
Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder defined by three pathological hallmarks: amyloid-β (Aβ) deposition, tau hyperphosphorylation leading to neurofibrillary tangles formation, and neuronal loss. Mounting evidence over the past decade has underscored that mitophagy deficiency and calcium dyshomeostasis play pivotal regulatory roles in AD pathological progression, with these two abnormalities persisting throughout the entire course of disease onset and development. Mitophagy impairment can result in the accumulation of dysfunctional mitochondria, thereby further exacerbating calcium dyshomeostasis which can suppress autophagic flux in turn. This reciprocal interaction establishes a vicious cycle that can synergistically accelerate Aβ plaque and neurofibrillary tangle formation, impair synaptic structure and function, and ultimately elicit neuronal programmed cell death and cognitive decline. This review systematically summarizes the biological basis of mitophagy and calcium homeostasis, as well as their mutual regulatory networks. It places particular emphasis on deciphering the pathological mechanisms through which concurrent impairments of these two pathways synergistically drive AD pathogenesis and progression. Furthermore, we propose intervention strategies targeting to modulate mitophagy deficiency and calcium dyshomeostasis, which hold great promise for providing novel insights and potential therapeutic targets for the clinical management of AD.
Introduction
Alzheimer disease (AD) is the most prevalent form of dementia worldwide. With the accelerating global aging process, its incidence has been on the rise annually. 1 Currently, there remains a lack of effective therapeutic interventions capable of slowing or reversing disease progression in the clinical setting.2–4 The classical “amyloid cascade hypothesis” and “tau hyperphosphorylation hypothesis” have clarified the primary pathological features of AD, yet they fail to fully account for early prodromal pathological alterations such as abnormal energy metabolism and synaptic dysfunction. 5 This implies that the pathological mechanism of AD is governed by a more complex regulatory network.
In recent years, the roles of mitophagy deficiency and calcium dyshomeostasis in AD have gradually become research hotspots. As the central hub of cellular energy metabolism, mitochondria also serve as key organelles regulating intracellular calcium homeostasis. Mitophagy, by selectively eliminating damaged mitochondria, directly modulates the functional integrity of mitochondrial calcium transport-related proteins, thereby participating in the precise regulation of calcium homeostasis and establishing a tight functional connection between the two processes.6,7
Emerging evidence suggests that decreased mitophagic activity and disrupted calcium signaling in critical cognitive brain regions such as the hippocampus and prefrontal cortex may precede the formation of amyloid-β (Aβ) plaques and neurofibrillary tangles in the AD patients.8,9 However, a consensus on this temporal sequence remains elusive. More importantly, these two abnormalities do not exist in isolation; instead, they interact through multiple molecular pathways to form a “pathological vicious cycle.” This cycle is closely crosstalked with neuroinflammation, synaptic impairment, and neuronal death, persisting throughout the entire disease course of AD from the preclinical stage to advanced dementia and serving as one of the key drivers of disease progression. Although there is ongoing debate about whether mitophagy impairment or Aβ deposition is the upstream initiating event in AD, this debate must be interpreted in the context of disease subtype heterogeneity between familial AD (FAD) and sporadic AD (SAD), which accounts for more than 95% of all clinical AD cases.10,11 For monogenic FAD driven by mutations in APP, PSEN1, or PSEN2, aberrant Aβ overproduction is the well-established upstream initiating event, which precedes and directly triggers mitophagy dysfunction and neuronal calcium dyshomeostasis. 12 Notably, PSEN1/2 mutations can also directly induce endoplasmic reticulum (ER) calcium leak and mitochondrial calcium overload independent of Aβ pathology, which in turn aggravate mitophagy impairment and further promote Aβ generation, thus establishing the mitophagy-calcium vicious cycle at the earliest stage of FAD. 13 In stark contrast, for age-related sporadic AD, accumulating preclinical and clinical evidence indicates that aging-induced mitophagy decline and neuronal calcium buffering dysfunction are very early initiating events, which occur prior to detectable cerebral Aβ plaque deposition and tau hyperphosphorylation. 14 These early subcellular impairments drive the abnormal accumulation of Aβ and tau pathology, which in turn further exacerbate mitophagy failure and calcium dyshomeostasis, continuously amplifying the pathological vicious cycle throughout the entire progression of AD. 15 Taken together, regardless of the differential initiating drivers in FAD and SAD, the reciprocal vicious cycle between mitophagy impairment and calcium dyshomeostasis is the core driving force for the progression of AD pathology. Therefore, in-depth dissection of their synergistic mechanism can not only refine the theoretical framework of AD pathological mechanisms and compensate for the limitations of classical hypotheses but also provide novel therapeutic strategies targeting the “mitophagy-calcium homeostasis” axis. This represents an urgent and critical research direction awaiting breakthroughs in the field of neurodegenerative diseases.
The biological basis of mitophagy and calcium homeostasis
Regulatory mechanisms of mitophagy
Mitophagy acts as a critical quality control mechanism for the selective clearance of damaged mitochondria in cells. Its major regulatory pathways are mainly classified into two categories: ubiquitin-dependent and ubiquitin-independent pathways, among which the PINK1/Parkin pathway is a classical ubiquitin-dependent regulatory pathway. 16 Under physiological conditions, when mitochondrial membrane potential (ΔΨm) is stable, PINK1 is cleaved and degraded by mitochondrial matrix proteases, maintaining a low-activity state.16,17 However, when mitochondrial damage leads to a decrease in ΔΨm, PINK1 fails to translocate into the mitochondrial matrix, accumulates on the outer mitochondrial membrane, and phosphorylates to activate the E3 ubiquitin ligase Parkin. 18 Activated Parkin ubiquitinates outer mitochondrial membrane proteins, such as mitochondrial fusion protein 1/2 (MFN1/2) and voltage-dependent anion channel 1 (VDAC1), thereby recruiting autophagy-related proteins p62/SQSTM1 and LC3 to promote autophagosome formation. Eventually, the autophagosome fuses with the lysosome to complete the degradation of damaged mitochondria.19–22
In contrast, non-ubiquitin-dependent pathways regulating mitophagy mainly include those mediated by mitophagy receptors such as NIX, BNIP3, and FUNDC1. These receptors contain a conserved LC3 Interaction Region (LIR), which enables direct binding to LC3 to initiate autophagic processes without relying on ubiquitination modification steps.23,24 Additionally, the AMPK/ULK1 pathway modulates mitophagy under energy stress conditions. AMPK can not only trigger the autophagic cascade by phosphorylating ULK1, a key autophagy-initiating kinase, but also phosphorylate mitochondrial fission factor (MFF) which facilitates mitochondrial fission into “easily cleared” small fragments, laying a structural foundation for subsequent autophagic clearance. 25 Moreover, the dynamic balance between mitochondrial fusion and fission is also integrated into the regulatory network of mitophagy. Outer mitochondrial membrane fusion is dependent on mitochondrial fusion protein 1/2 (MFN1/2), while inner membrane fusion is mediated by optic atrophy protein 1 (OPA1). The fission process is primarily co-mediated by mitochondrial fission protein 1 (Drp1) and MFF. Notably, excessive Drp1-mediated mitochondrial fission exacerbates the accumulation of damaged mitochondria, whereas ubiquitination of MFN1/2 inhibits mitochondrial fusion, further promoting the autophagic clearance of damaged mitochondria (Figure 1).26,27
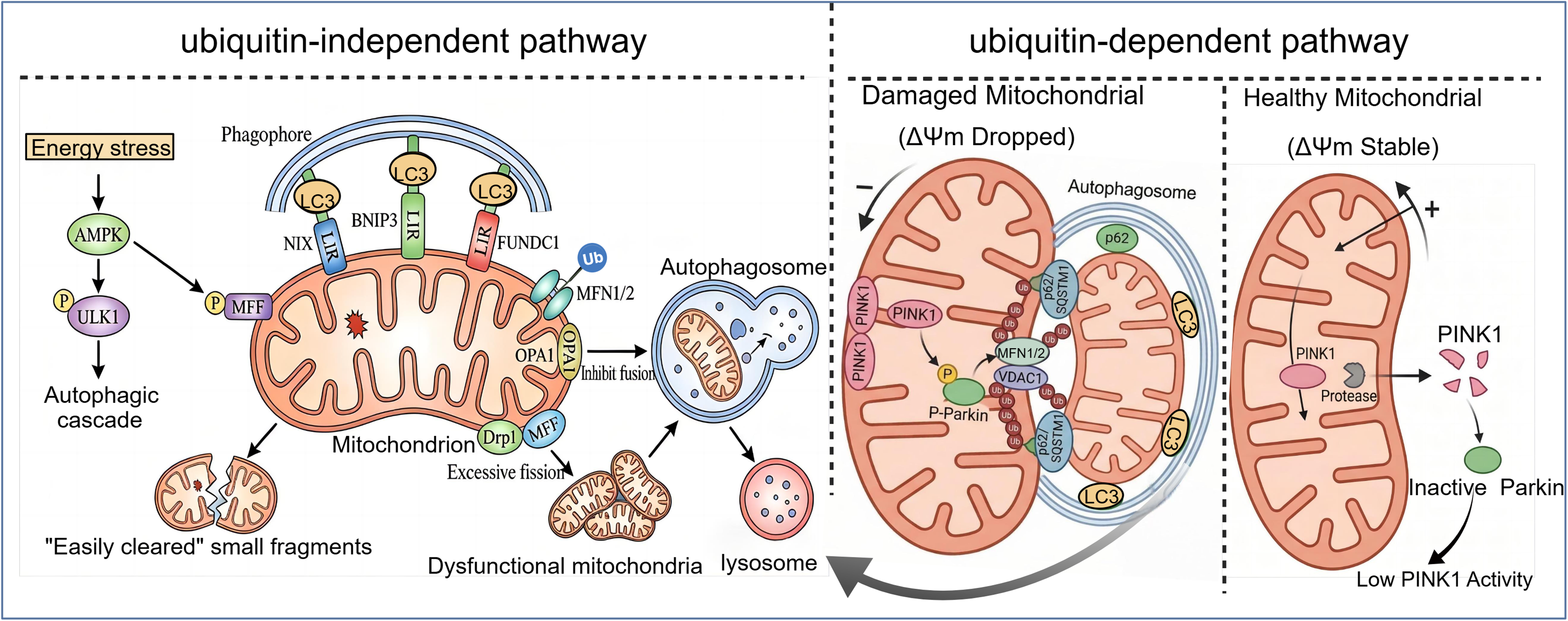
Ubiquitin-dependent (PINK1/Parkin) and ubiquitin-independent pathways in mitophagy regulation. Under stable mitochondrial membrane potential (ΔΨm), PINK1 is cleaved and degraded by mitochondrial matrix proteases, maintaining low basal activity. Mitochondrial damage-induced ΔΨm depolarization promotes PINK1 accumulation on the outer mitochondrial membrane, which phosphorylates and activates the E3 ubiquitin ligase Parkin. Parkin ubiquitinates outer mitochondrial membrane proteins, thereby recruiting autophagy-related proteins p62/SQSTM1 and LC3 to form autophagosomes. Under energy stress, cells selectively clear damaged mitochondria via the AMPK–ULK1 axis, activated AMPK phosphorylates ULK1 to initiate autophagy and mitochondrial fission factor (MFF). Phosphorylated MFF recruits Drp1 to mediate excessive mitochondrial fission, generating small “easily cleared” fragments, while ubiquitination of MFN1/2 inhibits mitochondrial fusion. On the outer membrane of damaged mitochondria, receptors NIX, BNIP3, and FUNDC1 bind LC3 on autophagic membranes via their LIR motifs, recruiting damaged mitochondria to expanding phagophores and driving autophagosome formation. Finally, autophagosomes loaded with damaged mitochondria fuse with lysosomes for degradation.
