genes, including clusterin/Apo J (CLU), sorting protein-related receptor 1 (SORL1), fermitin family member 2 (FERMT2) and APOE, are expressed mainly in astrocytes. During the development of AD, astrocytes undergo a series of morphological, molecular and functional changes, which indicates that astrocytes play important roles in the pathogenesis of AD. In an AD mouse model, changes in astrocytes may occur even before the appearance of Aβ. Studies have shown that induced pluripotent stem cell (iPSC)-derived astrocytes in AD patients also atrophy in vitro. 174 Furthermore, transcriptomic analysis of reactive astrocytes isolated from AD model mice revealed strong induction of the expression of inflammatory genes such as Cst7, CCl4, Il1b, Clec7a and Tyrobp, indicating that astrocytes are involved in the neuroinflammatory process of AD. 175
When microglia are stimulated, the emergence of A1-reactive astrocytes marks the beginning of AD neuronal death. Microglia induce these toxic A1 astrocytes by secreting interleukin-1α (IL-1α), tumor necrosis factor-α (TNF-α) and complement component 1q (C1q). 169 A1-reactive astrocytes further upregulate complement cascade genes, including complement component 3 (C3). Approximately 60% of astrocytes in the prefrontal cortex of AD mice express C3 and release an unknown neurotoxin that induces neuronal and oligodendrocyte death. In addition, A1-reactive astrocytes in mice lose their normal ability to support synaptic formation and function, engulf synaptic and myelin debris, and promote neuronal survival. 169 Neuroinflammation induces neurotoxic A1-reactive astrocytes, whereas ischemia induces protective A2-type astrocytes. These A2-type astrocytes upregulate the expression of neurotrophic genes such as cardiotrophin-like cytokine factor 1 (CLCF1), transglutaminase 1 (TGM1), pentraxin 3 (PTX3), calcium-binding protein A10 (s100a10 gene) and sphingosine kinase 1 (SPHK1). A2 astrocytes further secrete neurotrophic factors, promote neuronal survival and growth, and participate in synaptic repair. 176 Morphological analysis of reactive astrocytes revealed that in AD patients and mouse models, astrocytes around Aβ plaques exhibit phagocytic activity and can engulf malnourished neurites. 177 In addition, Aβ-activated astrocytes secrete transforming growth factor β, promote the uptake of Aβ by glial cells, and protect neurons from its toxic effects. 178
There is a complex relationship between astrocytes and Aβ. In an AD mouse model, Aβ promotes the activation of the NF-κB pathway by astrocytes, leading to the release of C3. Aβ-activated astrocytes also exhibit the A1 phenotype. 179 C3 binds to neurons through C3aR receptors and destroys dendritic morphology and function. The interaction between C3 and glial cells also affects the phagocytosis of Aβ. Both pathways may be involved in the pathogenesis of AD. 180 Additionally, reactive astrocytes upregulate the expression of AβPP and BACE1, which are involved in the production of Aβ in the AD brain. 181 On the other hand, astrocytes also participate in Aβ clearance through the secretion of factors such as ApoE, apolipoprotein J (Apo J, aggrecan), α1-antichymotrypsin, α2-macroglobulin, etc., and promote the transport of Aβ across the BBB through receptors such as low-density lipoprotein receptor-related protein-1and very low-density lipoprotein receptor. 182 Although astrocytes express a variety of Aβ-degrading enzymes, quantitative studies on the overall effect of astrocytes on Aβ levels in the brain are lacking. These complex interactions need to be further explored in the context of the pathological state of AD (Figure 2).
