
Abstract
Background
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by the accumulation of amyloid-β (Aβ) and the formation of neurofibrillary tangles. Although the amyloid cascade hypothesis underscores the centrality of Aβ accumulation, the precise initiators of this process remain unknown.
Objective
In this study, we investigate the potential role of Esophageal Cancer-Related Gene 4 (ECRG4) in AD. We hypothesized that ECRG4, which is associated with cognitive impairment and upregulated in AD, directly contributes to amyloid pathology.
Methods
We performed cell-based assays, co-immunoprecipitation, in vivo experiments using APPNL-G-F/NL-G-F knock-in mouse, and immunohistochemistry of human hippocampal sections.
Results
ECRG4(133–148) associated with the amyloid precursor protein (APP) intracellular domain (AICD), leading to increased APP/Aβ accumulation. Furthermore, intracerebral injection of synthetic ECRG4(133–148) into AD model mice significantly augmented APP/Aβ deposition. Notably, the co-localization of ECRG4(133–148)-containing peptides with AICD-containing peptides increased with AD severity in human hippocampal tissue.
Conclusions
Our findings establish that the carboxy-terminal fragment of ECRG4 acts as a potential initiator of amyloid pathology in AD through its interaction with AICD.
Keywords
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by specific pathological changes, including the formation of cytotoxic amyloid-β (Aβ) plaques, intracellular neurofibrillary tangles (NFT) caused by hyperphosphorylated tau protein, and neurovascular dysfunction.1–3 Because plaque formation has traditionally been regarded as the primary cause of AD pathogenesis, numerous molecular mechanisms have been elucidated, including genetic mutations in the amyloid precursor protein (APP) (e.g., Swedish, Dutch, and London mutations), Aβ clearance through both enzymatic pathways, such as neprilysin and metalloproteinases, and non-enzymatic pathways, such as apolipoprotein E (APOE)-dependent transcytosis and perivascular drainage, or a combination of these factors and mechanisms.1–4 However, despite extensive research, the precise mechanisms initiating Aβ accumulation and plaque formation remain elusive, underscoring the need to identify novel contributors to AD pathogenesis.
Recent studies have highlighted the multifaceted roles of Esophageal Cancer-Related Gene 4 (ECRG4). Although initially identified as a tumor suppressor gene with typically decreased expression in malignant tumors,5–7 ECRG4 exhibits a contrasting pattern in the brain, where its expression increases in aged mice and in the hippocampus of patients with AD.8,9 We and others have also shown that Ecrg4 is expressed in the epithelial cells of the choroid plexus and in both ependymal cells and neural stem cells.10,11 ECRG4 is a secreted hormone-like protein localized in the secretory vesicles of endothelial cells of the choroid plexus that produces cerebrospinal fluid (CSF). ECRG4 contains two predicted cleavage sites for furin and thrombin, which generate three fragments ECRG4(32-70), ECRG4(71-132), and ECRG4(133-148). 12 Furin, located in the Golgi apparatus, may cleave ECRG4(32-148) into ECRG4(32-70) and ECRG4(71-148) prior to secretion, 13 while thrombin, which has been highly detected in the CSF of AD patients, may further cleave ECRG4(71-148) into ECRG4(71-132) and ECRG4(133-148). Another potential mechanism is that ECRG4(133-148) could be incorporated into the cells. Previous study had reported that ECRG4(133-148) can be internalized into cells through interacting with the TLR4/CD14/MD2 receptor complex and localizes to perinuclear region. 13 It was also shown that Ecrg4 stimulates the production of inflammatory cytokines, such as interleukin 6 and tumor necrosis factor α, from microglia, which play crucial roles in both AD development and tumorigenesis.14–16 Furthermore, we have demonstrated that Ecrg4-deficient mice exhibit improved spatial learning abilities compared to wild-type mice, suggesting that Ecrg4 is involved in cognitive impairment. 11 Collectively, these findings indicate that ECRG4 may contribute to AD pathogenesis. However, it is totally unknown whether ECRG4 influences APP/Aβ accumulation and acts as an initiator for amyloid pathology.
Given the multifaceted roles of ECRG4, particularly its upregulation in AD and involvement in cognitive impairment, and the unresolved molecular mechanisms of Aβ accumulation, we hypothesized that ECRG4 may play a direct role in AD pathogenesis. To test this hypothesis, we investigated whether ECRG4 deposition correlates with AD progression and co-localizes with APP/Aβ in human hippocampal tissue. We further sought to identify the specific ECRG4 fragment responsible for influencing APP/Aβ accumulation and elucidate its interaction with APP. Finally, we explored the in vivo impact of ECRG4 on Aβ plaque formation using an AD mouse model, thereby evaluating ECRG4's potential as a novel initiator of Aβ pathology in AD.
Methods
Human tissue samples and definition of EAD and AD
Human hippocampal tissue sections were obtained from the Brain Bank for Aging Research, Tokyo Metropolitan Geriatric Hospital, and Institute of Gerontology. Samples were categorized as non-AD (NAD), Early AD (EAD), or AD based on neuropathological assessment according to Braak staging and CERAD scores.17,18
EAD is defined by neuropathological criteria, including Aβ plaque (also known as senile plaque (SP) stage 2 with NFT stage ≥3 or SP stage 3 with NFT stage 3, without other confounding dementia pathologies. In contrast, AD fulfills all established diagnostic criteria, including SP stage 3, NFT stage ≥4, and clinical dementia (Supplemental Table 1).
Animals
C57BL/6 mice were purchased from Charles River Japan, Inc. APPNL−G−F/NL−G−F mice, a knock-in model closely mimicking human AD pathology by expressing human APP with specific pathogenic mutations, 19 were kindly provided by Prof. Saido and maintained as heterozygote mice with C57BL/6 mice. All mouse experiments were conducted in accordance with the protocols approved by the Institutional Review Board of the Animal Care and Use Committee of the Hokkaido University (Protocol code: 21-0066, Date of approval: July 19, 2021).
Cell culture
Rat C6 glioma cells, chosen for their retention of both glial and neural stem cell characteristics and ability to express neuronal markers, 20 and HEK293 T (293 T) cells, used for their high transfection efficiency, were purchased from Tohoku University Cell Resource Center for Biomedical Research Cell Bank (TKG0589) and RIKEN BRC (RCB2202), respectively.
These cells were cultured in Dulbecco's Modified Eagle medium (DMEM) (Nacalai, 08458-16) supplemented with 10% Fetal Bovine Serum (FBS, BioWest, S1580) in a 5% CO2 humidified incubator at 37°C. For immunostaining, cells were cultured in chamber slides (Nunc, 177445) as described previously.21,22
Chemicals
Chemicals and growth factors were purchased from Sigma and PeproTech, respectively.
Vector construction
C99 cDNA was amplified from the human APP expression vector pCAX APP Swe/Ind (Addgene, #30145) using KOD plus-Ver.2 polymerase, according to the manufacturer's protocol (TOYOBO, KOD-201). C99 cDNA was used to generate Aβ42 AICD. The amplified fragments were cloned into p3xFLAG-CMV10 to create p3xFLAG-C99, -Aβ42, -AICD (C43-84), -C85-99, and -Aβ42 + C85-99.