Biological basis of calcium homeostasis
Calcium ions (Ca2+), as important intracellular second messengers, are extensively involved in multiple physiological processes, including the regulation of gene expression, cell cycle progression, initiation of apoptosis, and maintenance of neuronal plasticity. 28 Calcium homeostasis refers to a complex process by which cells maintain the dynamic balance of intracellular Ca2+ concentration through the synergistic action of various organelles such as the cell membrane, endoplasmic reticulum, and mitochondria, as well as specific ion channels, transmembrane transporters, and regulatory proteins. 29
On the molecular regulatory level, molecules such as N-methyl-D-aspartate receptors (NMDARs) and L-type voltage-dependent calcium channels (L-VDCCs) on the cell membrane mediate extracellular Ca2+ influx, while Na+-Ca2+exchangers (NCX) participate in Ca2+ efflux. The endoplasmic reticulum can complete Ca2+ release and storage via ryanodine receptors (RyRs) and inositol 14,5-trisphosphate receptors (IP3Rs). As a core intracellular Ca2+ store, mitochondria rely on the mitochondrial calcium uniporter complex for Ca2+ uptake, which consists of mitochondrial calcium uniporter (MCU), mitochondrial calcium uptake 1 (MICU1), mitochondrial calcium uptake 2 (MICU2), and essential MCU regulator (EMRE), enabling the selective uptake of Ca2+. 30 Meanwhile, mitochondria can also release Ca2+ through pathways such as the mitochondrial permeability transition pore (mPTP). Additionally, Ca2+ signals can further regulate various cellular physiological functions via downstream effector molecules, including calmodulin (CaM) and calmodulin-dependent protein kinases (CaMKs) (Figure 2).17,31 The aforementioned regulatory mechanisms of calcium homeostasis serve as an important foundation for ensuring the normal operation of neuronal synaptic plasticity, energy metabolism, gene transcription, and other functions.
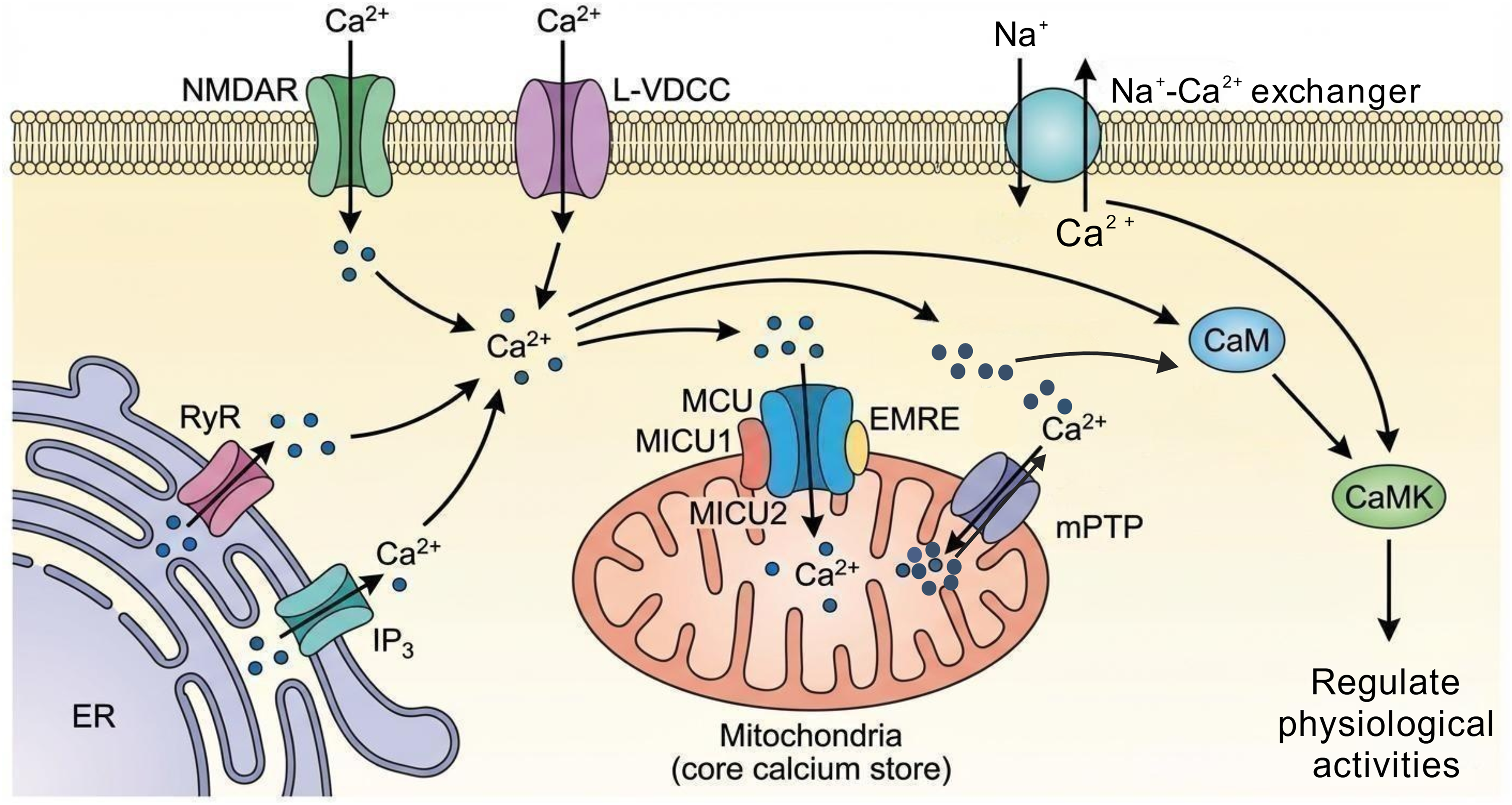
Mechanisms of cellular calcium homeostasis regulation. Extracellular calcium influx is mediated by N-methyl-D-aspartate receptors (NMDARs) and L-type voltage-dependent calcium channels (L-VDCCs) on the cell membrane, while the Na+-Ca2+ exchanger (NCX) mediates calcium efflux. The endoplasmic reticulum stores and releases calcium via ryanodine receptors (RyRs) and IP3 receptors. As the central calcium store, mitochondria selectively uptake calcium through mitochondrial calcium uniporter (MCU), mitochondrial calcium uptake 1/2 (MICU1/2), and essential MCU regulator (EMRE), and release calcium via the mitochondrial permeability transition pore (mPTP) and other pathways. Additionally, calcium signals regulate cellular physiological activities through downstream molecules such as calmodulin (CaM) and calmodulin-dependent kinases (CaMKs).