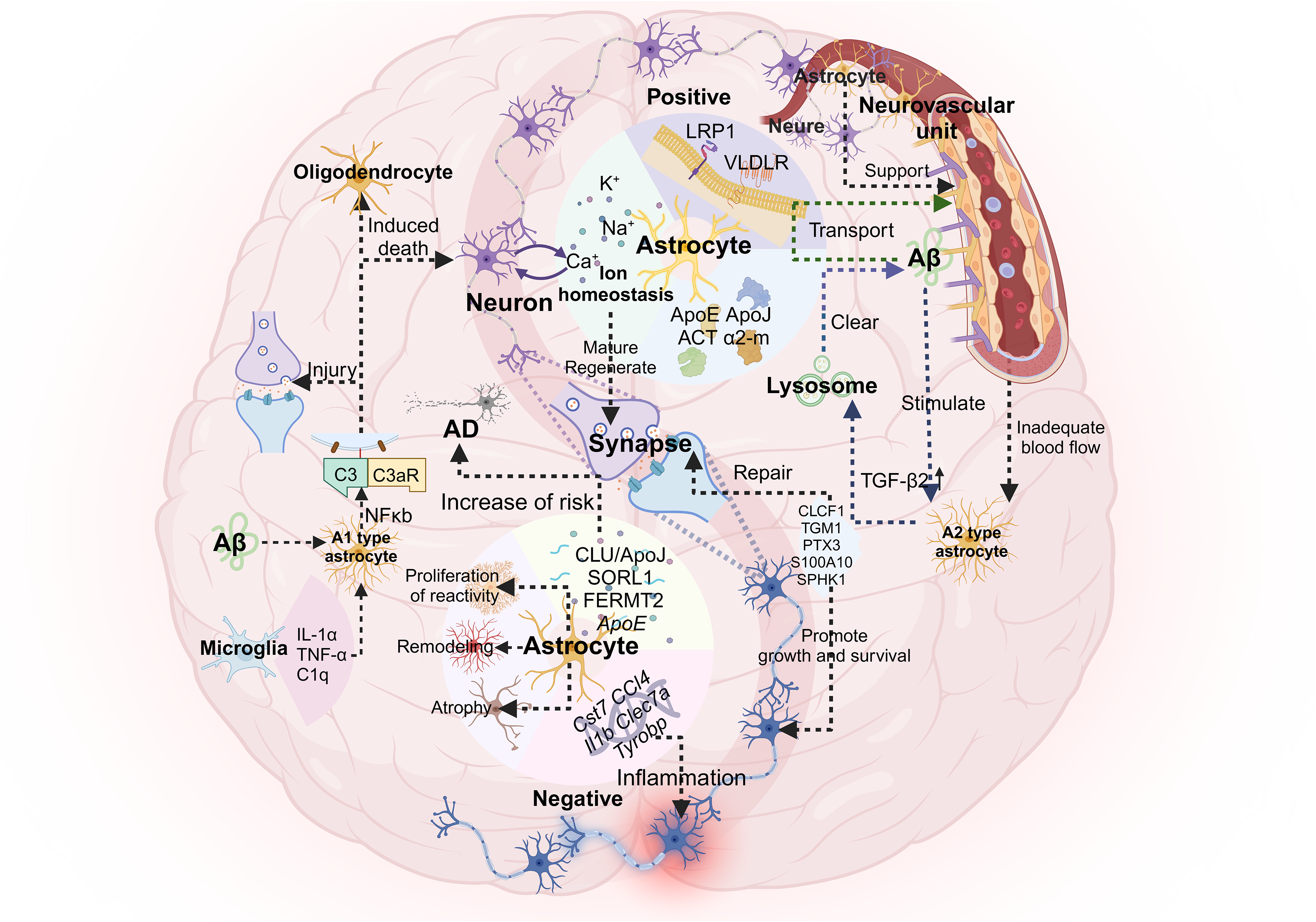
A1-reactive astrocytes are involved in the pathogenesis of AD. The emergence of A1-reactive astrocytes marks the beginning of AD neuronal death. Induced by factors secreted by microglia, A1 astrocytes upregulate complement cascade genes (e.g., C3), release an unknown neurotoxin, kill neurons and oligodendrocytes, and lose the ability to support normal neuronal function. In contrast, ischemia induces the formation of protective A2 astrocytes. These A2 astrocytes upregulate the expression of neurotrophic genes, secrete neurotrophic factors, and participate in synaptic repair. Astrocytes have a complex relationship with Aβ. In an AD mouse model, Aβ promotes the activation of the NF-κB pathway by astrocytes, resulting in C3 release and the A1 phenotype. C3 destroys neuronal function and impairs Aβ phagocytosis, thereby promoting the pathogenesis of AD. Reactive astrocytes upregulate AβPP and BACE1, which are involved in the production of Aβ. In turn, they also promote the clearance and transport of Aβ through the secretion of specific proteins.