The Ecrg4 protein contains a signal sequence at the N-terminus, encompassing amino acids (AA) 1-31, and two cleavage sites: furin at AA70 and thrombin at AA132. 12 These cleavage processes generate four peptides: AA1-31, 32-70, 71-132, and 133-148. cDNAs for each fragment, except the N-terminus, were amplified from pCMS-EGFP-Ecrg4 8 using KOD plus-Ver.2 polymerase. To generate Ecrg4(133-148), Sense (ATCGAGCCGGGAAAGCTTCAGGCATGGAGCCAGTGTCAACTATAATGACTATA) and anti-sense (GATCTATAGTCATTATAGTTGACACTGGCTCCATGCCTGAAGCTTTCCCGGCTCGAT) oligonucleotides were synthesized and annealed using a conventional method.
The amplified fragments and Ecrg4(133-148) cDNA were inserted into pFUSE-hIgG-Fc2 (pFUSE-hIgG, InvivoGen) to produce pFUSE-hIgG-Ecrg4(32-70), -Ecrg4(71-132), -Ecrg4(133-148), and -Ecrg4(32-148).
A set of human ECRG4 fragments, ECRG4(3-70), ECRG4(71-132), and ECRG4(133-148), were amplified from pEF-Fc-ECRG4(1-148) and inserted into pEF-Fc to create pEF-Fc-ECRG4(3-70), pEF-Fc-ECRG4(71-132), and pEF-Fc-ECRG4(133-148). The oligonucleotide DNA primers used to generate these fragments are listed in Supplemental Table 2.
RT-PCR
Reverse transcription (RT)-Polymerase chain reaction (PCR) was performed as previously described.23,24 Briefly, total RNA was extracted from the transfected cells with a set of expression vectors using the RNeasy Mini Kit (QIAGEN, 74104) and reverse-transcribed using ReverTraAce qPCR RT Master Mix (TOYOBO, FSQ-201), according to the supplier's instructions. PCR was performed on a thermal cycler (Biometra, TOne) using Ex Taq DNA polymerase (TAKARA, RR001A), according to the manufacturer's instructions. The cycle parameters were as follows: 10 s at 98°C, 30 s at 55°C, and 140 s at 72°C for 35 cycles. The cycles for gapdh were as follows: 1 s at 98°C, 30 s at 53°C, and 90 s at 72°C for 22 cycles. The following oligonucleotide DNA primers were synthesized for C99:5’ primer, 5’-ATGCTGCCCGGTTTGGCAC-3’; 3’ primer, 5’-CTAGTTCTGCATCTGCTCAAA-3’. The primers for GAPDH were as described by DeMonte et al. 25
Co-immunoprecipitation and western blotting
To examine the association between Ecrg4 and C99, HEK293 T cells were transfected with p3xFLAG vectors and either pFUSE-hIgG-Ecrg4(133-148) or pEF-Fc-ECRG4(133-148) using the polyethylenimine (PEI) method at a DNA:PEI ratio of 1:6. Two days after transfection, the cells were solubilized in RIPA buffer containing a protease inhibitor mix according to the manufacturer's protocol (Nacalai Tesque, 25955). The lysates were then subjected to immunoprecipitation using a mouse anti-FLAG M2 Ab (10 µg/mL, Sigma-Aldrich, F1804). Immunocomplexes were analyzed using standard SDS-PAGE and western blotting methods with mouse anti-FLAG M2 (1:100), followed by HRP-conjugated anti-mouse Ab (1:1000, Santa Cruz Biotechnology, sc-2005), HRP-conjugated anti-hIgG (1:1000, Bethyl Laboratories, A80-104P), and HRP-conjugated mouse anti-GAPDH (1:1000, Proteintech, HRP-60004). Protein bands were visualized using Clarity Western ECL substrate (Bio-Rad, 1705061) and a ChemiDocTM MP imaging system (Bio-Rad). Co-immunoprecipitation assays were performed in three independent co-transfection experiments.
Dot-blot analysis
Dot blot analysis was performed as described previously. 24 Briefly, the immunoprecipitated samples were spotted onto a nitrocellulose membrane (0.45 µm, Bio-Rad, 1620117) and allowed to dry at room temperature (RT). The membrane was incubated with a blocking buffer containing 5% skim milk and 0.1% Tween 20 in Tris-buffered saline at RT for 1 h. The membrane was then incubated with either rabbit anti-ECRG4 (1:200, Sigma, HPA008546) or mouse anti-APP/Aβ (1:200, Cell Signaling Technology, NAB228) Abs suspended in blocking buffer at 4°C overnight. Abs were detected using either HRP-conjugated anti-mouse IgG (1:1000, Santa Cruz Biotechnology, sc-2005) or HRP-conjugated anti-rabbit IgG (1:1000, Santa Cruz Biotechnology, sc-2004) at RT for 1 h. Clarity western ECL substrate (Bio-Rad, 1705061) was used for detection.
Histological analyses
Immunostaining of paraffin-embedded human hippocampal sections (6 μm thick) and frozen sections of the human hippocampus and mouse brain (8 and 14 μm thick, respectively) was performed as described previously.26,27 The sections were treated with HistoVT One (Nacalai Tesque, 06380-05), according to the supplier's instructions. The cells were permeabilized with 0.3% TritonX-100 in PBS, treated with a blocking solution (5% skim milk, 0.3% Triton X-100, and PBS) for 2 h at RT, and incubated with primary antibodies overnight at 4°C.
DAB staining was performed using the DAB peroxidase substrate (Vector Laboratory, SK-4100), according to the manufacturer's protocol. In brief, sections of paraffin-embedded hippocampus of NAD, EAD, and AD with Apolipoprotein E (APOE)3/3 and 3/4.
The following antibodies were used to detect antigens: rabbit anti-ECRG4, mouse anti-APP/Aβ, rabbit anti-APP (1:200, Invitrogen, PA5-17829), rabbit anti-Aβ42 (1:500, Invitrogen, H31L21), mouse anti-ECRG4(133-148) (20 µg/ml) (1H4 28 ), mouse anti-APP/Aβ, mouse anti-Nestin (1:200, Beckton Dickinson, 611658), mouse anti-CD31 (1:200, Cell Signaling Technology, 89852), and mouse anti-NG2 (1:200, R&D Systems, MAB2585). Ulex Europaeus Lectin 1 (UEA1)-Biotin (1:100, J-OIL Mills, J219) was used to detect endothelial cells. Antibodies and Biotin were detected with Alexa 488-conjugatedgoat anti-mouse -rabbit (1:1000; Molecular Probe, #A-11001 respectively), Alexa 594-conjugated goat anti-mouse or -rabbit IgG (1:1000 Molecular Probe #A-11032 or #A-11012, respectively), and Streptoavidine-Cy3 (1:500; Invitrogen, 434315). Cells were counterstained with Hoechst33342 (1:1000, Invitrogen, #H3570) to visualize the nuclei.