Reciprocal regulation between mitophagy and calcium homeostasis
Close bidirectional regulatory crosstalk exists between mitophagy and calcium homeostasis, and they synergistically maintain the stability of cellular energy metabolism and the balance of signal transduction networks. As an essential node in the calcium signal regulatory network, mitochondria are not only precisely regulated by calcium signals in their autophagic processes, but their functional integrity and quantity homeostasis alsodirectly determine the overall calcium buffering capacity of cells. Accumulating evidence has confirmed that the activation of mitophagy can selectively eliminate damaged mitochondria induced by calcium overload, thereby restoring cellular calcium buffering function; conversely, mitophagy deficiency can exacerbate calcium dyshomeostasis and amplify its pathological effects. 32
A normal mitochondrial membrane potential (ΔΨm) is a prerequisite for mitochondrial Ca2+ uptake which drives Ca2+ into the mitochondrial matrix through the MCU, while the membrane potential of damaged mitochondria decreases significantly, resulting in the loss of Ca2+ uptake capacity.33,34 By selectively eliminating damaged mitochondria, mitophagy can reduce abnormal Ca2+ release, thereby directly participating in the regulation of calcium homeostasis. After damaged mitochondria are cleared by autophagy via the activated PINK1/Parkin pathway, the membrane potential of the remaining normal mitochondria is restored to normal, and the Ca2+ uptake function is maintained, preventing cytoplasmic Ca2+ overload due to the failure of mitochondrial buffering. 19 Calcium homeostasis also serves as a key signaling hub regulating mitophagic activity. Physiological levels of Ca2+ signals can activate the PINK1/Parkin pathway to promote mitophagy. Meanwhile, calcium signals can indirectly affect mitophagic processes by influencing mitochondrial morphology and function. Under low Ca2+ concentrations in the mitochondrial matrix, the biological activity of mitochondrial fusion proteins is significantly enhanced, enabling mitochondria to form larger networks. In this state, mitochondria exhibit complete physiological functions and are generally not recognized and degraded by autophagic pathways. In contrast, when Ca2+ concentrations in the mitochondrial matrix increase, calcium signals can regulate the activity of mitochondrial fission-related factors, inducing mitochondrial fragmentation. Such fragmented mitochondria are usually the target substrates for mitophagic recognition and clearance. Additionally, calcium signals may directly regulate mitophagic processes by affecting the formation and maturation of autophagosomes. These mechanisms can collectively form a complex network by which calcium signals regulate mitophagy, which is crucial for maintaining the balance of mitochondrial quality and quantity in cells. 35
Mitophagy deficiency and calcium dyshomeostasis form a vicious cycle
Once mitophagy deficiency occurs, it will establish a stable vicious cycle with calcium dyshomeostasis through bidirectional regulation involving “mitochondrial dysfunction - autophagic pathway inhibition - calcium overload - mitophagy deficiency”.
In the context of mitophagy deficiency, the accumulation of damaged mitochondria will impair mechanisms of calcium uptake and release. Reduced mitochondrial ATP production relating to mitophagy deficiency can inhibit the functional activity of Na+-Ca2+ exchangers, exacerbating cytoplasmic calcium overload. 36 On the other hand, abnormal opening of the mitochondrial permeability transition pore (mPTP) can cause calcium store leakage. The mPTP is a non-specific channel on the mitochondrial membrane that opens transiently under physiological conditions to regulate mitochondrial calcium concentration. However, mitochondrial calcium overload induced by calcium dyshomeostasis triggers prolonged and irreversible opening of mPTP,35,36 leading to increased mitochondrial matrix osmotic pressure, mitochondrial swelling, and outer membrane rupture—further disrupting calcium homeostasis. 28
Abnormal calcium concentrations can interfere with mitophagic processes through multiple pathways. 37 Chronic calcium overload disrupts mitochondrial membrane potential, induces the expression of apoptosis-related proteins, and simultaneously inhibits the initiation of mitophagy, resulting in the massive accumulation of damaged mitochondria that cannot be promptly cleared. Studies have shown that high concentrations of calcium ions in mitochondria can damage mitochondrial DNA (mtDNA) and functional proteins by activating reactive oxygen species (ROS) signaling pathways. In turn, damaged mitochondria further reduce calcium buffering capacity, forming a positive feedback loop between calcium overload and mitochondrial damage.38,39
During calcium dyshomeostasis, cytoplasmic calcium overload abnormally activates Drp1 through two pathways. First, calcium can directly bind to the regulatory domain of Drp1, enhancing its guanosine triphosphate (GTP) enzyme activity and promoting Drp1 recruitment to the mitochondrial membrane. 17 Second, calcium can also activate calmodulin (CaM), which further phosphorylates Drp1 to maintain its sustained activation. 40 Excessive activation of Drp1 will induce excessive mitochondrial fission, generating a large number of fragmented mitochondrial fragments. Some of these fragments with mild damage may be mislabeled, leading to “overload” of the autophagic system that fails to prioritize the clearance of dysfunctional mitochondria. 32 Meanwhile, the mitochondrial network structure is destroyed, losing the synergistic calcium buffering effect between mitochondria and exacerbating local calcium overload. Fragmented mitochondria are more prone to membrane integrity impairment, and the released pro-apoptotic factors such as cytochrome C can preferentially initiate apoptotic programs, antagonizing the cytoprotective function of autophagy.8,33 Calcium ions and pro-apoptotic factors released from damaged mitochondria into the cytoplasm can activate the caspase family, 41 which will block autophagosome maturation by cleaving autophagy-related proteins. 17 Additionally, abnormal opening of mPTP will lead to the leakage of mitochondrial contents such as mtDNA fragments and ROS into the cytoplasm, triggering inflammatory responses and activating the NLRP3 inflammasome. 42 Inflammatory signals can disrupt lysosomal membrane stability, causing the release of lysosomal enzymes. This can not only prevent the fusion of degrade damaged mitochondria with autophagosomes but also exacerbate cellular damage, further inhibiting the degradation process of mitophagy.32,33,43 Furthermore, abnormal activity of calcium-dependent enzymes, such as calcineurin (CaN) and calmodulin-dependent protein kinase II (CaMKII), can also affect mitophagy by regulating the transcription of autophagy-related genes (Figure 3). 35
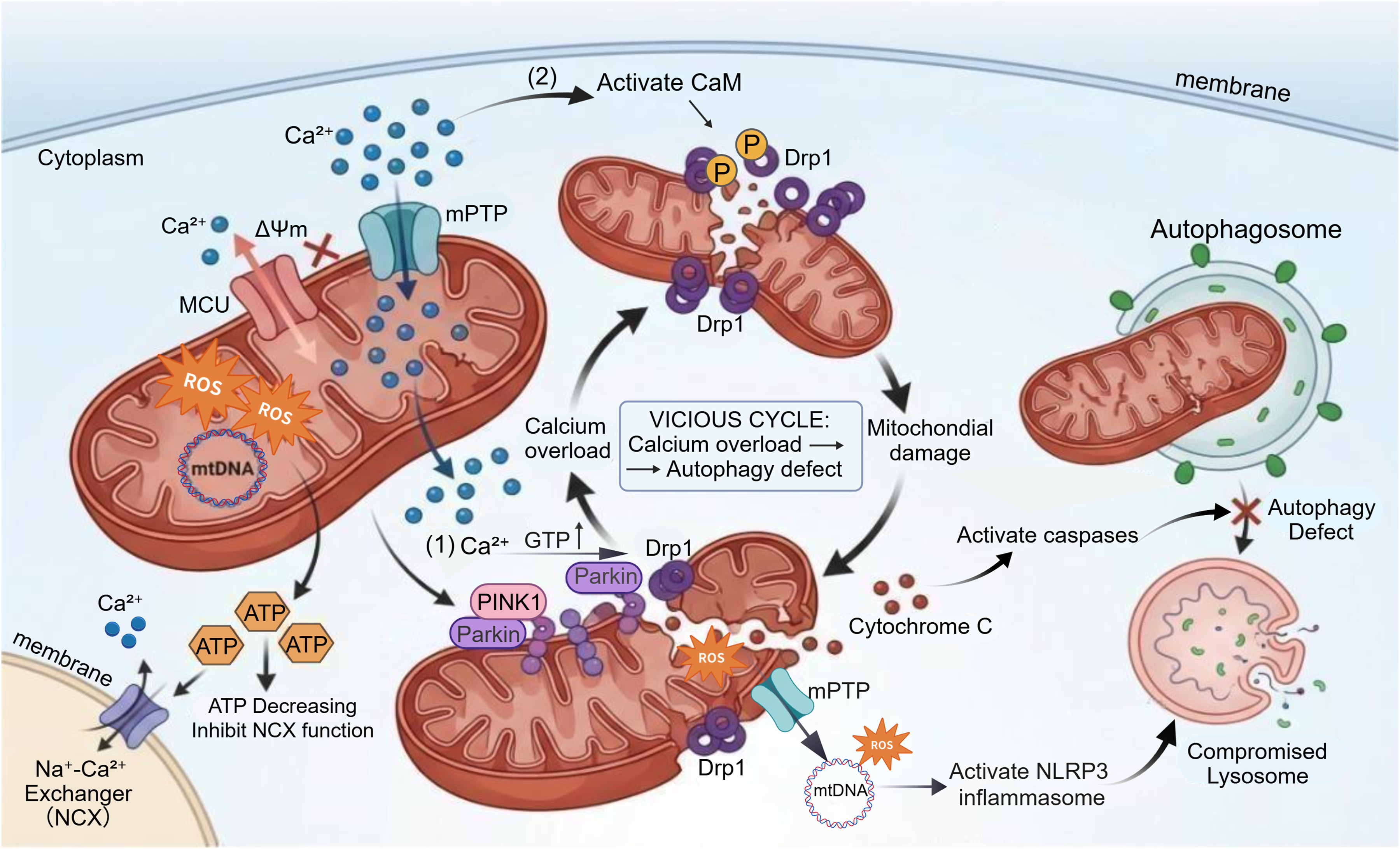
Defective mitophagy and calcium dyshomeostasis form a reciprocal vicious cycle. Pathologically, calcium overload triggers ΔΨm collapse, ROS burst, and mtDNA damage. Diminished ATP production impairs NCX activity, exacerbating cytosolic calcium overload. Calcium excess promotes mitochondrial fragmentation via two mechanisms: (1) Direct binding to Drp1 boosts its GTPase activity and mitochondrial membrane recruitment; (2) Calcium activates CaM, which phosphorylates Drp1 to maintain its persistent activation, further enhancing mitochondrial fragmentation. The PINK1/Parkin pathway is activated to clear damaged mitochondria, but defective autophagy abrogates efficient clearance. Ultimately, mitochondrial damage elicits cytochrome C release, which activates caspases and suppresses autophagosome maturation. Furthermore, aberrant opening of the mPTP causes leakage of mtDNA fragments and ROS into the cytosol, activating the NLRP3 inflammasome. Coupled with lysosomal dysfunction, these processes synergistically promote cellular damage progression.
