
Abstract
Background
Takotsubo cardiomyopathy is an uncommon, reversible disease process of the left ventricle that typically occurs in the setting of physiological or psychological stress. Cases following neurological injury are becoming increasingly recognized, with development of takotsubo cardiomyopathy reported in as many as 28% of intensive care patients with traumatic brain injuries (TBI) during their hospital course.
Observations
We present a case involving a patient hospitalized following TBI with subsequent development of Takotsubo cardiomyopathy complicating the management of her primary traumatic injuries.
Discussion
This case demonstrates the importance of early recognition of this entity in patients with TBI and suggests the role for management by interdisciplinary teams. Given the incidence, risk factors, and complexity of stress cardiomyopathy in patients with TBIs, future prospective studies are needed to develop evidence-based guidelines to define treatment standards for takotsubo cardiomyopathy in patients with traumatic injuries.
Background
Traumatic brain injuries (TBI) affect approximately 10 million people annually, and often pose numerous debilitating sequelae. 1 The incidence of post-traumatic cardiac dysfunction is poorly understood among these patients as there is a paucity of prospective studies in the literature. Case series and retrospective data describe cardiac dysfunction measured by echocardiography in 22%–28% of moderate-to-severe TBI patients.1,2 Takotsubo cardiomyopathy (TC) is an uncommon, typically reversible, disease of the left ventricle (LV) that typically occurs in the setting of severe physiological and/or psychological stress.3,4 TC has been noted as a rare complication of acute psychiatric illness as well as a sequela of neurologic disease such as ischemic and hemorrhagic strokes, status epilepticus and intracranial- 5 as well as non-neurological- trauma. 6 One prospective study identified up to 28% of patients admitted to the intensive care setting develop some component of TC during their hospitalization. 6 However, the true incidence of TBI-associated TC remains unclear.
The pathophysiology of TC is believed to derive from “neurogenic stunning,” a phenomenon in which neurologic insults result in dysregulation of the autonomic nervous system, leading to the release of inflammatory cytokines and catecholamine modulators from the brain. 3 Catecholaminergic surge can produce cardiac dysfunction due to microvascular ischemia and myocardial intracellular calcium overload. 3 Incidence is disproportionately higher among post-menopausal patients, suggesting a role played by estrogens in catecholamine synthesis and activity. 7 Approximately 85% of patients diagnosed with TC are women with a mean age of 62–75 years at time of presentation. 8
This case report describes a young female patient with a severe TBI who developed TC complicating her primary traumatic injuries, demonstrating the importance of early recognition and the value of interdisciplinary management.
Case presentation
A 22-year-old woman with a past medical history significant for opioid use disorder presented to a Level I Trauma Center following a motor vehicle collision. On arrival she was acutely encephalopathic with an initial Glasgow Coma Scale (GCS) of 6 and decerebrate posturing. She had a left gaze deviation, expressed only incomprehensible groaning, and demonstrated uncoordinated extremity movement. Physical examination was significant for a heart rate of 107 beats per minute, blood pressure of 93/50 mmHg, respiratory rate of 24, and room air oxygen saturation of 100%. Computed tomography (CT) of the head revealed a right thalamic and internal capsule intraparenchymal hemorrhage (Figure 1). Chest CT revealed a left apical pneumothorax that did not necessitate intervention. Following initial evaluation in the Emergency Department, she was admitted to the trauma intensive care unit (ICU) for further management.