Stereotactic injection
Two-month-old APPNL−G−F/NL−G−F and C57BL/6 mice were anesthetized with a mixture of medetomidine (Kyoritsu Seiyaku), midazolam (Maruishi Pharmaceutical Co., Ltd), and butorphanol (Meiji Animal Health Co., Ltd) administered intraperitoneally at a dose of 10 ml/kg body weight. A midline skin incision was made to expose the mouse skull. Stereotactic injection was performed 2 mm anterior to the lambda, 2 mm lateral to the sagittal suture, and 3 mm deep into the brain. Five microliters of 1.7 mM synthetic ECRG4(133-148) (purity >95% by HPLC, GenScript) in PBS was gradually infused into the parenchyma using a microliter syringe (HAMILTON, #87930) over 1 min, and the needle was left in place for 1-min post-injection to avoid backflow. The mice were injected once every two days for five injections. Two weeks after the last injection, the mice were transcardially perfused and sacrificed for analysis. The brains were then embedded in Fluorescence Mounting Medium (DAKO, S3023) and stored at −80°C.
Statistical analysis
All statistical analyses were performed using GraphPad software. Data are presented as mean ± standard error (SD). Data were analyzed by a Kruskal-Wallis test with Dunn's multiple comparison test, two-way ANOVA with Tukey's multiple post hoc comparison test, and unpaired two-tailed t-test for two-group comparison, as indicated in the figure legends. Statistical significance was defined as p < 0.05. All experiments were conducted more than three times and yielded consistent results.
Results
ECRG4 co-localizes with Aβ plaques in the AD hippocampus
Initially, we investigated the localization of ECRG4 within the hippocampus of patients with AD possessing an APOE3/4 genetic background, given that ECRG4 expression is known to increase in this context. 9 Our analysis confirmed significant ECRG4 expression, which predominantly co-localized with APP/Aβ signals in the hippocampus of patients with AD (Figure 1A, right panels), whereas signals of both ECRG4 and APP/Aβ were minimal in NAD specimens (Figure 1A, left panels). Subsequently, we explored whether elevated ECRG4 expression in the hippocampus of AD patients was contingent on the APOE genotype. Immunostaining of the AD hippocampus with APOE3/3, E3/4, and E4/4 demonstrated that ECRG4 was expressed in all samples and co-localized with APP/Aβ (Figure 1B). Moreover, we observed an increase in the number of merged signals that correlated with disease progression (Figure 1C). These findings suggest that ECRG4 accumulates in Aβ plaques within the hippocampus of AD patients, irrespective of the APOE background.
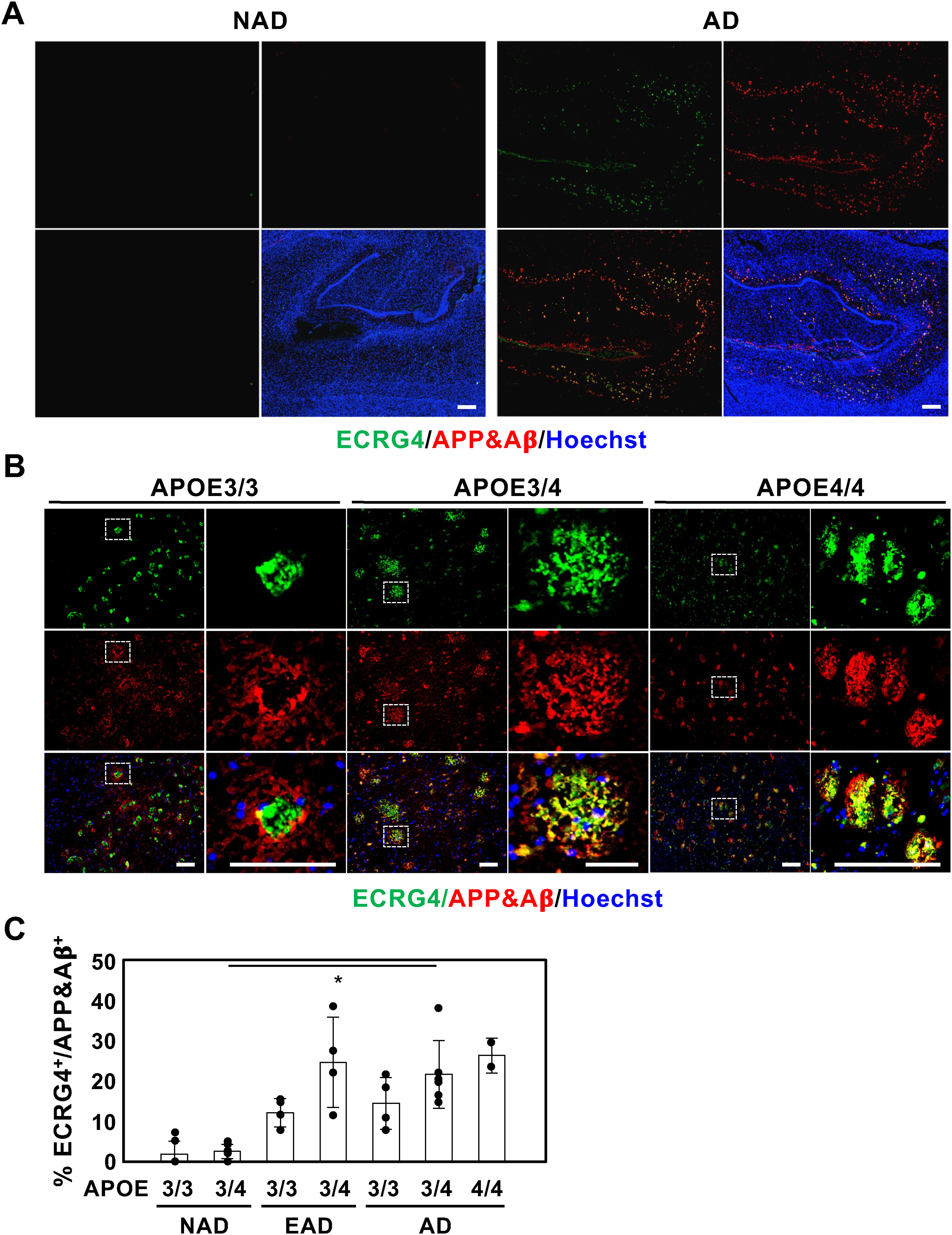
ECRG4 increased in hippocampus of EAD and AD with co-localization with APP/Aβ. (A) Representative images of hippocampal sections of NAD and AD with APOE3/4 immunolabeled for ECRG4 (green) and APP/Aβ (red). (B) Representative images of hippocampal sections of AD with APOE3/3, 3/4, and 4/4 immunolabeled for ECRG4 (green) and APP/Aβ (red). The higher magnification images are shown on the right of each figure surrounded by white dotted lines in the left images. (C) Percentage of co-localization of ECRG4 with APP/Aβ in hippocampal sections of NAD (4 and 6 samples of E3/3 and E3/4, respectively), EAD (4 samples of E3/3 and E3/4), and AD (3, 5, and 2 samples of E3/3, E3/4, and E4/4, respectively). All nuclei were counterstained with Hoechst33342 (blue). Scale bar: 200 μm (A) and 100 μm (B). Kruskal-Wallis test with Dunn's Multiple comparison test was used to determine statistical significance. Error bars indicate SD. *p < 0.05 (Color figure available online).