Mitophagy deficiency and calcium dyshomeostasis synergistically promote the onset and progression of Alzheimer's disease
Mitophagy deficiency and calcium dyshomeostasis run through the entire process of the onset and progression of AD. Instead of acting independently, they form a vicious cycle via the pattern of “mitophagy deficiency - calcium dyshomeostasis - autophagy inhibition”. Their synergistic effect is far stronger than the independent effect of a single factor, which may be the cardinal reason for the irreversibility of AD. The pathological toxic effects of traditional proteins such as Aβ and tau are also largely achieved by impairing these two factors.44,45 However, the academic community still has many controversies regarding the causal sequence and spatiotemporal-specific regulatory mechanisms of their interaction. 46 Moreover, the role and regulatory logic of pyroptosis pathways mediated by neuroinflammation in this cycle have not been fully elucidated, and these issues constitute key bottlenecks in the research on AD pathological mechanisms.
Mitophagy deficiency and mild calcium dyshomeostasis interact with the formation of Aβ plaques, tau phosphorylation and neuroinflammation during early AD
Mitophagy deficiency exhibits pleiotropy in its pathological manifestations in AD, including energy metabolism disorders, calcium dyshomeostasis, enhanced oxidative stress, mitochondrial dynamics imbalance, and synaptic dysfunction. These functional impairments are interrelated and collectively drive the pathological process.47,48 As the pathological hallmarks of AD, the interaction mechanisms between Aβ oligomer accumulation, tau hyperphosphorylation and mitophagy deficiency have been widely confirmed, but controversies remain regarding their causal relationship. 49 One viewpoint holds that Aβ can directly target mitochondria by binding to mitochondrial fusion protein 2 (MFN2), and inhibit mitochondrial fusion and simultaneously suppress the activity of the PINK1/Parkin pathway, leading to impaired clearance of damaged mitochondria, which in turn triggers ROS accumulation and decreased membrane potential.32,50 Recent human neural model and post-mortem brain data further validate this causal cascade in FAD and advanced SAD, demonstrating that sustained Aβ exposure drives progressive mitochondrial dysfunction from early redox imbalance to terminal mitophagy failure, 51 with single-neuron metabolomics mapping Aβ oligomer enrichment at mitochondria-associated membranes (MAMs) as the proximal trigger of electron transport chain inhibition and mitophagy blockade. 52 Spatial proteomic analysis of postmortem SAD brains further confirms that Aβ-mitochondria interactions precede widespread loss of mitophagy markers in late-stage pathogenesis. 53 However, another line of research has found that in the preclinical stage of AD transgenic animal models, abnormal mitochondrial membrane potential and increased autophagic markers precede the detectable accumulation of Aβ oligomers, suggesting that mitophagy deficiency may be an upstream event initiating abnormal Aβ production. 54 Recent studies using artifact-free knock-in AD models refine this hypothesis, identifying ALKBH3-mediated PINK1 mRNA hypomethylation as a driver of mitophagy failure that occurs prior to Aβ plaque formation, with mitophagy restoration sufficient to reduce Aβ burden via normalization of AβPP processing.55,56 Notably, this upstream mitophagy defect is consistently observed in preclinical SAD models, rather than in FAD models driven by germline APP/PSEN mutations. 57 Collectively, these latest findings support a stage-dependent dual initiator framework, rather than a mutually exclusive linear cascade, in which mitophagy deficiency acts as a preclinical initiating trigger in SAD, while Aβ aggregation becomes the dominant pathological driver that perpetuates mitophagy failure once established pathology emerges.53,56,57 In addition, some studies have confirmed that abnormal tau aggregation can disrupt microtubule structure, block the transport and binding of autophagosomes to mitochondria, and exacerbate dynamics imbalance40,58; yet other studies point out that mild tau phosphorylation can initiate protective mitophagy by activating the AMPK pathway, and only hyperphosphorylation exerts toxic effects, the threshold and regulatory conditions for this “bidirectional effect” have not been clarified.59–61 Recent mechanistic studies have resolved this bidirectional paradox, demonstrating that the effect of tau phosphorylation on mitophagy is determined by site specificity, phosphorylation stoichiometry, and subcellular localization, rather than the mere presence of phosphorylation. Site-specific tau phosphorylation at T231, an early AD pathological marker, directly inhibits mitophagy via suppression of ULK1 complex assembly independent of microtubule disruption, 62 while low-level tau phosphorylation at S396 activates AMPK-dependent adaptive mitophagy to clear damaged mitochondria in early neuron models. 63 Quantitative phosphoproteomic work further defines a clear phosphorylation threshold, at which tau hyperphosphorylation exceeding 3.2-fold over physiological levels switches mitophagy regulation from adaptive activation to pathological inhibition, with synaptic mitochondria exhibiting selective vulnerability to this effect. 64
As a “regulatory hub” of AD pathology, calcium dyshomeostasis forms a bidirectional synergistic effect with Aβ/tau pathology, but the regulatory mechanisms of its early mild imbalance remain unclear. 65 In the early stage of AD (preclinical phase), the minimal production of Aβ oligomers can initiate the cycle through two pathways. 66 It can directly inhibit the accumulation of PINK1 on damaged mitochondria, blocking the initiation of mitophagy and resulting in the initial accumulation of damaged mitochondria. It also binds to calcium channel proteins on the cell membrane and endoplasmic reticulum, triggering a mild increase to 0.3–0.5 μmol/L in cytoplasmic calcium concentration.67–69 At this time, the dual effects of calcium signals are controversial. Some studies suggest that calcium at this concentration can initiate protective autophagy via the CaMKK2-AMPK pathway to alleviate early damage. However, other studies have found that Aβ can simultaneously inhibit the phosphorylation of downstream AMPK substrates, weakening the compensatory effect of this pathway and leading to “insufficient autophagy activation”, the balance mechanism and regulatory factors of these two effects have not been elucidated. 70 Mitophagy deficiency reduces the phagocytic clearance capacity of microglia for Aβ, while mild calcium overload can activate the NF-κB pathway in microglia, promoting the release of pro-inflammatory cytokines such as IL-1β and TNF-α and transforming microglia from protectors into creators of the pathological microenvironment.71,72 This process not only impairs the protective effect of glial cells on neurons but also promotes AD pathological progression through chronic inflammation and disruption of the synaptic microenvironment. Questions such as how calcium homeostasis in microglia itself regulates mitophagic activity, whether there is calcium signal-mediated cross-talk in autophagy regulation between neurons and microglia, and whether differences in mitophagy among microglia with different activation phenotypes (M1/M2) have distinct effects on calcium homeostasis have not been systematically resolved.73–75 Based on the emerging insights from single-cell RNA sequencing (scRNA-seq) studies in AD, several unresolved questions regarding the microglial calcium homeostasis and its intricate linkage to mitophagic activity have been further highlighted. ScRNA-seq has enabled the high-resolution dissection of microglial heterogeneity, revealing distinct transcriptionally defined subpopulations within disease-associated microglia (DAM) that exhibit unique calcium-handling signatures.76–78 Current evidence suggests that calcium homeostasis in microglia is not only a core regulator of mitophagic flux but also a key mediator of neuron-microglia cross-talk in the regulation of autophagy.79,80 For instance, specific calcium-permeable channels, such as the ORAI family of store-operated calcium entry (SOCE) components, have been shown to be differentially expressed in activated microglia (M1/M2 phenotypes), where they modulate the clearance of damaged mitochondria via mitophagy and reciprocally impact intracellular calcium dynamics.81–83 Although calcium dyshomeostasis at this stage does not directly induce neuronal death, it can promote tau phosphorylation by over activating CaMKII and disrupt the balance between long-term potentiation (LTP) and long-term depression (LTD), triggering early memory impairment (Figure 4). 65
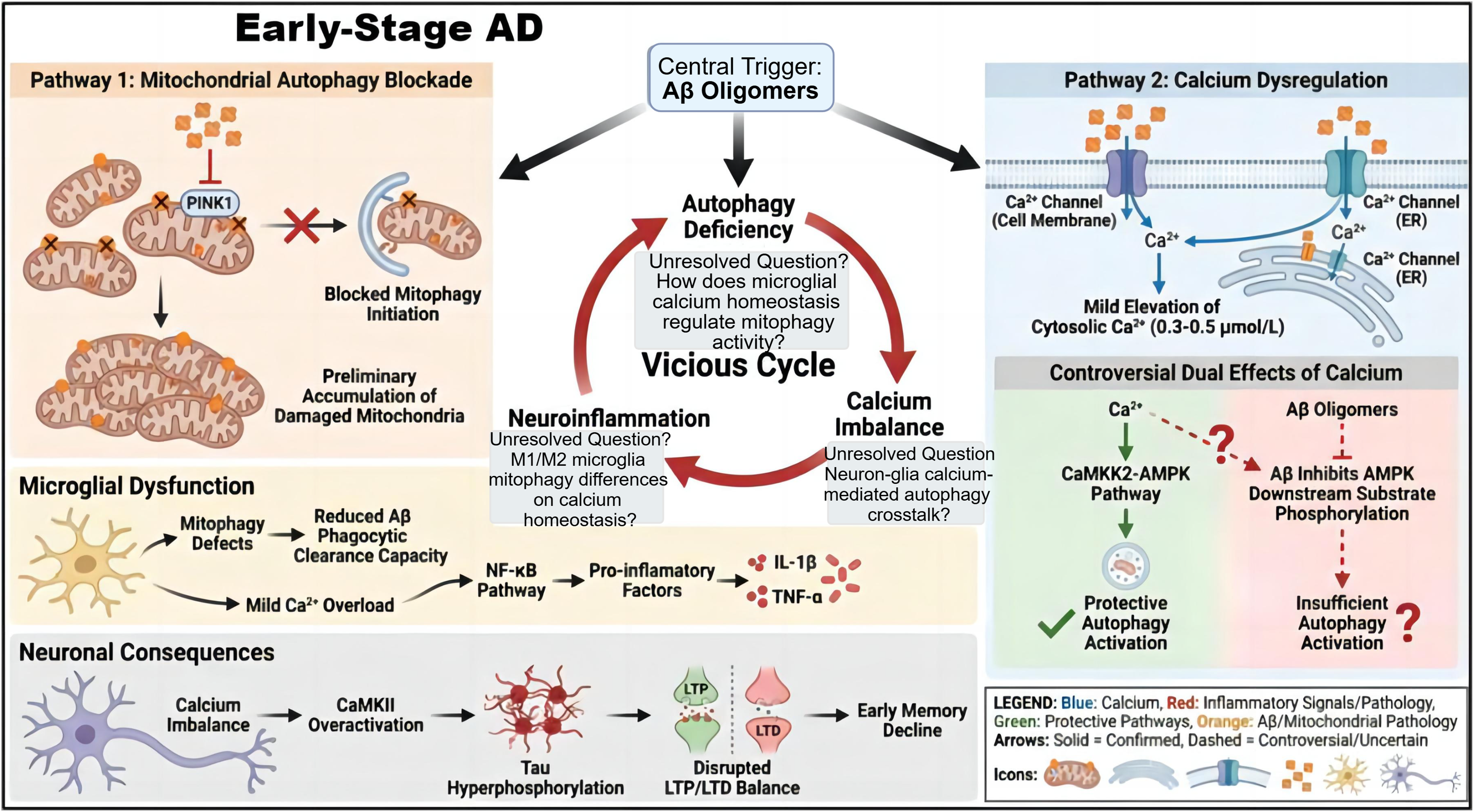
Aβ oligomer-driven vicious cycle linking mitophagic defects, calcium dyshomeostasis, neuroinflammation, and early neuronal impairment in early-stage AD. Aβ oligomers inhibit PINK1 accumulation on damaged mitochondrial membranes, which blocks mitophagy initiation and causes the accumulation of dysfunctional mitochondria, thereby impairing microglial mitophagy. When combined with mild calcium overload, this impairment activates the NF-κB pathway, mediating the release of pro-inflammatory factors (IL-1β, TNF-α). In the calcium dyshomeostasis pathway, Aβ binds to calcium channels on the cell membrane and ER, inducing extracellular calcium influx and ER calcium release, which moderately elevates cytosolic calcium concentration to 0.3–0.5 μmol/L. Notably, calcium exerts controversial regulatory effects on autophagy which activation of the CaMKK2-AMPK pathway promotes protective autophagy, whereas Aβ inhibits the phosphorylation of AMPK downstream substrates, leading to insufficient autophagy activation. These pathways synergize to amplify mitophagic defects, calcium dyshomeostasis, and neuroinflammation, collectively driving excessive CaMKII activation, tau hyperphosphorylation, disrupted LTP/LTD balance, and subsequent early memory decline.