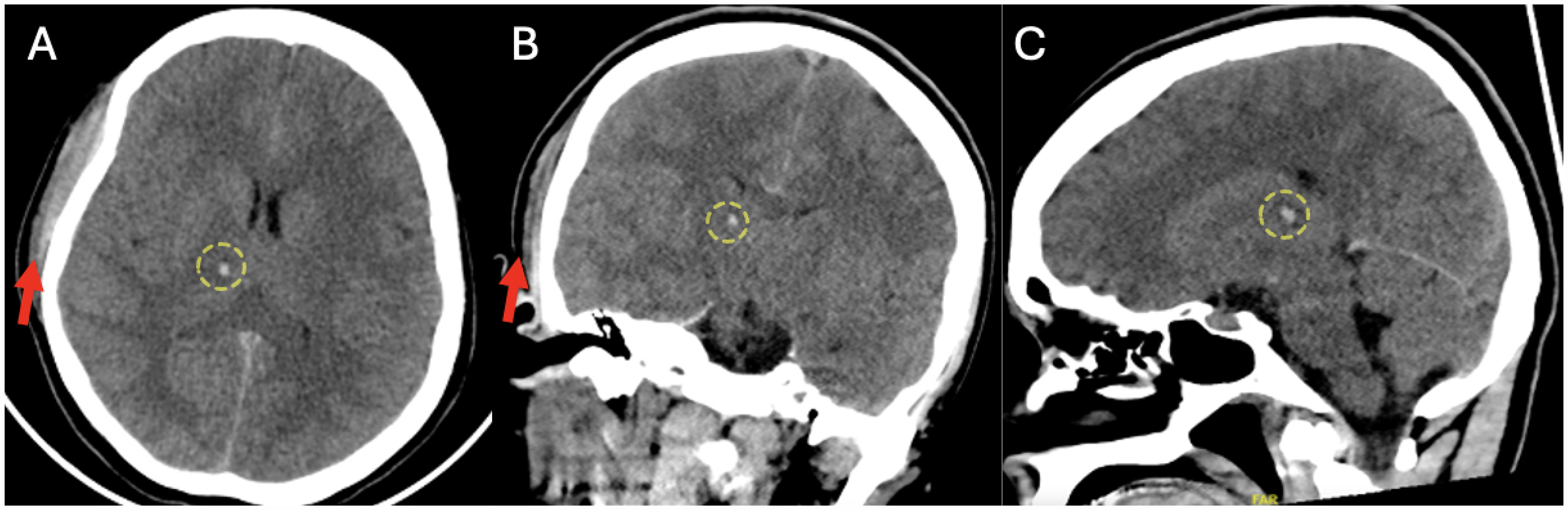
CT scan of head. (A) Axial view, (B) coronal view, and (C) sagittal view demonstrating right scalp hematoma (red arrows) and small right subthalamic hemorrhage (yellow circles).
Approximately five hours after initial presentation, she experienced a generalized tonic-clonic seizure, confirmed with placement of a continuous electroencephalographic monitor. She was sedated with propofol, intubated for airway protection, and administered levetiracetam.
On hospital day one, the patient became febrile to 38.2 °C with a white blood cell count of 23 × 10−3 µL. She developed hypotension with a blood pressure of 89/40 mmHg and was administered intravenous fluids and placed on a norepinephrine infusion. Repeat head CT showed diffuse cerebral edema with sulcal effacement. An intracranial transducer was placed to monitor intracranial pressure (ICP) and cerebral perfusion pressure (CPP).
Throughout the course of the first 10 days, the patient remained sedated and was administered intravenous vasopressors and hypertonic saline with frequent readjustments to maintain blood pressure, ICP, and CPP. The patient's vital signs were labile during the first four days with frequent episodes of hypotension and hypoxemia requiring increased supplemental oxygen. These episodes decreased in severity and frequency by day 5, and sedation and supplemental oxygen were weaned. Head CT repeated at this time demonstrated diffuse cerebral edema and signs of evolving diffuse axonal injury without brainstem herniation.
On hospital day 10, the patient developed worsening hypoxemia. Physical exam revealed pulmonary rales, abdominal ascites, and lower extremity edema. She required increasing doses of norepinephrine to maintain mean arterial pressure above 65 mmHg. Chest X-ray at this time demonstrated bilateral pleural effusions (Figure 2). Brain natriuretic peptide (BNP) was found to be elevated to 1474 pg/mL (reference range <100 pg/mL). Electrocardiography demonstrated sinus rhythm with intermittent sinus tachycardia, without evidence of ischemia. Transthoracic echocardiogram (TTE) revealed LV ejection fraction (LVEF) estimated at 20%–25%, abnormal LV strain, hypokinesis, apical ballooning, and mitral regurgitation (Figure 3). Cardiology was consulted and recommended continuing vasopressors and adding furosemide twice daily with a daily urine output goal of 2–3 liters.