Accumulation of ECRG4 in hippocampus blood vessels is a novel hallmark for EAD
In addition to its co-localization with Aβ plaques, we examined the spatiotemporal accumulation of ECRG4 during the progression of AD. To determine the timing and location of ECRG4 increases during AD progression, we performed immunostaining on hippocampal sections from NAD, EAD, characterized by AD traits but not meeting all diagnostic criteria, and AD for ECRG4. We observed Aβ plaque-like ECRG4 signals in both the EAD and AD hippocampi (Supplemental Figure 1).17,18
Unexpectedly, we identified strong ECRG4 signals distinct from Aβ plaques in the EAD sections (blue arrowheads, Supplemental Figure 1A, B). These signals were more pronounced in APOE3/3 EAD than in APOE3/4 and were significantly diminished in the AD sections, irrespective of the APOE genetic background (Supplemental Figure 1). The morphology of the strong ECRG4 signals in the EAD sections suggests ECRG4 accumulation in the blood vessels.
To investigate this possibility, we immunolabeled hippocampal sections with anti-ECRG4 Ab, anti-APP/Aβ Ab, and UEA1, which preferentially binds to endothelial cells in the blood vessels. We observed that ECRG4 levels increased in both the EAD and AD sections, localizing with UEA1+ endothelial cells, whereas small dot signals were also detected in the NAD sections (Supplemental Figure 2A, B). Notably, ECRG4 signals on UEA1+ endothelial cells faintly merged with APP/Aβ, particularly as ECRG4 primarily co-localized with APP/Aβ (arrows in Supplemental Figure 2A, B). We confirmed that ECRG4 was strongly present on Nestin+ and CD31+ endothelial cells and weakly on NG2+ pericytes (arrows) in the EAD sections (Supplemental Figure 2C-E). These findings are consistent with the Human Protein Atlas data, which show that ECRG4 is expressed in both pericytes and endothelial cells. Together, these data suggest that ECRG4 protein is present not only within APP/Aβ aggregates but also in proximity to endothelial cells and pericytes or is upregulated in these cells during the initial stages of AD.
Overexpression of Ecrg4 induces accumulation of endogenous APP/Aβ in culture
Having established the presence and accumulation of ECRG4 in the hippocampus of patients with AD, we next sought to investigate the direct mechanistic link between ECRG4 and APP/Aβ accumulation using in vitro cell models. We overexpressed Ecrg4-human Fc fusion protein (Ecrg4-Fc) and FLAG-tagged C99 (FLAG-C99), which encodes a pre-Aβ fragment, either separately or together in 293 T cells, and then immunolabeled the cells for Ecrg4 and APP/Aβ. When both Ecrg4-Fc and FLAG-C99 were overexpressed, over 70% of Ecrg4+ cells were positive for APP/Aβ (Supplemental Figure 3A). Unexpectedly, overexpression of Ecrg4-Fc alone also induced the accumulation of endogenous APP/Aβ (over 70% in Ecrg4-overexpressing cells), whereas no such effect was observed in cells transfected with control vectors (Supplemental Figure 3A).
We confirmed the accumulation of endogenous APP/Aβ in Ecrg4-hFc-overexpressing cells by immunolabeling them with APP/Aβ and hFc (Supplemental Figure 3B), but not for FLAG and hFc (Supplemental Figure 3C). 293 T cells consistently express APP mRNA, and the expression level was not significantly affected by the overexpression of either Ecrg4-hFc or FLAG-C99 (Supplemental Figure 3D), these results suggest that the overexpression of Ecrg4 induces the accumulation of endogenous APP/Aβ through its association rather than by increasing APP/Aβ expression.
To verify the interaction between Ecrg4 and APP/Aβ, we overexpressed Ecrg4-Fc in 293 T cells, immunoprecipitated Ecrg4-Fc, and examined the precipitate for APP/Aβ using dot blotting. As shown in Supplemental Figure 3E, Ecrg4 is associated with endogenous APP/Aβ in 293 T cells. These data suggest that Ecrg4 initiates APP/Aβ accumulation in the brain.
Ecrg4(133-148) associated with the APP intracellular domain (AICD)
We conducted an in-depth investigation to determine which domain of Ecrg4 is responsible for inducing the accumulation of endogenous APP/Aβ in brain cells. Ecrg4 undergoes cleavage into four distinct fragments: the N-terminal signal sequence (1-31), 32-70, 71-132, and 133-148, facilitated by furin and thrombin. 12 We engineered a series of expression vectors encoding these fragments—Ecrg4(32-70), Ecrg4(71-132), and Ecrg4(133-148)—fused with hFc and subsequently overexpressed them in C6 glioma cells that retain the characteristics of both glial cells and neural stem cells and can express neuronal markers. 20 Our findings revealed that the overexpression of Ecrg4(133-148)-hFc and Ecrg4(32-148)-hFc led to the accumulation of APP/Aβ (Figure 2A). Over 70% of the cells expressing either Ecrg4(133-148)-hFc or Ecrg4(32-148)-hFc were positive for APP/Aβ, whereas cells expressing the other fragments were not (Figure 2B). Notably, no polyclonal ECRG4+ signals were detected in ECRG4(133-148)-overexpressing cells (Figure 2A). These results identified Ecrg4(133-148) as the fragment responsible for this effect.
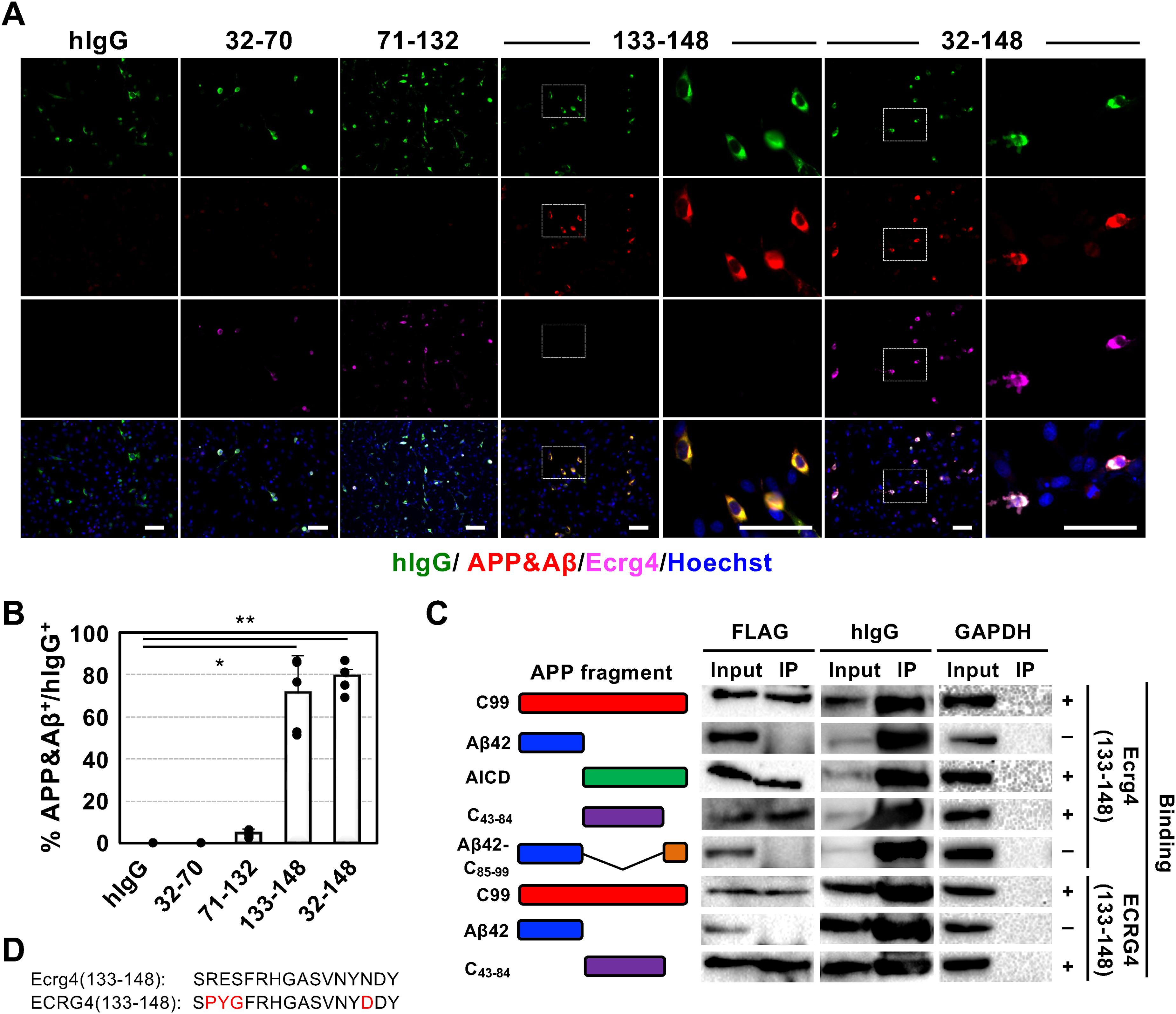