Mitophagy deficiency and moderate calcium dyshomeostasis aggravate synaptic structure and function impairment during middle-stage AD
Synaptic loss is an important pathological basis for the progression of cognitive impairment in patients with AD, and its occurrence is closely associated with abnormal synaptic mitochondrial function. The synergistic effect of mitophagy deficiency and calcium dyshomeostasis serves as the key driver of synaptic damage. 84 Regarding the mechanism underlying synaptic mitophagy deficiency, there are two mainstream hypotheses. It maybe result from the specifical downregulation of the PINK1/Parkin pathway in the postsynaptic density (PSD) region, leading to impaired clearance of synaptic mitochondria. 85 Some believe that it is due to tau hyperphosphorylation disrupts microtubule transport function, preventing autophagosomes from recruiting to the synaptic region and blocking the degradation process of synaptic mitochondria.33,86 Currently, the primary-secondary relationship and interaction between these two mechanisms remain unclear. The core controversy in the AD field is not limited to the independent contributions of these two mechanisms, but centers on their causal hierarchy, spatiotemporal specificity, and functional interplay across the full pathological trajectory of AD. To reconcile this long-standing debate, we propose a spatiotemporal bidirectional cascade reconciliation model synthesizing the most recent preclinical data and findings from human postmortem studies, which frames the functional relationship between these two pathways specifically within middle-stage AD progression.87–89 This model further integrates spatial specificity to resolve the apparent discrepancy between the two competing hypotheses. Specifically, within the PSD region, downregulation of the PINK1/Parkin pathway is the predominant driver of mitophagy impairment, whereas tau-mediated transport dysfunction exerts the dominant effect in presynaptic terminals and axonal compartments. 90 Moreover, there are differences in the initiation time and degree of synaptic mitophagy deficiency among different AD models, resulting in a lack of unified pathological staging criteria. 91 In addition, controversies exist regarding changes in the expression of calcium regulation-related molecules. Some studies have shown that MCU is overactivated in the middle stage, leading to increased mitochondrial calcium uptake.35,92 However, other studies have found no significant change in MCU expression in the middle stage of the 3×Tg mouse model, and calcium overload mainly arises from abnormal RyR channels in the endoplasmic reticulum. Whether this difference is related to model background and pathological progression rate requires further verification. At this stage, the synergistic damage of mitophagy deficiency and calcium overload presents an additive effect. On the one hand, synaptic mitophagy deficiency can cause insufficient energy supply, while excessive ROS production impairs the neurotransmitter release mechanism of the presynaptic membrane, and calcium overload directly inhibits LTP induction in the postsynaptic membrane. Together, these factors result in decreased synaptic transmission efficiency and loss of dendritic spines. 93 On the other hand, ROS and calcium overload can jointly activate proteases such as glycogen synthase kinase-3β (GSK-3β) and Calpain, further promoting Aβ production and tau phosphorylation, and accelerating pathological protein accumulation. However, there are still prominent unknown areas such as the spatiotemporal expression pattern of autophagy-related proteins in the PSD region, how mitochondrial-associated endoplasmic reticulum membrane (MAM) structural abnormalities regulate the interaction between calcium exchange and mitophagy at synaptic sites, and whether inflammatory factors in the synaptic cleft directly affect the calcium buffering capacity of synaptic mitochondria. 94 In addition, the role of astrocytes in middle-stage synaptic damage remains unclear, and there is a lack of direct experimental evidence for whether their mitophagy deficiency affects synaptic trophic support function through abnormal calcium signals (Figure 5).
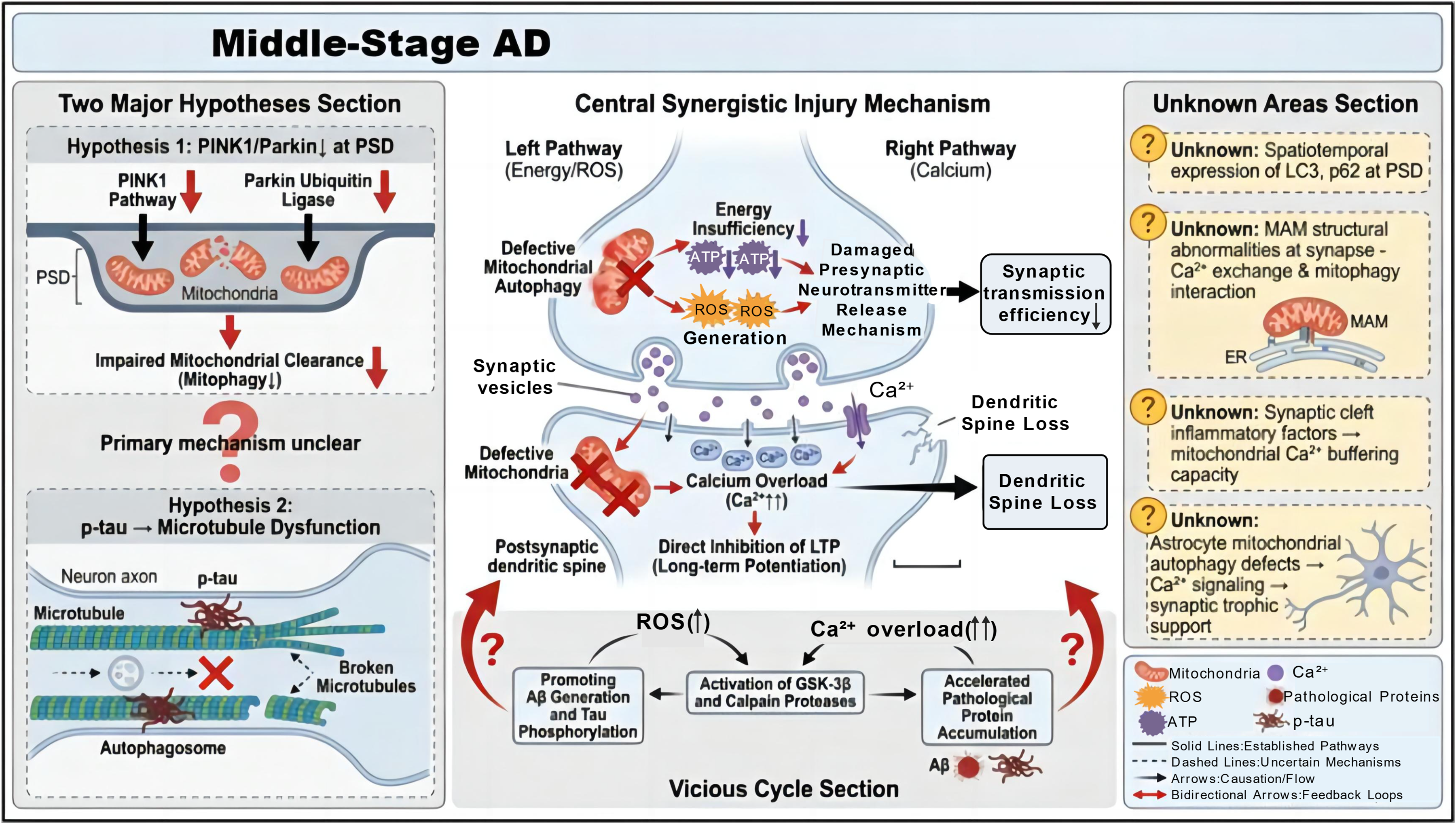
Core synergistic pathological cascades in middle-stage Alzheimer's disease. Two mainstream hypotheses exist in the field regarding the mechanism of synaptic mitophagic defects. one is that impaired PINK1/Parkin signaling at the postsynaptic density (PSD) blocks mitophagic clearance of mitochondria, and the other is that hyperphosphorylated tau (p-tau) disrupts microtubule function, which impairs autophagosome trafficking, while the hierarchical overload inhibits LTP and induces relationship and crosstalk between these two mechanisms remain unclear. In the energy/ROS pathway, defective mitophagy leads to energy insufficiency and excessive ROS production, which interferes with presynaptic neurotransmitter release. In the calpain pathway, calcium dendritic spine loss, while activating GSK-3β/calpain proteases, a process that further exacerbates synaptic damage. These cascades synergize to promote Aβ generation, tau hyperphosphorylation, and pathological protein accumulation, forming a self-amplifying vicious cycle. Additionally, key factors including the spatiotemporal expression of LC3/p62 at the PSD, structural abnormalities of mitochondria-associated membranes (MAM) at synapses, synaptic calcium buffering capacity, and the role of astrocytes in middle-stage synaptic injury all of which are critical for elucidating the underlying pathology remain undefined.