Microglial dysfunction in AD pathogenesis
In the brain, microglia belong to the mononuclear phagocyte system, accounting for 10–15% of all brain cells, and are the main sentinels for immune defense against pathogen invasion. 183 Under normal circumstances, microglial activation is essential for tissue repair and maintenance of homeostasis in the brain through the engulfment of harmful substances and the secretion of anti-inflammatory factors. However, abnormal activation of these cells increases the levels of inflammatory mediators, impairs immune clearance mechanisms, and triggers neurological disorders. 184
Microglia are divided into two phenotypes, “resting M0” and “activated M1/M2”, according to their activation status. They can rapidly change their shape and function in response to changes in the brain's microenvironment. By secreting neurotransmitters (such as GABA, acetylcholine, and norepinephrine) that bind to microglial cell membrane receptors, neurons help maintain the resting M0 state, allowing microglia to continuously monitor the surrounding brain environment. Under the stimulation of various factors, microglia are transformed into activated M1 or M2 types, which are characterized by larger cell bodies and thicker protrusions. Different activation phenotypes have different immune functions in the nervous system. Stimulation with ligands such as lipopolysaccharide, interferon-γ, Aβ, and α-synuclein polarizes microglia to the classically activated M1 phenotype. M1 microglia secrete inflammatory mediators such as TNF-α, nitric oxide, and reactive oxygen species (ROS) to damage nerve cells. In contrast, interleukin (IL) 4/13 induced microglia to alternatively activate M2 polarization. M2 microglia mainly promote the production of arginase-1 (Arg-1) and chitinase-3-like proteins (Chil31 and YKL-40), thereby exerting anti-inflammatory effects, promoting tissue repair, and contributing to extracellular matrix remodeling. In addition, IL-10 and transforming growth factor-β can promote the transition to an inactivated M2 phenotype, which is aimed mainly at inhibiting the production of proinflammatory cytokines and reducing acute inflammation, thereby facilitating inflammation resolution and wound healing.183,185–187
In AD, abnormal accumulation of Aβ and tau proteins leads to chronic activation of microglia, causing dysfunction and an imbalance of M1/M2 subtypes. This leads to the release of a large number of inflammatory mediators and accelerates the phosphorylation of tau. Increased levels of ROS and reactive nitrogen species also drive microglia to polarize to the M1 phenotype, further increasing the secretion of proinflammatory cytokines and other inflammatory factors and exacerbating neuronal damage. 188 Additionally, tau overexpression induces synaptic dysfunction, increases the phagocytic burden of microglia, and promotes the formation of neurofibrillary tangles. 189 The absence of CX3C chemokine receptor 1 (CX3CR1) on microglia leads to increased IL-1β levels, increased neuronal AβPP expression, and increased Aβ deposition. 190 In AD, IL-1β can cause neuronal calcium overload, neuronal loss or death, and increased neurofibrillary tangles. 191 A study by Shi et al. 192 revealed that the elimination or control of microglial activation can block the phosphorylation of the tau protein, suggesting that the AD risk gene APOE may regulate tau pathology by affecting the polarization of M1/M2 microglia. Neurons form abundant synaptic connections to generate signals during development. 193 Microglia perceive dynamic changes in the extracellular environment through the processes and secretion of nerve growth factor, basic fibroblast growth factor, brain-derived neurotrophic factor, and other factors to maintain cell survival, promote the phagocytosis of apoptotic neurons, and prune synapses.183,194 In a variety of neurodegenerative diseases, the balance between M1 and M2 microglial polarization is disrupted, resulting in decreased or excessive phagocytosis, which is a common pathological feature.