Ecrg4(133-148) induces accumulation of APP/Aβ by interacting with amino-terminus of AICD. (A) Representative immunostaining images of C6 cells overexpressing C99 (red) and a set of Ecrg4 fragments (purple) with hIgG (green), hIgG alone (hIgG), Ecrg4(32-70)-hIgG, Ecrg4(71-132)-hIgG, Ecrg4(133-148)-hIgG, or Ecrg4(32-148)- hIgG. The higher-magnification images of the inlets in both 133-148 and 32-148 are shown in the right panel. Scale bar: 100 μm. (B) Quantitative data of the percentage of APP/Aβ-positive cells from the experiments shown in (A). n = 5 for each group. Kruskal-Wallis test with Dunn's Multiple comparison test was used to determine statistical significance. Error bars indicate SD. *p < 0.05, **p < 0.01. (C) Co-immunoprecipitation assay for identifying the binding region in C99 with Ecrg4(133-148). A set of 3×FLAG tagged C99 and its fragments, Aβ42, AICD, C43-84, and Aβ42 + C85-99, were co-expressed with either Ecgr4(133-148)-hIgG or ECRG4(133-148)-hIgG in 293 T cells. Total cell extracts were immunoprecipitated with anti-FLAG Ab. Cell extracts (input) and immunoprecipitants (IP) were analyzed by western blotting using anti-FLAG, anti-hIgG (precipitation control), and anti-GAPDH (internal control) antibodies. The schematic image on the left shows C99 and the fragments used in the assay. The plus and minus signs in the right panel indicate whether the two proteins interact. (D) The amino acid sequence of ECRG4(133-148) and Ecrg4(133-148). The amino acids that differ between species are shown in red (Color figure available online).
Subsequently, we examined which domain of APP/Aβ associates with Ecrg4(133-148). Given that the Aβ precursor C99 accumulates in the brains of individuals with AD and contributes to inflammatory responses, synaptic dysfunction, and neuronal cell death,29,30 we initially investigated whether Ecrg4(133-148)-hFc interacts with C99. Through co-immunoprecipitation (Co-IP) assay, we confirmed the interaction between Ecrg4(133-148) and C99 (Figure 2C).
Given that C99 is cleaved into Aβ42 (C1-42) and AICD (C43-99), we further explored the target region of Ecrg4(133-148). Based on our previous immunohistochemical and in vitro studies showing colocalization between ECRG4 and APP/Aβ, we initially hypothesized that ECRG4(133–148) might interact directly with Aβ. However, we unexpectedly identified AICD as the binding target of Ecrg4(133-148). In contrast to the extensively studied Aβ, there have been limited investigations into factors such as Fe65 and Tip60, which are associated with the C-terminus (C85-99) of AICD through a binding motif sequence GYENPTY.31–33 Utilizing a Co-IP assay, we observed that the N-terminus (C43-84) of AICD associates with Ecrg4(133-148), while we were unable to detect any expression of C85-99 in transfected cell extracts. Given that very short peptides containing fewer than 24 amino acid residues are structurally unstable and rapidly degraded by aminopeptidase, 34 we fused C85-99 to Aβ42 to stabilize it. After confirming with Alphafold3 35 that the fusion of C85-99 with Aβ42 does not alter the native structure (Supplemental Figure 4), we investigated the association of the fusion protein with Ecrg4(133-148) and found no association between the proteins (Figure 2C).
Considering that mouse and human ECRG4(133-148) differ by four amino acid residues (Figure 2D, shown in red), we further examined the binding of ECRG4(133-148) with C99 and its fragments. We confirmed that ECRG4(133-148) binds to C43-84 but not to Aβ42 (Figure 2C). The data revealed an association between Ecrg4(133-148) and C43-84 and revealed that the nine conserved amino acids in both species, FRHGASVNY, contributed to this association.
Administration of ECRG4(133-148) accelerated the accumulation of APP/Aβ in the brains of AD model mouse APPNL−G−F/NL−G−F
Considering that ECRG4 signals increase in the brain with age, accumulates APP/AICD by direct binding, and associates with Aβ plaques in the hippocampus of patients with EAD and AD, these observations suggest that ECRG4 may be involved in the initiation of plaque formation. To investigate this hypothesis, we intracerebrally injected the ECRG4 (133-148) peptide into C57BL/6 mice every two days for a total of five injections (Supplemental Figure 5A). Brain sections were collected at 2, 7, 14, and 22 days post-injection and immunolabeled for APP/Aβ. As shown in Supplemental Figure 5B, none of the sections exhibited APP/Aβ labeling.
Given that Aβ accumulation is a lifelong process in the brain, inducing plaque formation in a normal brain within a short timeframe may be challenging. Consequently, we examined whether ECRG4(133-148) injection accelerated plaque formation in APPNL−G−F/NL−G−F mice, in which the APP gene was replaced with human APP containing three mutations: Swedish (KM670/671NL), Arctic (E693G), and Beyreuther/Iberian (I716F). 17 We intracerebrally injected the ECRG4 (133-148) peptide into APPNL−G−F/NL−G−F mice, following our protocol utilized for wild-type mice, and immunolabeled brain sections for APP/Aβ two weeks after the final injection (Figure 3A, B). An increase in APP/Aβ plaques was observed surrounding the peptide-injected site compared to that in the PBS-injected brains (Figure 3C, D). Furthermore, we observed increased number of glial fibrillar acidic protein (GFAP) positive astrocytes in the ECRG4(133-148) injected brain (Figure 3E, F).