Mitophagy deficiency and severe calcium dyshomeostasis induce neuronal programmed cell death during late-stage AD
In the late stage of AD with moderate to severe dementia, the regulatory systems of mitophagy and calcium homeostasis are completely decompensated. Coupled with the continuous activation of pyroptosis pathways driven by neuroinflammation, this leads to irreversible massive neuronal loss and disruption of brain circuit structure, thereby resulting in comprehensive cognitive decline in patients.36,68 Previous studies have mostly focused on regulatory mechanisms such as apoptosis and ferroptosis, 95 while the role of neuroinflammation-mediated pyroptosis in the pathological process of late-stage AD and its upstream and downstream regulatory networks remain key scientific issues that urgently need in-depth clarification and systematic analysis. 96
Calcium dyshomeostasis in the late stage exhibits a characteristic of systemic dysregulation. NMDAR on the cell membrane, RyR on the endoplasmic reticulum, and mitochondrial MCU are all in an abnormally activated state, accompanied with cytoplasmic calcium concentration exceeding 1.5 μmol/L. Mitochondrial calcium overload triggers sustained opening of mPTP, leading to mitochondrial rupture and cytochrome C release, which initiates the intrinsic apoptotic pathway.7,67 Meanwhile, calcium overload and mitophagy deficiency synergistically activate the ferroptosis pathway because massive ROS production can induce lipid peroxidation, while autophagy deficiency prevents the clearance of damaged mitochondria, further exacerbating iron ion accumulation and forming a synergistic effect of apoptosis and ferroptosis.97,98 However, controversies exist regarding the mutual relationship between these two modes of cell death. Some studies suggest that ferroptosis is the main form of late neuronal death, and inhibiting ferroptosis can significantly reduce neuronal loss.99,100 Other studies have confirmed that apoptosis inhibitors are more effective in improving cognitive function in AD models. This difference may be related to brain region specificity, but its regulatory mechanism remains unclear. In addition, persistent neuroinflammation can activate the NLRP3 inflammasome to initiate neuronal pyroptosis, forming a vicious cycle of “inflammation - pyroptosis - inflammation amplification”. 101 Specifically, ROS and DNA fragments released by damaged mitochondria due to mitophagy deficiency can act as damage-associated molecular patterns (DAMPs) to activate the NLRP3 inflammasome in microglia, 102 promoting the maturation and release of pro-inflammatory cytokines (IL-1β, IL-18), and simultaneously inducing the cleavage of GSDMD to form membrane pores, leading to neuronal lysis and death. 71 Pyroptosis can not only directly cause neuronal loss but also activate surrounding glial cells through its released pro-inflammatory cytokines and cellular contents, exacerbating calcium dyshomeostasis and mitophagy deficiency, 103 thus forming a multidimensional pathological loop of “mitophagy deficiency - calcium overload - neuroinflammation - pyroptosis” (Figure 6).
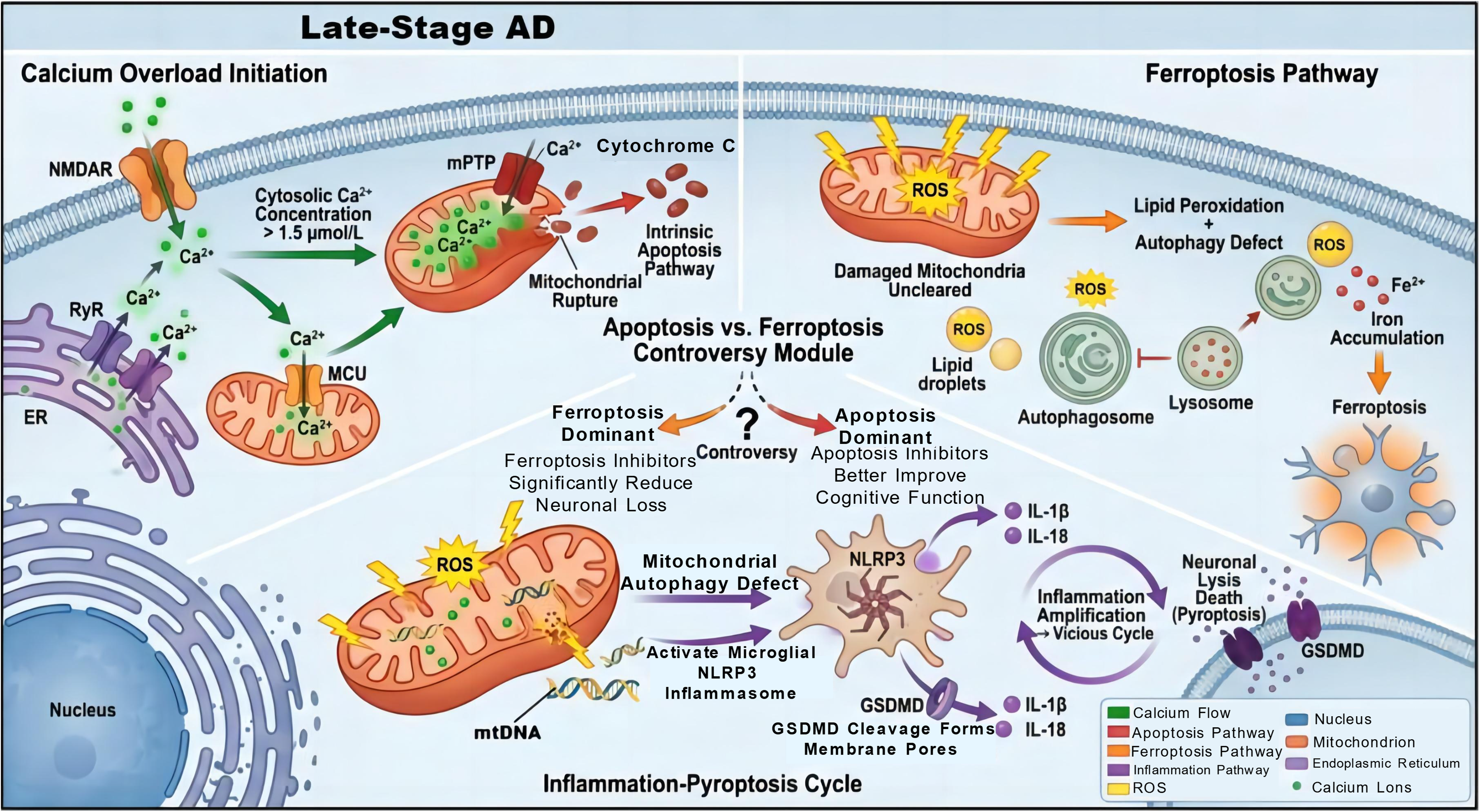
Dominant pathogenic mechanisms in late-stage AD. Excessive calcium influx mediated by N-methyl-D-aspartate receptors (NMDARs), ryanodine receptors (RyRs), and mitochondrial calcium uniporters (MCUs) elevates cytosolic calcium concentration to 1.5 μmol/L, thereby triggering opening of the mPTP, cytochrome C release, and the intrinsic apoptotic pathway. Notably, the dominant cell death pathway of apoptosis and ferroptosis remains controversial. when ferroptosis predominates, ferroptosis inhibitors substantially attenuate neuronal loss; conversely, apoptosis inhibitors improve cognitive function if apoptosis serves as the primary pathway. Concurrently, defective mitophagy drives the accumulation of damaged mitochondria, excessive ROS production, lipid peroxidation, and Fe2+ enrichment, ultimately inducing ferroptosis. Furthermore, mitochondrial dysfunction and mitochondrial DNA (mtDNA) release activate the NLRP3 inflammasome, which cleaves Gasdermin D (GSDMD) to form membrane pores; the released IL-1β/IL-18 further amplifies the inflammatory response, establishing a self-amplifying inflammation-pyroptosis cycle that ultimately induces neuronallysis.