Microglia release different signals according to their surface complement receptors, which can promote or inhibit phagocytosis. In the AD brain, Aβ deposition induces abundant C1q production, resulting in increased levels of downstream C3. C3 signaling is transmitted to microglia and binds to complement receptor 3, triggering excessive phagocytosis of synapses by microglia and eventually leading to synaptic loss and neuronal death. 195 Cluster of differentiation 47 (CD47) binds to signal regulatory protein α (SIRPα) on microglia to inhibit synaptic phagocytosis. 196 However, the absence of SIRPα in microglia impairs the recognition of CD47, thereby increasing synaptic phagocytosis and exacerbating cognitive impairment. 197 In an AD mouse model, knockdown of CX3CR1 can reduce the excessive phagocytosis of synapses by microglia and reduce synaptic loss. 198 Therefore, regulating the phagocytic activity of microglia is essential for maintaining synaptic homeostasis.
Therefore, excessive microglial activation significantly exacerbates cognitive dysfunction in patients with AD. Therefore, therapeutic strategies may include inhibiting the activation of microglia by upregulating the expression of anti-inflammatory cytokines and downregulating the expression of proinflammatory cytokines, regulating inflammation of the systemic immune system and the central nervous system, inhibiting the excessive activation of microglia, inducing M2 polarization and the secretion of anti-inflammatory substances, enhancing antioxidant capacity, regulating mitophagy, and reducing brain Aβ deposition and neurofibrillary tangles, thereby slowing the progression of AD.
Oligodendrocytes
Oligodendrocytes, together with astrocytes, microglia and neurons, form crucial collections in the central nervous system of vertebrates. Oligodendrocytes play a crucial role in maintaining homeostasis in the brain, and their unique function is the myelination of neuronal axons.199,200 However, increasing evidence has shown that oligodendrocytes, in addition to their role as the shell of neuronal axons, play an indispensable metabolic support role in maintaining the metabolic homeostasis of neuronal axons. 201 Because axon formation requires more energy, these processes are highly dependent on the metabolites provided by oligodendrocytes through myelin channels. Notably, highly lipid-rich myelin can provide key lipid and energy substrates for axons to maintain synaptic survival under early metabolic stress induced by cerebral ischemia or AD. 202
In the process of myelination, oligodendrocyte progenitor cells (OPCs) proliferate and migrate and ultimately differentiate into mature oligodendrocytes, forming myelin around axons. They play key roles in myelination and remyelination under pathological conditions.203,204 Oligodendrocytes not only facilitate the rapid transmission of signals but also provide metabolic support for axons. 205 ALP is the core hub for maintaining oligodendrocyte function and myelin homeostasis. Glial cells undergoing myelin sheath formation are highly dependent on autophagy to continuously remove aging-damaged myelin debris and lipid peroxides. 206 Under physiological conditions, OPCs must be reprogrammed by precise autophagic metabolism to successfully differentiate into mature oligodendrocytes and undergo myelination. 207 Recent mononuclear RNA sequencing studies revealed that in the pathological microenvironment of AD, ALP damage can lead to a large accumulation of undegraded oxidized lipids and Aβ, which promotes the transformation of healthy oligodendrocytes to disease-related oligodendrocytes.208,209 When the ALP is damaged, undegradable toxic proteins and abnormal lipids accumulate in oligodendrocytes, which not only directly hinders the differentiation of OPCs and the regeneration of the white matter myelin sheath but also cuts off the metabolic supply of oligodendrocytes to axons.210,211 Therefore, the occurrence of AD, multiple sclerosis, ALS, and other neurodegenerative diseases is closely related to the dysfunction of oligodendrocytes. 209 Recent studies have shown that Aβ deposition is associated with myelin dysfunction and demyelination in AD mouse models. Myelin sheath injury can aggravate Aβ deposition, affect AβPP metabolism, and impair the ability of microglia to remove Aβ aggregates. 212 Crucially, ALP defects in oligodendrocytes form a vicious cycle with Aβ pathology. On the one hand, Aβ toxicity and local neuroinflammation directly attack oligodendrocytes and induce demyelination. 213 On the other hand, recent breakthrough studies have confirmed that myelin dysfunction due to impaired ALP itself directly drives the accelerated deposition of Aβ, further worsening cognitive decline.212,214