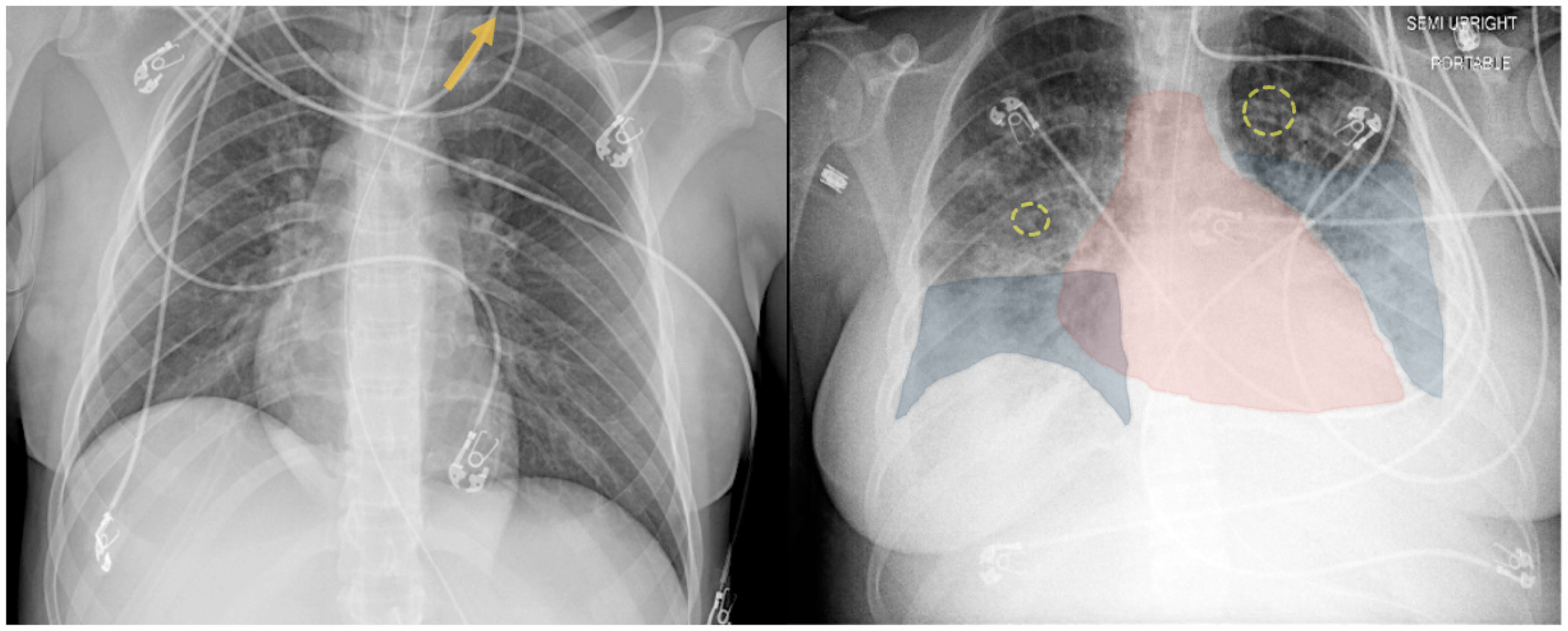
Left: Chest X-Ray on hospital day 1 demonstrating a small left pneumothorax (yellow arrow). Right: Chest X-Ray on hospital day 9 demonstrating enlarged cardiac silhouette (red highlight), bilateral interstitial opacities with peribronchial cuffing (yellow circles), and pleural effusions (blue highlight).