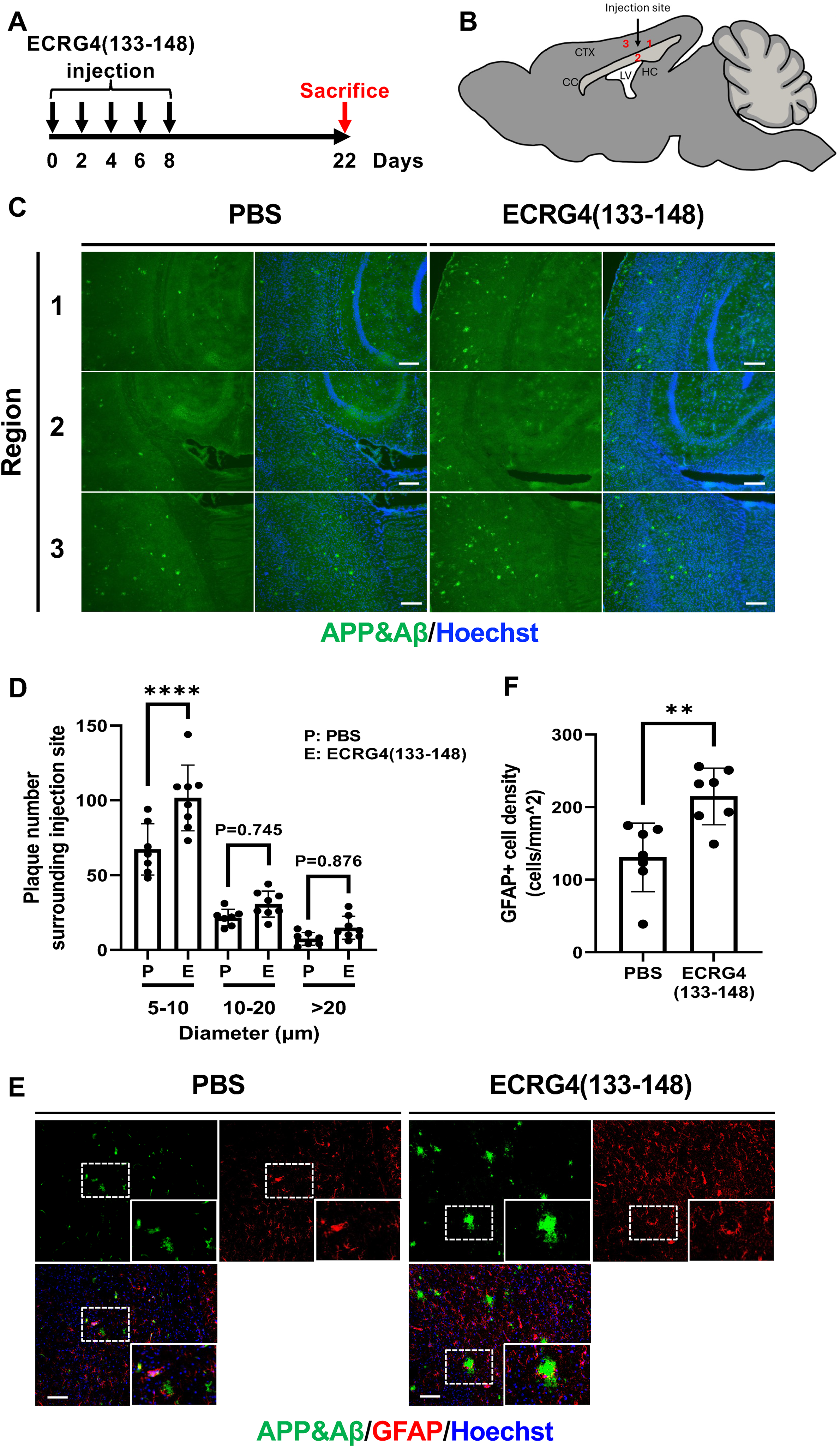
Injection of ECRG4(133-148) peptides accelerates APP/Aβ deposition. (A) APPNL−G−F/NL−G−F mice were injected with ECRG4(133-148) every 2 days, five times in total, and sacrificed 2 weeks after the last injection. (B) Schematic representation of the injection sites. Regions 1, 2, and 3 around the injection site were chosen for the APP/Aβ deposition area analysis. Black arrows indicate the injection sites. CTX: Cortex. CC: Corpus callosum. HC: Hippocampus. LV: Lateral ventricle. (C) Representative images of brain sections immunolabeled for APP/Aβ (green). (D) Quantification of (C). Two-way ANOVA followed by Tukey's multiple post hoc comparison test was used to determine statistical significance among all groups. n = 7 for PBS and n=8 for ECRG4(133-148). (E) Representative images of brain sections immunolabeled for APP/Aβ (green) and GFAP (red). The inset in each figure provides a detail view of the area delineated by the dotted box. (F) Quantification of GFAP cell density in (E). Statistical significance was determined using unpaired two-tailed Student's t-test. n = 7 for each group. All nuclei were counterstained with Hoechst33342 (blue). Scale bar: 200 μm (C) and 100 μm (E). Data are presented as mean ± SD. Each data point represents an individual animal. For plaque number analysis, regions 1–3 were analyzed and summed to obtain a single value. And overlapping plaques were excluded. For GFAP quantification, a single region within regions 1-3 with prominent GFAP immunoreactivity was selected for quantification in each mouse to assess peak astrocytic activation surrounding the injection site. **p < 0.01, ****p < 0.0001 (Color figure available online).
ECRG4(133-148)-containing peptide increased with AD progression and largely co-localized with Aβ plaques in AD patient brain
We investigated the localization of ECRG4(133-148)-containing peptides within the hippocampus of patients with NAD, EAD, and AD across three APOE genotypes, utilizing mouse anti-ECRG4(133-148), which specifically recognizes ECRG4(133-148) that the rabbit Ab did not (Supplemental Figure 6A-C), and rabbit anti-Aβ42 Abs, which exclusively recognize Aβ42 (Figure 4A). Immunohistochemical analysis demonstrated that mouse anti-ECRG4(133-148) signal was extensively detected in the hippocampus of patients with EAD and AD (Figure 4B), compared to rabbit anti-Aβ42 signal (Figure 4C), and significantly overlapped with the anti-Aβ42 signal, irrespective of the APOE subtype (Figure 4D, Supplemental Figure 7A), as illustrated in Supplemental Figure 2A and 2B.