Controversies persist in the academic community regarding the interaction between pyroptosis, apoptosis, and ferroptosis. Some studies hold that pyroptosis is the upstream initiator of late neuronal death, which can activate apoptotic and ferroptotic pathways. 104 Other studies propose that pyroptosis and the latter two are independent parallel pathways, only exhibiting synergistic effects in the final pathological stage. 105 In addition, whether GSDMD directly regulates mitophagy or calcium transporter activity, how inflammatory factors released during pyroptosis specifically affect the calcium signal differences between neurons and glial cells, and whether targeting pyroptosis pathways such as inhibiting NLRP3 and GSDMD will affect the balance between mitophagy and calcium homeostasis are all directions for subsequent research. Meanwhile, there is a lack of in-depth research on whether mitophagy deficiency and abnormal calcium signals can indirectly promote pyroptosis by affecting the synaptic microenvironment in glial scars formed by astrocyte proliferation in the late stage of AD. The specific regulatory mechanisms of the mitophagy-calcium axis, differential calcium/mitophagy-related triggers, and AD stage specificity of pyroptosis, ferroptosis, and apoptosis are systematically summarized and compared in Table 1.
Comparison of programmed cell death subtypes regulated by the mitophagy-calcium axis in AD.
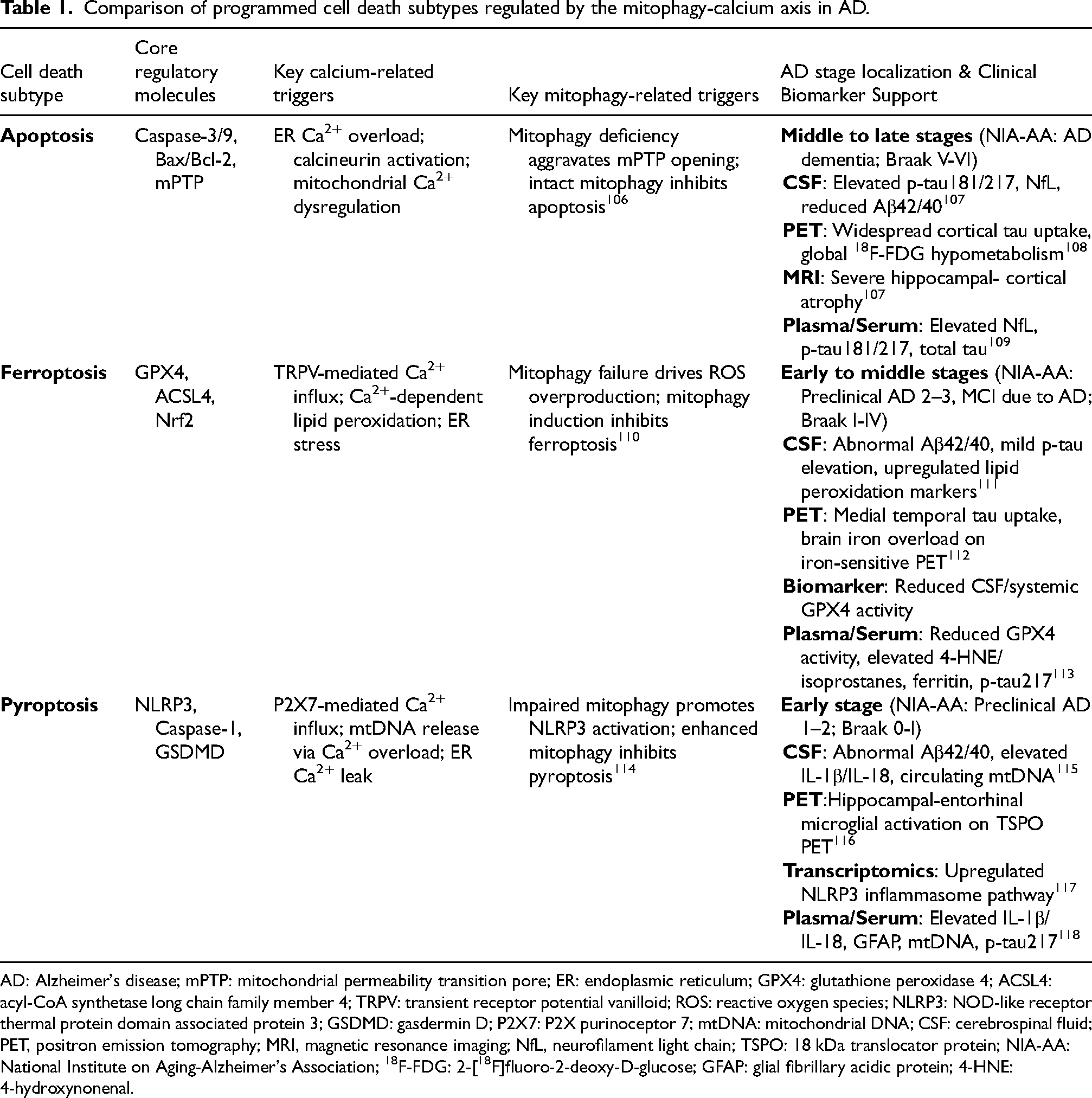
AD: Alzheimer's disease; mPTP: mitochondrial permeability transition pore; ER: endoplasmic reticulum; GPX4: glutathione peroxidase 4; ACSL4: acyl-CoA synthetase long chain family member 4; TRPV: transient receptor potential vanilloid; ROS: reactive oxygen species; NLRP3: NOD-like receptor thermal protein domain associated protein 3; GSDMD: gasdermin D; P2X7: P2X purinoceptor 7; mtDNA: mitochondrial DNA; CSF: cerebrospinal fluid; PET, positron emission tomography; MRI, magnetic resonance imaging; NfL, neurofilament light chain; TSPO: 18 kDa translocator protein; NIA-AA: National Institute on Aging-Alzheimer's Association; 18F-FDG: 2-[18F]fluoro-2-deoxy-D-glucose; GFAP: glial fibrillary acidic protein; 4-HNE: 4-hydroxynonenal.
In summary, mitophagy deficiency and calcium dyshomeostasis form a central pathological vicious cycle through bidirectional regulatory disorders, thereby synergistically amplifying the pathology of Aβ oligomer accumulation and tau hyperphosphorylation, damaging neuronal energy metabolism and synaptic structure and function, and inducing neuroinflammation as well as multiple forms of programmed cell death such as neuronal apoptosis, ferroptosis, and pyroptosis. This process permeates the entire course of AD, from early pathological changes to middle-stage synaptic damage and late-stage cognitive decline. The synergistic damage effect of them is the key driver of AD pathological progression. Targeting to simultaneously improve mitophagy function and calcium homeostasis is expected to become an effective strategy for blocking the progression of AD.
AD treatment strategies based on mitophagy deficiency and calcium dyshomeostasis
During the progression of AD, mitophagy deficiency and calcium dyshomeostasis form a synergistic pathological loop that exacerbates neuronal damage. Therefore, AD treatment needs to break through the limitation of single target and take multi-target synergistic regulation as the crucial to simultaneously activate mitophagy and repair calcium dyshomeostasis, thereby achieving pathological loop blockage and neuroprotection.
Therapeutic strategies for activating mitophagy
Mitophagy inducers can target and clear damaged mitochondria, showing efficacy in preclinical AD models. 119 Urolithin A (UA) can initiate mitophagy by activating the AMPK/ULK1 pathway, reducing Aβ deposition by 40% and tau phosphorylation by 35% in the brains of APP/PS1 mice. It has entered Phase I clinical trials, with good safety and high bioavailability.120–122 Nicotinamide riboside (NR)/nicotinamide mononucleotide (NMN) can activate the SIRT1/PGC-1α pathway by increasing NAD⁺ levels, improving mitochondrial function and energy metabolism in AD mice. 9 Clinical studies have shown that oral administration of NR in AD patients can reduce Aβ42/Aβ40 ratio in the cerebrospinal fluid.123,124 Metabolic modulators such as coenzyme Q10 and metformin can repair mitochondrial function, laying a foundation for autophagy activation.125–127
Despite these encouraging preclinical findings and early clinical indications, critical translational obstacles need to be resolved to establish realistic therapeutic prospects for Alzheimer's disease. Prominent among these are persistent challenges related to blood–brain barrier delivery and the consistent lack of success of previous antioxidant and mitochondrial-targeted therapies in late-stage clinical trials for human AD.
First, the blood–brain barrier constitutes a fundamental bottleneck limiting central nervous system bioavailability for mitophagy modulators. For urolithin A, favorable systemic absorption is observed in preclinical settings, yet pharmacokinetic investigations in rodents reveal that brain parenchymal concentrations of urolithin A represent only 0.1%–1% of corresponding plasma levels. This limited distribution arises from restricted passive diffusion across the blood–brain barrier and active efflux mediated by P-glycoprotein and other ATP-binding cassette transporters.128,129 In addition, biosynthesis of urolithin A relies entirely on the composition of the host gut microbiota, resulting in considerable interindividual variation in systemic exposure and further impeding consistent central nervous system delivery in human populations. 130 For nicotinamide riboside and nicotinamide mononucleotide, oral supplementation effectively elevates circulating NAD⁺ concentrations in humans. However, evidence from preclinical models and clinical studies demonstrates that central nervous system NAD⁺ elevation is substantially blunted relative to peripheral tissues, with only minimal increases observed in human cerebrospinal fluid even following high oral doses.131,132 This challenge is amplified in patients with Alzheimer's disease, as progressive blood–brain barrier impairment and pericyte degeneration represent well-characterized pathological features that disrupt selective molecular transport and reduce the reliability of drug delivery to vulnerable brain regions.133,134
Second, the extensive record of unsuccessful late-phase clinical trials involving antioxidant and mitochondrial-targeted agents in AD offers essential contextual insight for current therapeutic development. Over the past two decades, numerous agents with related mechanisms of action including the mitochondrial antioxidant idebenone, the targeted reactive oxygen species scavenger MitoQ, coenzyme Q10 and high-dose vitamin E have failed to reach predefined primary clinical endpoints in Phase II/III trials for mild-to-moderate AD despite strong preclinical evidence of efficacy.135–139 A systematic meta-analysis of 24 randomized controlled trials evaluating mitochondrial-targeted antioxidants in AD verified no significant improvements in cognitive performance or global clinical status among treated individuals.140,141 Although UA and NR/NMN exert effects through more specific mitophagy modulation rather than non-selective antioxidant activity, they remain vulnerable to the same core translational limitations. Preliminary biomarker alterations detected in early-phase trials have yet to be validated as correlates of clinically meaningful cognitive benefits in larger and sufficiently powered clinical cohorts.