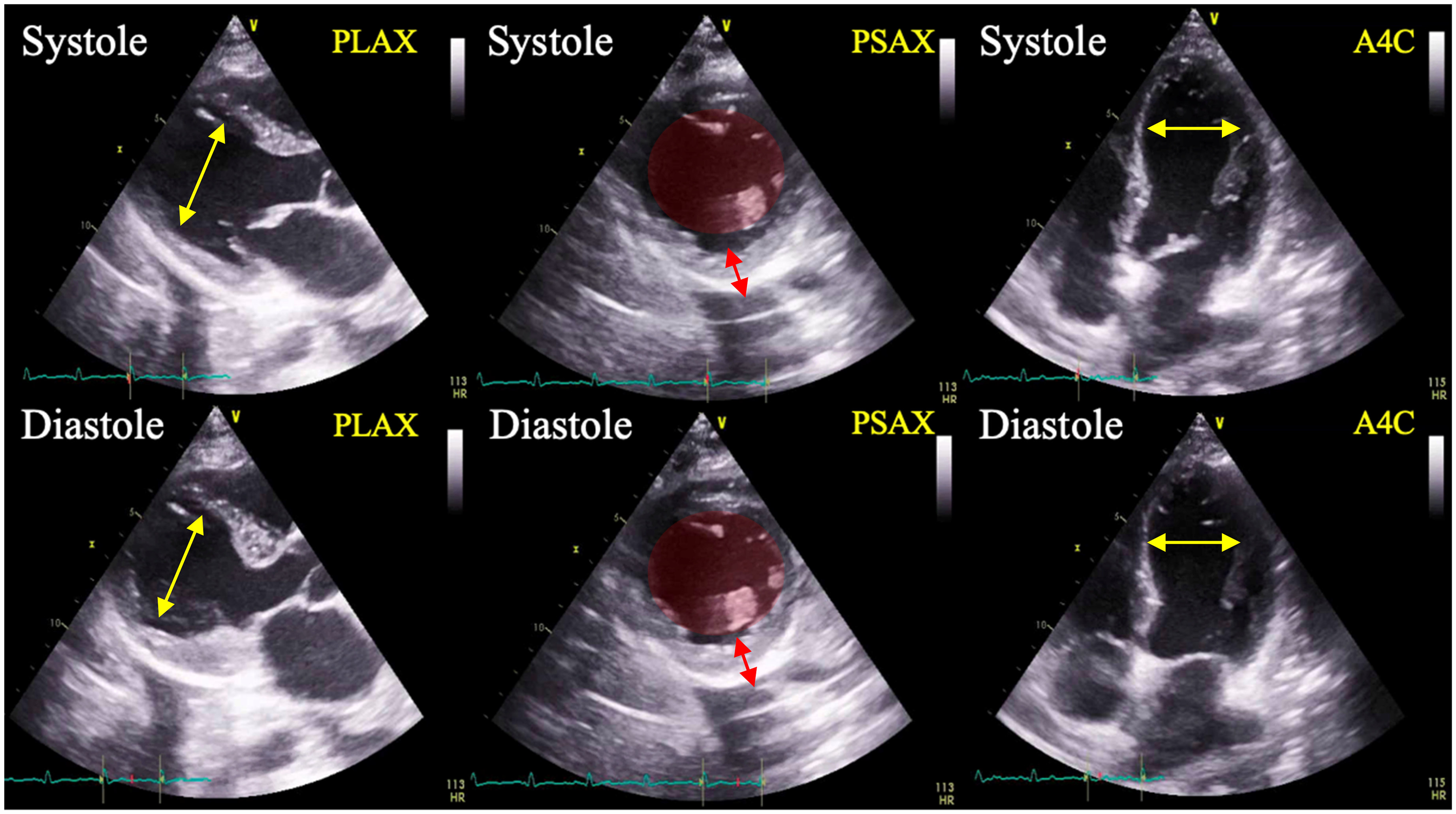
Transthoracic echocardiogram demonstrating hypokinesis and apical ballooning of the left ventricle during systole. Yellow arrows: apical ballooning. Red circles: minimal variation in left ventricle chamber size between systole and diastole. Red arrows: Minimal contracility and variation in left ventricle wall thickness between systole and diastole. PLAX: parasternal long axis view; PSAX: parasternal short axis view; A4Capical four chamber view.
On hospital day 23, nearly two weeks following initial recognition of TC, repeat TTE demonstrated LVEF recovered to 55%. By hospital day 40, the patient had resolution of all pulmonary and cardiovascular complications with neurologic improvement to a GCS of 11 T (E4V1M6). She was discharged to a long-term acute care facility to continue rehabilitation. After extensive neurological and physical rehabilitation, she was able to return home with complete neurologic recovery.