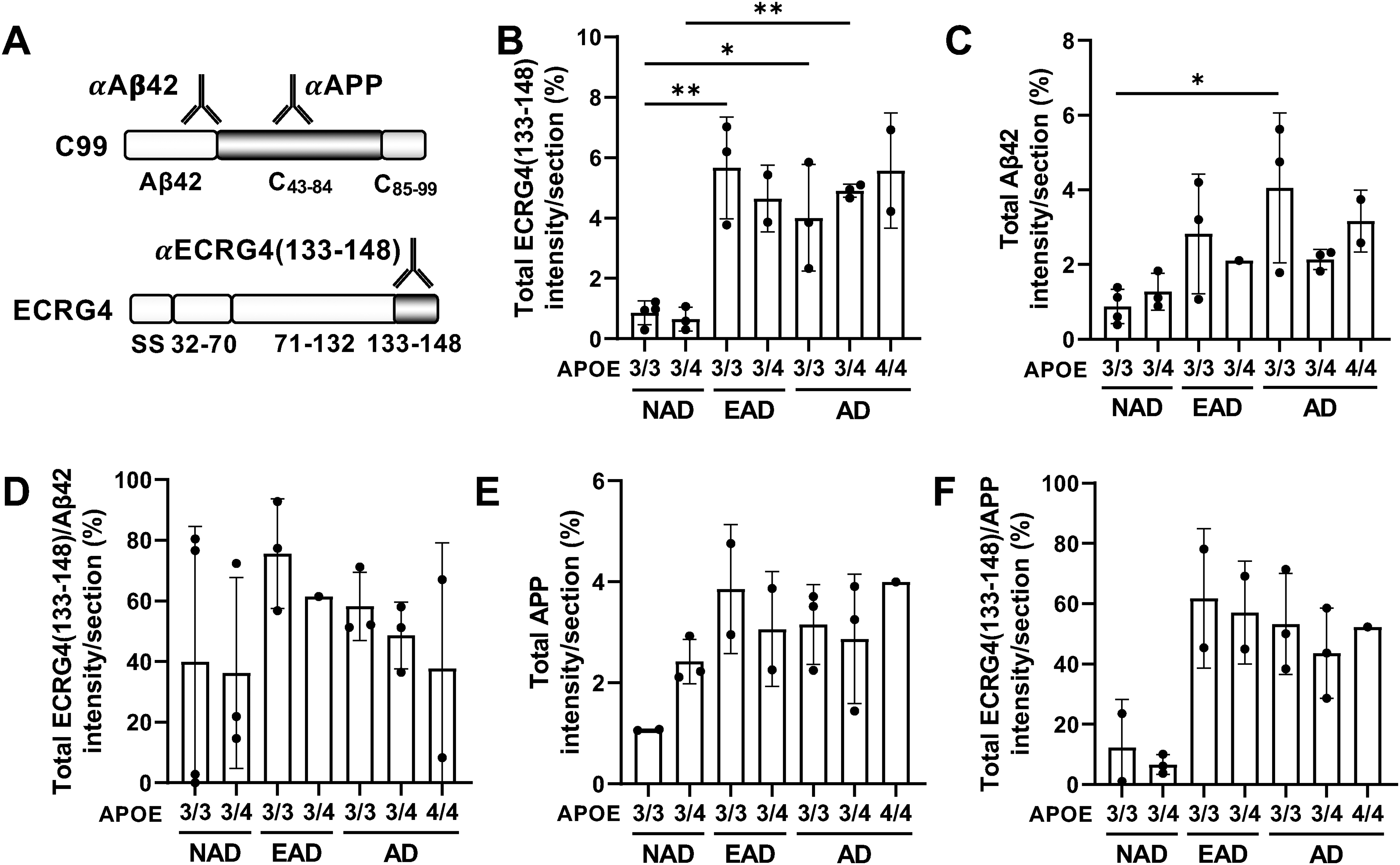
ECRG4(133-148)- and C43−84-containing peptides are co-localized in human hippocampus and increase with the disease progression. (A) Schematic representation of antibody recognition sites. αAβ42 and αAPP recognize Aβ42 and AICD region in C99, respectively (upper panel), while αECRG4(133–148) recognizes both full-length ECRG4 and its C-terminal fragment ECRG4(133–148) (lower panel). (B, C) Quantification of ECRG4(133–148)+ area (B) and Aβ42 + area (C) in hippocampal sections from NAD, EAD, and AD patients with APOE 3/3, 3/4, and 4/4 genotypes. (D) Percentage of ECRG4(133–148)+ area within Aβ42+ area, calculated from (B) and (C). (E) Quantification of APP+ area in hippocampal sections from the same cohorts. (F) Percentage of ECRG4(133–148)+ area within APP+ area, calculated from (B) and (E). Sample sizes (n) per group are as follows:
Whereas the rabbit anti-ECRG4 antibody identified only a limited number of ECRG4 signals in NAD brains (Figure 1A, Supplemental Figure 2A-C), numerous mouse anti-ECRG4(133-148)+ cells were observed in the same brain (Figure 4B, Supplemental Figure 7). The findings indicated that ECRG4(133-148) levels are elevated in the aged hippocampus, even in the absence of AD symptoms.
We next utilized another anti-APP antibody whose epitope is located at the AICD region (Figure 4A). Immunohistochemical analysis further demonstrated that the rabbit anti-APP+ signal, indicative of AICD-containing peptides, was extensively observed in the NAD hippocampus, corresponding to endogenous APP expression in brain cells (HPA: https://www.proteinatlas.org/ENSG00000142192-APP/brain) (Figure 4E, Supplemental Figure 7B). Although the overall intensity of the rabbit anti-APP+ signal in sections did not significantly increase with AD progression, its accumulation pattern was altered (Figure 4E, Supplemental Figure 7B). In addition, the mouse anti-ECRG4(133-148)+ signal overlapped with rabbit anti-APP+ signal increased with disease progression (Figure 4F). These findings imply that the augmented presence of ECRG4(133-148)-containing peptides facilitates the accumulation of AICD-containing peptides as AD advances in the brain. Collectively, these data suggest that ECRG4(133-148)-containing peptides play a role in the initiation and enhancement of plaque formation, comprising Aβ42 and AICD-containing peptides.
Discussion
Our study revealed that ECRG4 increased as AD progresses, and co-localized with APP/Aβ signals in the hippocampus of patients, irrespective of the APOE phenotype. Evidence suggests that the overexpression of ECRG4(133-148) promotes the accumulation of APP/Aβ by directly binding to AICD in cultured cells. Moreover, the introduction of ECRG4(133-148) into the brains of APPNL−G−F/NL−G−F mice led to a more rapid accumulation of APP. In the NAD hippocampus, we identified peptides containing ECRG4(133-148) alongside those containing Aβ or AICD, with their signals intensifying as AD advanced. Given the robust expression of ECRG4 in the brain, as documented in the Human Protein Atlas (https://www.proteinatlas.org/ENSG00000119147-ECRG4/tissue), these findings suggest that ECRG4(133-148) may play a pivotal role in initiating plaque formation, predominantly within the brain.
The molecular mechanism by which ECRG4(133-148) leads to APP/Aβ accumulation is not yet fully understood, but several mechanisms could be proposed. One is that ECRG4(133-148) may enhance Aβ production by facilitating the dimerization of C99 through Gly x-x-x-Gly motifs, which are conducive to γ-secretase cleavage, hence increasing extracellular Aβ level.36,37 ECRG4(133-148) may also regulate the stability, localization, or AICD-dependent transcription, depending on the interacting proteins, such as transcriptional regulators Fe65 and Tip60, resulting in either enhancement or inhibition of this process. 38 Therefore, acquiring deeper molecular insights is vital for the creation of innovative therapeutic approaches for AD that focus on reducing Aβ accumulation by targeting ECRG4(133-148).
It is unclear the cellular context in which ECRG4(133-148) and AICD would interact remains unclear. We demonstrated that ECRG4(133-148) directly associates with AICD using a co-immunoprecipitation assay. However, given that ECRG4(133-148) is an extracellular peptide and AICD is an intracellular peptide, the question of how these proteins interact under physiological conditions arise. As illustrated in Figure 2A, we observed that all Ecrg4 fragment-hIgG fusion proteins were present in the cytoplasm, possibly within the endoplasmic reticulum (ER), suggesting that nascent ECRG4(133-148) may associate with APP in the ER compartment prior to secretion.