Acupuncture at Baihui (GV20) and Taichong (LR3) acupoints can upregulate the expression of Beclin-1 and LC3 to activate mitophagy, improving cognitive function in APP/PS1 rats. 142 It can optimize calcium store function by regulating mitochondrial dynamics, repairing membrane potential, and inhibiting mPTP opening. With multi-target effects and no side effects, it is suitable for long-term intervention.143,144
Therapeutic strategies for restoring calcium homeostasis
Calcium homeostasis modulators represent an important direction for AD treatment. Knockdown of MCU can significantly improve mitochondrial function and neuronal survival in APP/PS1/tau mice, 145 but the targeted mitochondrial delivery of small-molecule MCU inhibitors such as Ru360 remains a major challenge for clinical translation. 60 Memantine, an approved drug, memantine reduces calcium influx through non-competitive inhibition of NMDAR, can improve cognitive impairment in patients with moderate to severe AD, and its bidirectional regulation property enhances medication safety. 146 Combination of RyR inhibitors and SERCA activators can restore endoplasmic reticulum calcium homeostasis and reduce tau phosphorylation. 68 NR can exert a synergistic effect by enhancing Sirtuin activity, which not only clears damaged mitochondria but also improves calcium uptake efficiency in healthy mitochondria.147–149
Combined therapeutic strategies for synergistically regulating mitophagy and calcium homeostasis
Combined therapy targeting to mitophagy deficiency and calcium overload can break the pathological loop in AD model. The combination of UA and MCU inhibitors have exerted a significantly better effect on improving cognitive function in APP/PS1 mice than monotherapy,92,125 making it a key direction to break through the bottleneck of AD treatment. Multi-target small-molecule compounds such as CaMKK2-AMPK pathway modulators can simultaneously regulate mitophagy and calcium homeostasis, simplify medication regimens, and have shown efficacy in in vitro models.25,150
In addition, lifestyle intervention is also a potential strategy for AD prevention and treatment. 151 Studies have shown that in AD mouse models, regular exercise can reduce cytoplasmic calcium overload by 35%, increase the expression of mitophagy-related proteins by 50%, and simultaneously decrease tau phosphorylation levels, resulting in significant improvement in cognitive function. 40 Exercise can also enhance the activity of Na+-Ca2+ exchangers on the cell membrane to promote cytoplasmic calcium efflux; meanwhile, it can improve the function of endoplasmic reticulum SERCA pumps, enhance endoplasmic reticulum calcium storage, and directly restore calcium homeostasis. 152 Improved calcium homeostasis will reduce mitochondrial calcium overload and avoid ROS-induced damage to autophagic pathways. At the same time, exercise can activate mitophagy through the AMPK pathway, clear a small number of damaged mitochondria, and retain the calcium store function of healthy mitochondria. Exercise can also enhance cerebrovascular dilation and microglial activity, promote Aβ clearance, and reduce the inhibitory effect of Aβ on mitophagy and calcium homeostasis. 153
As the main genetic risk factor for sporadic AD, apolipoprotein E4 (ApoE4) exacerbates the pathological process through multiple mechanisms. 126 At the subcellular level, ApoE4 preferentially localizes to and disrupts MAMs, the structural and functional hub mediating calcium (Ca2+) signaling crosstalk between the ER and mitochondria. Specifically, ApoE4 downregulates the expression and assembly of the IP3R-Grp75-VDAC1 Ca2+ transport complex at MAMs, impairing the spatiotemporal precision of ER-to-mitochondria Ca2+ transfer. This disruption directly drives excessive mitochondrial Ca2+ uptake and persistent mitochondrial Ca2+ overload. 154 This ApoE4-induced Ca2+ dyshomeostasis is a core upstream trigger of mitochondrial dysfunction, and it establishes a self-reinforcing vicious cycle with mitophagy impairment. On the one hand, ApoE4 directly suppresses the canonical PINK1/Parkin-mediated mitophagy pathway by blocking the mitochondrial outer membrane translocation of PINK1 and the subsequent recruitment of Parkin, thereby preventing the recognition and clearance of damaged mitochondria by autophagosomes.155–157 On the other hand, persistent mitochondrial Ca2+ overload driven by ApoE4 promotes sustained opening of the mPTP, exacerbates mitochondrial depolarization and oxidative stress, and further impairs mitophagy flux. This defective mitophagy in turn worsens the collapse of neuronal Ca2+ buffering capacity and aggravates Ca2+ homeostasis dysregulation. 158 Furthermore, ApoE4 amplifies dysregulation of the mitophagy-Ca2+ axis via synergistic interactions with core AD pathological hallmarks. Specifically, ApoE4 impairs Aβ clearance and promotes the generation of soluble Aβ oligomers, which in turn further activate ryanodine receptor (RyR)-mediated ER Ca2+ release and exacerbate mitochondrial Ca2+ overload. Meanwhile, ApoE4-induced Ca2+ dyshomeostasis accelerates tau hyperphosphorylation; hyperphosphorylated tau in turn suppresses mitophagy via binding to mitochondrial outer membrane proteins, thereby establishing a feedforward pathological cascade that amplifies neuronal injury. 159
Given the genotype-specific regulatory effects of ApoE4 on the mitophagy-calcium axis, individualized therapeutic strategies for AD must be developed and adjusted based on ApoE genotype stratification. For ApoE4 carriers, the core pathological defect lies in the dual disorder of calcium homeostasis and mitophagy function, thus requiring dual-target drugs that simultaneously correct mitochondrial calcium overload and restore impaired mitophagy, combined with antioxidant therapy to block the above-mentioned vicious cycle 160 ; for non-ApoE4 carriers, the dysregulation of the mitophagy-calcium axis is mostly secondary to Aβ and tau pathologies, so the treatment focus should be on maintaining the balance of mitochondrial dynamics, while avoiding non-specific excessive autophagy activation that leads to the clearance of normal mitochondria. 161 Meanwhile, a biomarker system based on ApoE genotype, combined with circulating or cerebrospinal fluid biomarkers reflecting mitochondrial calcium homeostasis and mitophagy activity, should be established to dynamically monitor treatment response and adjust the treatment regimen in real time, so as to maximize the clinical benefit of AD patients.162–164
In summary, synergistic intervention targeting mitophagy and calcium homeostasis will provide a new direction for AD prevention and treatment. The combination of drug and non-drug therapies, as well as the optimization of individualized and combined treatments, are the urgent and important directions for future research. 165 It is necessary to deepen mechanism research and clinical translation to provide precision regimens for patients.
Summary and outlook
Mitophagy and calcium homeostasis jointly maintain the steady state of normal neuronal physiological functions through a synergistic regulatory mechanism of mitochondrial quality control—calcium buffering. In the AD pathological microenvironment, functional defects of them can form a vicious cycle of mutual reinforcing effect and pathological amplification. They become the core pathological pathway driving the progression of AD through multiple pathways, such as synergistically promoting Aβ deposition and tau hyperphosphorylation, inducing synaptic structure damage and functional disorders, exacerbating neuroinflammatory responses, and triggering neuronal death.
Existing studies have confirmed that intervention strategies targeting the mitophagy-calcium homeostasis regulatory axis have shown significant neuroprotective effects in preclinical AD models, providing an important breakthrough for AD treatment. However, the clinical translation and application in this field still face multiple challenges. First, it is necessary to further analyze the dynamic interaction network between mitophagy deficiency, calcium dyshomeostasis, and neuronal programmed cell death, clarify the differences in the weight of each pathway in different pathological stages of AD, and then construct stage-specific intervention regimens. Second, the inefficiency of mitochondrial targeting delivery systems remains the main bottleneck for clinical translation. Existing drugs generally have problems such as low blood-brain barrier penetration efficiency and weak neuronal mitochondrial targeting enrichment ability. The research and application of novel nanocarriers, cell-penetrating peptides, and mitochondrial targeting ligands such as triphenylphosphine are expected to break through this technical bottleneck and significantly improve drug delivery efficiency. Third, the pathological mechanism of AD involves complex network regulation of multiple pathways and nodes. Single-target drugs are difficult to break through the treatment bottleneck, so there is an urgent need to develop multi-target synergistic regulatory drugs to achieve simultaneous regulation of vital pathways such as mitochondrial quality control, calcium signal transduction, and neuroinflammation. Integrating artificial intelligence-assisted drug design and multi-omics data analysis technology can provide technical support for the efficient discovery and structural optimization of novel multi-effect compounds, accelerating the research and development process. Fourth, the clinical translation process urgently needs to be accelerated. Although a number of mitophagy activators and calcium homeostasis modulators have achieved positive results in preclinical models, their targeting specificity, clinical safety, and long-term tolerance still need to be further verified by large-sample, long-term human clinical trials.
In the future, AD-related research should focus on the spatiotemporal regulatory mechanisms of mitophagy and calcium homeostasis, and clarify their primary regulatory nodes in the AD pathological process; further improve the precision intervention system based on pathological staging to achieve optimal matching of intervention timing and intervention regimens; meanwhile, strive to develop novel drugs and delivery systems with higher targeting, safety and efficiency to break the existing treatment predicament and provide new strategies and scientific basis for the early prevention, diagnosis and clinical treatment of AD.
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Horizontal Cooperation Project of Shaodong People's Hospital with Guilin Medical University (Grant No. 2023SDHX02).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.