Discussion
Despite significant disease burden, risk for complications, and unique clinical challenges posed by cardiac dysfunction in the setting of TBI, guidelines for the care of TBI patients offer few recommendations for prediction, prognostication, or management of this patient subset. This absence of recommendations leaves care teams to make decisions on an individual basis depending on the severity of injuries and the severity of cardiac dysfunction without guidance on monitoring and treating sequelae.
The Brain Trauma Foundation published updated evidence-based guidelines for management of severe TBI in 2016 that focused care on direct treatment to improve neurological function, prophylactic treatments to avoid common complications, and pertinent vitals to monitor for cerebral status. 9 In terms of direct treatment, therapeutic guidelines were described for decompressive craniectomy, neuroprotective hypothermia, hyperosmolar therapy, cerebrospinal fluid drainage, anesthetics, analgesics, sedatives, steroids, nutritional therapy, and ventilation therapy. Recommendations are given for prophylaxis against infection, venous thrombosis, and seizures. The guidelines describe the importance of monitoring ICP and CPP and noted advanced cerebral monitoring techniques and blood pressure thresholds as key tools during this process. While cardiac function is crucial to the success of many of these therapies, stress cardiomyopathy and neurogenic stunned myocardium and the potential severe burden these conditions place on cardiovascular function in this patient population are not mentioned.
As the medical field works towards reinvestigating and reestablishing updated universal protocols for these patients, Takotsubo cardiomyopathy should take precedence as a common post-traumatic occurrence with life-threatening complications. With a recent prospective cohort study finding TC to affect more than a quarter of patients with TBI, monitoring cardiovascular complications in these patients could be significant in preventing life-threatening complications. 6
Despite extensive research conducted to navigate the etiology of TC, the exact pathophysiology of the syndrome remains a subject of active investigation. The most widely accepted pathogenesis involves a cascade event with trauma-induced sudden release of neurohormones including norepinephrine, epinephrine, and dopamine, with consequent coronary artery vasospasm, transient myocardial ischemia, pathological changes to the myocardium and coronary microvasculature, and stunning of the basal myocardium. 10 Because the basal myocardium contains a greater density of sympathetic receptors than the apical myocardium and the LV apex contains a higher concentration of adrenoceptors, the pathophysiology of TC may reflect hyper-localized catecholaminergic toxicity with decreased basal β-adrenoceptor responsiveness, leading to stunning of the basal LV with loss of contractility and apical hyperkinesis. This increases mechanical wall stress at the apex leading to increasing LV end-diastolic pressure (LVEDP), pathognomonic echocardiographic “ballooning,” and rising BNP levels.11–16 Acutely elevated LVEDP results in the clinical manifestations of heart failure, with pulmonary vascular congestion, alveolar edema, and resultant hypoxemia, as well as coronary and systemic hypoperfusion.