Another potential mechanism is that ECRG4(133-148) could be incorporated into the cells. Previous study had reported that ECRG4(133-148) can be internalized into cells through interacting with the TLR4/CD14/MD2 receptor complex and localizes to perinuclear region. 13 Since the internalization did not activate the canonical NF-κB pathway, suggesting that ECRG4(133–148) may undergo noncanonical intracellular trafficking. Although direct evidence for ECRG4(133-148) undergoing endosomal escape is currently lacking, endosomal trafficking is a dynamic process involving membrane remodeling events. Notably, intraluminal vesicles within the endosome can undergo back-fusion with the endosomal membrane, leading to the release of vesicular contents into the cytoplasm. 39 Thus, it is plausible that a fraction of internalized ECRG4(133-148) may access the cytosolic compartment and interact with AICD. In addition, neural stem cells and neurons have been reported to express the TLR4, CD14 and MD2, suggesting that such cells may be capable of internalizing ECRG4(133-148)40,41 (https://www.proteinatlas.org/ENSG00000170458-CD14/single±cell/brain, https://www.proteinatlas.org/ENSG00000154589-LY96/single+cell/brain). Nevertheless, mechanisms underlying ECRG4(133-148) internalization, including cell-type specificity, other potential receptors or transporters, remain to be fully characterized.
Beyond its intracellular effects, our immunohistochemical analysis revealed pronounced ECRG4 signals around endothelial cells in EAD, particular APOE3/3 hippocampus but not detect both ECRG4 and APP/Aβ on UEA1+ vascular endothelial cells (Supplemental Figure 2A, B). Given that ECRG4 induces the expression of many inflammatory cytokines in macrophages and microglia, blood-brain-barrier containing endothelial cells enclosed by ECRG4 may suffer damage through the activated residential macrophages or microglias, enhancing the disease progression. This hypothesis is supported by the observations that the upregulation of ECRG4 expression in intravascular monocytes within the brains of AD patients 42 and the elevated ECRG4 levels in the sera of patients with EAD and AD. 27 However, the specific mechanism by which ECRG4 accumulates in and around blood vessels and its functional role at these sites remain to be elucidated.
Despite these insights, several limitations should be considered. First of all, regarding the ECRG4(133-148) in vivo injection experiment, although we demonstrated that the administration of ECRG4(133-148) facilitated the accumulation of APP/Aβ in the brain of AD model mice, the precise diffusion range of ECRG4(133-148) within the brain tissue remains difficult to define. The movement of small peptides in the brain is generally described within the framework of effective diffusion in the interstitial space, which is influenced by molecular size, extracellular tortuosity, and interactions with receptors, as well as enzymatic degradation. Previous studies of extracellular peptides, including Aβ monomers (40AA), EGF (53AA) and βNGF (118-120AA) have reported effective diffusion coefficients on the order of 0.623 × 10−6 cm2/s, 1.7 × 10−6 cm2/s and 1.3 × 10−6 cm2/s, respectively, in brain tissue.43,44 Given that ECRG4(133-148) is a smaller peptide (16AA), it would be expected to exhibit comparable or somewhat potentially broader diffusion compared to these proteins within the extracellular space; however, its actual distribution in vivo is likely constrained by binding interactions and proteolytic processing.
Second, although repeated administration of ECRG4(133–148) increased plaque burden in vivo, the concentrations used likely exceed physiological levels. Given that the endogenous concentrations of ECRG4 and its derived fragments, including ECRG4(133–148), remain largely unknown, the dosing strategy and concentrations adapted in this study were used for previous Aβ peptide injection paradigms to model the cumulative effects of prolonged peptide exposure within a feasible experimental timeframe.45–47 Therefore, our findings should be interpreted as demonstrating the potential capacity of ECRG4(133–148) to promote APP/Aβ accumulation, rather than reflecting its physiological concentration in vivo. Furthermore, investigating whether the administration of ECRG4(133-148) results in subsequent pathological outcomes, such as neuroinflammation, neuronal degeneration, or cognitive impairment, similar to human AD would contribute to establishing ECRG4(133-148) as a significant new therapeutic target for AD treatment.
Third, while the immunohistochemical analysis demonstrated that ECRG4 accumulates and colocalizes with APP/Aβ in human hippocampal sections, a thorough investigation into the specific cell types, aside from endothelial cells, that express or accumulate ECRG4 in the brain of AD patients remains to be conducted. Determining the primary cellular sources of ECRG4 and ECRG4(133-148) in human AD pathology is essential for understanding their proposed role as initiators.
Finally, mouse anti-ECRG4(133-148) utilized in immunohistochemical studies is capable of detecting both full-length ECRG4 and ECRG4(133–148) fragment, which prevents the specific identification of ECRG4(133–148) in human tissue samples. To accurately determine the physiological levels and spatial distribution of ECRG4(133–148) in the human brain, the development of antibodies that are specific to this fragment is imperative.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261462890 - Supplemental material for Carboxy terminal of ECRG4 is a potential initiator for amyloid pathology in Alzheimer's disease through interacting with APP intracellular domain
Supplemental material, sj-docx-1-alz-10.1177_13872877261462890 for Carboxy terminal of ECRG4 is a potential initiator for amyloid pathology in Alzheimer's disease through interacting with APP intracellular domain by Michael Lai Kit Chung, Shuji Takeda, Junnan Ryu, Shigeo Murayama, Uichi Koshimizu and Toru Kondo in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
We thank Kenju Miura for his initial support of the study. We also thank Prof. Shigekazu Nagata for the pEF-Fc plasmid, Prof. Takaomi C. Saido for the APPNL−G−F/NL−G−F mice, Dr Toshihiro Hata for immunohistochemical analysis of patient brains, Dr Maho Morishima for general support to proceed this research, and all members in Kondo Lab for their helpful comments to this research.
Ethical considerations
The study was conducted in accordance with the principles set forth in the Declaration of Helsinki and received ethical approval from the Ethics Committee of the Tokyo Metropolitan Institute for Geriatrics and Gerontology (Protocol code: R2, Date of approval: April 15, 2019) and the Ethics Committee of Hokkaido University Institute for Genetic Medicine (Protocol code: 12-0001(6), Date of approval: December 21, 2021) for research involving human participants.
Consent to participate
All included subjects gave informed consent.
Consent for publication
Not applicable
Author contribution(s)
Funding
This work was partly supported by collaborative research grants from ASUBIO Pharma Corporation (to T.K.), JSPS KAKENHI (grant number JP22H04923 (CoBiA) to S.M.), AMED (grant number JP21wm0425019 to S.M.), the Joint Research Program of the Institute for Genetic Medicine, Hokkaido University (to T.K.), and JST SPRING (grant number JPMJSP2119, to M.L.K.C). Japan Agency for Medical Research and Development, Japan Society for the Promotion of Science, Japan Science and Technology Corporation.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S.T. and U.K were employees of ASUBIO pharma Co. Ltd. The remaining authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data produced and/or analyzed during the current study is available from the first and corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.