Myelin basic protein (MBP) is a protein that is rich in basic amino acids and is essential for maintaining the structural and functional stability of the myelin sheath in the central nervous system. However, Chen et al. reported that the level of MBP protein in the hippocampus of APP/PS1 mice was decreased and that the MBP-positive areas in the cortex and hippocampus of AD patients were significantly reduced, indicating that there is a potential correlation between myelin loss and cognitive impairment. 215 The differentiation of OPCs seems to be affected by muscarinic receptor 1 (M1R). The downregulation of M1R expression can promote OPC differentiation into mature oligodendrocytes, thereby improving myelination. 216 Wang et al. further confirmed that enhanced myelination and regeneration can improve memory function and reduce synaptic loss in the hippocampus of aged mice. 217 Nevertheless, the specific pathways and mechanisms through which myelin dysfunction leads to neurodegenerative diseases are still unclear.
A recent study revealed that microglial dysfunction affects normal myelin development. 218 Neurodegenerative diseases are not caused by a single factor but involve a series of cascade reactions. Neurodegenerative diseases are not caused by a single factor but involve a series of cascade reactions.
Although the complex relationship between oligodendrocytes and AD is still unclear, targeted restoration of autophagic flux in oligodendrocytes not only protects white matter structure but also rests metabolic support of damaged axons. For example, enhanced myelin renewal has been shown to reverse cognitive dysfunction in AD mouse models. 215 These findings provide a new pathological basis for the intervention of AD from the perspective of oligodendrocyte metabolism.207,211,214 However, the molecular mechanism of ALP damage in oligodendrocytes and its direct contribution to AD are still relatively unclear, and future research needs to fill this gap. To elucidate the causal relationship between the two and elucidate its potential mechanism, follow-up studies can be carried out on the basis of the following two key dimensions: (1) Construction of a cell-specific autophagy loss model: Cre tool mice driven by oligodendrocyte-specific promoters (such as MBP or Plp1) were crossed with conditional knockout mice of key autophagy regulatory genes (such as Atg7 or Tfeb) and introduced into the AD genetic background (such as APP/PS1 or 5xFAD). This model can specifically block lysosomal degradation flow in oligodendrocytes to directly evaluate the specific mechanism through which ALP failure in oligodendrocytes affects the maintenance of myelin integrity and the effect on surrounding Aβ plaques. (2) Multidimensional omics combined with in vitro simulation verification: A coculture model was established by using oligodendrocytes derived from human iPSCs and neurons, and spatial transcriptomics technology was used to analyze the metabolic abnormalities between oligodendrocytes and neurons in the AD environment. This method helps to identify how oligodendrocyte lineages in different states evolve and participate in disease progression in the context of ALP damage. 219 By analyzing spatial location-related gene expression characteristics, we can further explore whether oligodendrocytes release proinflammatory mediators or metabolic toxic products through secretome remodeling under cell stress caused by impaired ALP function, thereby inducing axonal degeneration and synaptic dysfunction through paracrine effects. 220 This in-depth mechanistic analysis provides key evidence for the role of oligodendrocytes in the pathological evolution of AD.