Other neurohormonal factors may play a role in pathophysiology; most notably estrogens, as TC more commonly occurs in postmenopausal women. Estrogens are not only cardioprotective in their direct effects on vasculature through vasodilation and vascular protection, but also play a key role in the endocrine stress response through downregulation of catecholamine biosynthesis and beta-adrenergic receptors, and modulation of the hypothalamus-pituitary-adrenal axis.17–19 Finally, glucocorticoids sensitize myocardial and vascular smooth muscle calcium channels, increasing inotropy and modulating the vascular response to beta-agonists.20,21 Reversible cardiac dysfunction is widely described in patients with primary and secondary adrenal insufficiency, with volume contraction and low cardiac output recoverable with fluid resuscitation and corticosteroid replacement therapy.21,22 One important implication of the neurocardiogenic stunning pathophysiology hypothesized to undergird TC is that many transplant donor hearts become available as the result of catastrophic neurologic injury, secondary to traumatic or toxicologic etiologies, such as opioid overdose. One case report in the literature describes TC identified in a transplanted heart from a donor who had suffered a fatal opioid overdose. 23
The most common early symptoms of TC are acute substernal chest pain, dyspnea, and syncope. 18 In patients with TBIs, however, symptoms may not be identified expeditiously, as these patients often have impaired consciousness secondary to injuries and/or may be sedated and intubated, eliminating the ability of patients to communicate symptoms of cardiac dysfunction. In these patients, common findings include laboratory, imaging, and/or clinical exam evidence of volume overload, increasing oxygen requirements to maintain normoxemia, and increasing vasopressor requirements to maintain perfusion. The window where development of TC following a TBI is most prevalent is within five days of the inciting trauma, with a literature review in 2017 finding nearly 85% of reported cases to have occurred within this window.24,25
TC is typically diagnosed by TTE identifying hypokinesis, dyskinesis, and/or akinesis in one or more segments of the LV, typically involving the cardiac apex. This is best visualized as decreased or absent movement in one of the LV walls or as apical ballooning. In some cases, basal hyperkinesis has also been identified and can progress to LV outflow obstruction. 26 Most patients also have a reduced LVEF typically ranging between 20% and 49%. 18 ECG changes may be seen and most commonly show ST-segment elevation and/or T-wave inversions, but abnormal ECG findings are highly variable. Thirdly, levels of serum cardiac troponin, BNP, and norepinephrine may be significantly elevated. To ensure accurate diagnosis, significant coronary artery disease, pheochromocytomas, and myocarditis should be ruled out. 7 It is critical to rule out acute coronary syndrome (ACS) in these patients as this is a life-threatening emergency with distinct risk factors, pathophysiology, evaluation, treatment, and long-term management. While TC has a very different set of risk factors, natural history, and pathophysiologic mechanism from ACS, the two syndromes share significant overlap in presentation.7,27 Early indicators that suggest TC is the etiology underlying cardiac dysfunction include normal serum troponin levels and ECG findings inconsistent with ACS. While ST-segment elevation may be seen in the setting of TC, it is typically less pronounced than in MI, and there is no concurrent ST-segment depression seen in other ECG leads. T-wave inversion and Q-wave formation can be seen. 27 The mainstay of the treatment of ACS involves anticoagulation, antiplatelet agents, coronary angiography and revascularization. Treatment of TC is predominantly supportive with treatment focused on reducing volume overload, decreasing afterload, and promoting inotropy. When clinically appropriate, beta blockers, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers have been shown to reduce symptom severity and promote disease resolution. Resolution of cardiac dysfunction usually occurs within 4 weeks and can be measured by echocardiography.3,24 When appropriate, anticoagulation should be used throughout the disease course as thromboembolism prophylaxis. 28
Complications of TC include progression to chronic heart failure, LV outflow tract (LVOT) obstruction, cardiac arrhythmias including atrial fibrillation and ventricular tachycardia, cardiogenic shock, and thromboembolism.18,28 In patients with TBI there are added risks as fluids for treatment of hemorrhage and medications such as vasopressors increase cardiovascular stress. Sympathomimetic vasopressors can worsen TC, as inotropes can increase basal hyperkinesis which may in turn produce or worsen LVOT obstruction. Many of these patients suffer from concomitant traumatic injuries to other organ systems that are subsequently more vulnerable to ischemia in the setting of cardiac dysfunction. Finally, anticoagulation to prevent thrombus formation or embolization of ventricular mural thrombi may be prohibitively dangerous in the setting of active or recent intracranial bleeding and/or recent surgical procedures.
Conclusion
Most documented cases of TC are secondary to psychological and physiological stress. However, occurrence following neurologic injury is becoming more recognized. It is well understood that poor cerebral perfusion worsens outcomes in TBI, and TBI patients often suffer concomitant traumatic injuries to other organ systems vulnerable to ischemia in the setting of cardiac dysfunction. Case series describe TC occurring during the hospitalization of as many as 28% of intensive care patients with neurologic injury, however the literature base is limited with few prospective studies estimating the true incidence of this phenomenon. While TBI patients typically receive care in trauma ICU settings, the potential incidence of TC reflects a need for interdisciplinary care of these complex patients. Updated TBI guidelines should reflect the potentially high burden of cardiac dysfunction following neurotrauma.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.