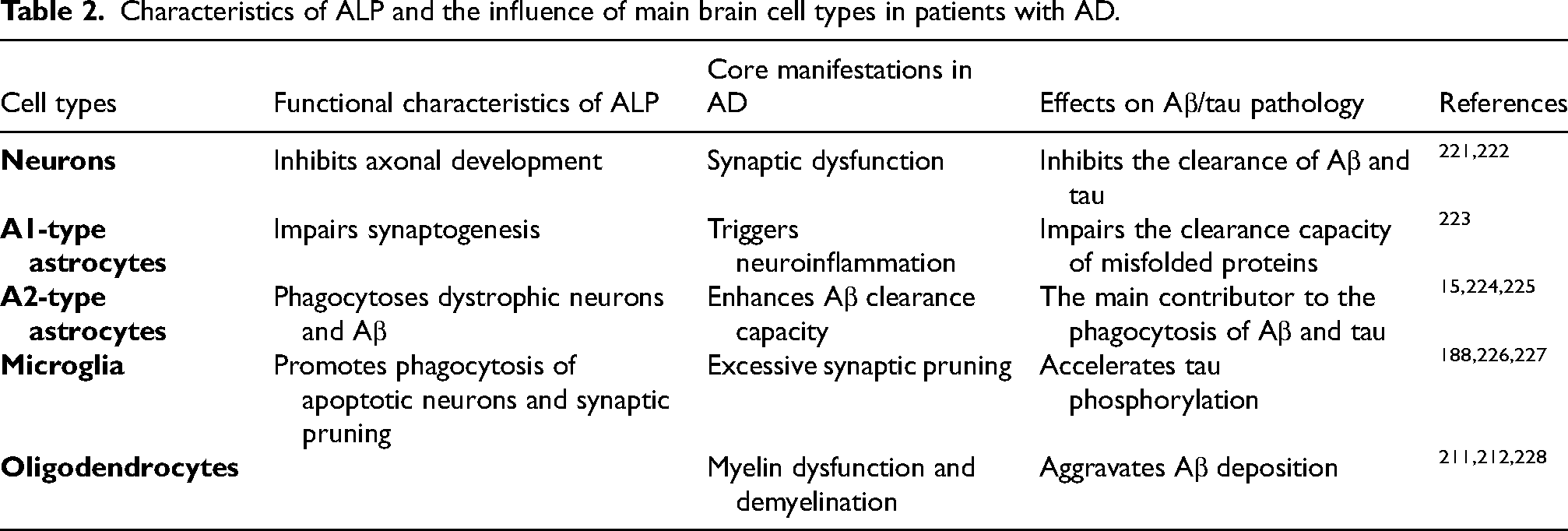
Finally, we summarized the cell type-specific changes and effects of ALP dysfunction in different brain cells in patients with AD (Table 2), providing a systematic framework for understanding the multiple roles of ALP in the pathogenesis of AD.
Characteristics of ALP and the influence of main brain cell types in patients with AD.
Challenges of clinical transformation and new therapeutic strategies targeting autophagy
Although many basic studies have confirmed that inducing autophagy is a promising strategy for the treatment of AD, 229 in past clinical trials, the use of candidate drugs for the ALP has not achieved effective results in clinical treatment, which has forced us to examine the limitations of existing drug development strategies. 230
First, the low permeability of the BBB is the main reason that existing therapeutic drugs are ineffective. Taking a classic mTOR inhibitor as an example, although it strongly induces autophagy and has neuroprotective effects in vitro, 231 it is difficult for macromolecular compounds to effectively cross the BBB to achieve effective therapeutic drug concentrations in the brain. Moreover, long-term systemic inhibition of mTOR can lead to serious side effects, such as immunosuppression and metabolic disorders. In addition, although some peripherally administered autophagy modulators have been shown to be safe for peripheral nerve regeneration, 232 the central nervous system has a very low tolerance to fluctuations in autophagic flux, which further reduces the therapeutic window of drugs.
Second, enhancing autophagy is highly limited in the treatment of AD. In AD (especially in patients with the APOE4 allele), owing to inherent defects in the endosomal–lysosomal compartment, downstream lysosomal acidification and degradation functions are often severely impaired. 233 In this case, if the drug is used alone to forcibly activate the upstream autophagy initiation stage, it will not only fail to clear Aβ but also lead to a large number of abnormally accumulated autophagosomes containing undegraded toxic substrates in neurons. This imbalance in protein homeostasis places great metabolic pressure on cells and ultimately triggers nonapoptotic forms of autophagic cell death.234,235
In the face of these challenges, the future clinical transformation strategy is shifting to drug repurposing and natural product regulation therapy. First, drug reuse treatment strategies have great clinical therapeutic potential because of the long development cycle and high failure rate of new drugs. Drugs that can penetrate the BBB and regulate autophagy have been approved by the FDA.236,237 Given its known safety and clear pharmacokinetic data, drug reuse treatment strategies can greatly accelerate the advancement of clinical trials. Second, compared with high-intensity single-target chemical synthetic drugs, natural herbal extracts have the advantages of multiple targets and low toxicity. 238 Resveratrol, for example, has been shown to act as a natural autophagy modulator that can effectively improve neurodegeneration in AD models by gently activating AMPK signaling without causing serious systemic side effects. 239
In summary, future AD treatment should not be limited to inducing enhanced autophagy but should focus on the use of highly penetrating, highly safe drugs or natural molecules to accurately restore autophagic flux, with the goal of achieving a real breakthrough in clinical practice.
Discussion
The relationship between ALP damage and the pathogenesis of AD has been increasingly supported by various studies. Lysosomes are the main organelles that remove cell waste, such as misfolded proteins and damaged organelles. Normal lysosomal function, as well as the lysosomal pathway and other related autophagy processes, is essential for maintaining cell homeostasis. 240 ALP damage not only affects the pathogenesis of AD but is also related to changes in tau secretion in tau lesions, increases the secretion of proteins, and can cause other metabolic disorders. 241 As mentioned above, the clearance of Aβ and tau is dependent on ALP. In AD pathology, ALP is impaired, leading to excessive accumulation of Aβ and tau, which in turn drives the development of the disease.242,243 Autophagy occurs at various stages of neuronal development. Impaired ALP can cause neuronal dysplasia and functional defects, thereby inducing neuroinflammation and neurodegeneration. 244 Although the current research on the relationship between oligodendrocytes and lysosomes is limited, oligodendrocyte defects are known to be associated with neurodegenerative diseases such as AD, which may represent a new research direction. As an important way to remove waste such as damaged organelles and abnormal protein aggregates, ALP plays an important role in the pathology and diagnosis of AD. Although many studies have established this relationship, it is still challenging to translate this knowledge into clinical AD treatment. Even before the significant accumulation of Aβ, ALP clearance failure occurs. Therefore, improving autophagy function in clinical practice is a promising therapeutic strategy for alleviating AD.
Footnotes
Acknowledgements
The authors acknowledge the use of Doubao (May 2024 Version), an AI-powered language assistance tool, during the preparation of this manuscript. Doubao was exclusively utilized for nonscientific tasks, including correcting grammatical errors, refining sentence structure, and polishing language expressions to enhance the clarity and readability of the text. This manuscript was polished using Rubriq AI. All AI-generated suggestions were carefully reviewed, validated, and edited by the authors to ensure alignment with the original scientific content. All graphical illustrations were developed utilizing BioRender, an online tool for scientific illustration.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Shandong Provincial Medical and Health Science and Technology Development Program (grant no. 202502051089), the Scientific Project of Weifang Health Commission (grant no. WFWSJK-2025-064) and the Scientific Project of Weifang Health Commission (grant no. WFWSJK-2025-083).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.