
Abstract
Electrospray ionization of mixtures of succinic acid (here denoted H2Su) and magnesium chloride in water/methanol give rise to ions of the type ESu− (E = H or ClMg). The unimolecular dissociation of these ions was studied by collisionally induced dissociation mass spectrometry and interpreted by quantum chemical calculations (density functional theory and the composite Gaussian-4 method) of relevant parts of the potential energy surfaces. The major dissociation pathways from HSu− were seen to be dehydration and decarboxylation, while ClMgSu− mainly undergoes decarboxylation. The latter reaction proceeds without barrier for the reverse reaction; addition of CO2 to a Grignard type structure ClMg(CH2CH2CO2)–. In contrast, addition of CO2 to the analogous H(CH2CH2CO2)– ion has a substantial barrier. Dehydration of HSu− gives rise to deprotonated succinic anhydride via a transition state for the key intramolecular proton transfer having an entropically favorable seven-member ring structure. The succinate systems studied here are compared to the previously reported analogous maleate systems, providing further insight to the structure–reactivity relationship.
Keywords
Introduction
Reductive fixation of CO2 is a key step in photosynthesis and in methodologies for artificial photosynthesis.1–5 In principle, two alternative approaches for C–C bond formation can be imagined—either first activating CO2 reductively by making it into a carbon nucleophile capable of reacting with an electrophilic carbon of a substrate molecule RX (equation (1)) or first activating the substrate reductively and then let the activated substrate react with CO2 (equation (2))
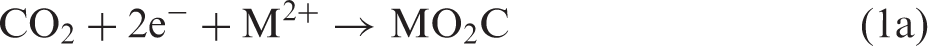
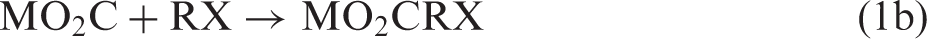
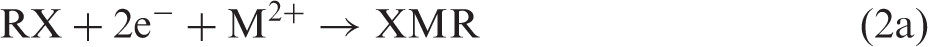
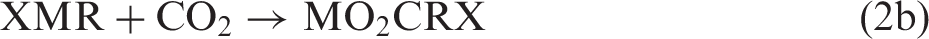
For M = Mg and X = halide, reaction 2 corresponds to the well-known Grignard reaction,2,6 while more recently, the carbenoic MO2C species formed in reaction 1a by umpolung has been spectroscopically characterized and shown to be a highly reactive carbon nucleophile. 7
The gas phase is the ideal environment for probing reactivity in the absence of solvents, and the mass spectrometer is ideally suited for exploring elementary reaction steps with well-defined reactants and products.6–10 Further details of the potential energy surface governing a chemical reaction can be obtained by combining mass spectrometric evidence with quantum chemical calculations. Systematic studies of this kind provide valuable information on structure/reactivity relationships of relevance also to condensed phase chemistry. A particular feature of the mass spectrometer as a chemical laboratory is that, under favorable circumstances, a reaction and its reverse reaction can be studied. For example, by activating a molecule by energy transfer (e.g. collisionally induced dissociation, CID), it may dissociate into well-defined fragments. In separate experiments, the association of the fragments may be studied. In any instance, and in particular when the details of the potential energy surfaces are known from quantum chemical calculations, insight into the reverse reaction is obtained from CID studies. Based on CID studies of carboxylate anions, Graul and Squires
11
realized that R− fragments may add to carbon dioxide, a reaction considered to be without an energy barrier, e.g. microscopic reversibility can be established for the following equilibria

From CID studies of carboxylate-metal anions, Rodríguez-Blanco et al. 12 found that formate (HCOO−) and acetate (CH3COO−) anions complexed with CaCl2 dissociate by decarbonylation and loss of CH2CO, while the analogous ZnCl2 and MnCl2 adduct anions decarboxylate. This was rationalized by Ca2+ having higher affinity for OH− than for H−, while the opposite is true for Zn2+.
Dicarboxylic acids are of interest in this respect because several are central in essential biochemical cycles involving CO2.13–15 In addition, two carboxylic acid functions present in the same molecule allows for a metal cation to bind simultaneously to both groups, thereby influencing the molecular conformation and thereby the reactivity. 16 Systematic study of the gas phase fragmentation of deprotonated dicarboxylic acids was conducted by Grossert et al. 17 showing an intimate relationship between carbon chain length and reactivity. The loss of CO2 was the most prevalent reaction for the shortest dicarboxylic acids, while H2O loss abundancy increased with chain length, the latter being attributed to increased conformational freedom. CID of the simplest dicarboxylic acid, oxalate, leads to concurrent loss of CO and CO2 as reported by Soldi-Lose et al. 18
Miller et al.
2
investigated the unimolecular dissociation of deprotonated maleic acid (HMa−) and ClMgMa–. Both anions readily undergo two consecutive decarboxylations. The first decarboxylation is more facile for ClMgMa– than for HMa–. It was also seen that HMa– loses water. In addition to losing a second CO2 molecule, the fragment ion produced by decarboxylation of ClMgMa–, ClMg(O2C)(HCCH)−, was seen to lose ethyne giving rise to ClMg(η2-O2C)− without having a barrier for the reverse reaction. The latter ion corresponds to a carbenoic species of the type MCO2 mentioned above. In other words, in the reverse reaction, ClMg(η2-O2C)− is able to form a C–C bond by addition to ethyne (Scheme 1).
Addition of ClMg(η2-O2C)− to ethyne.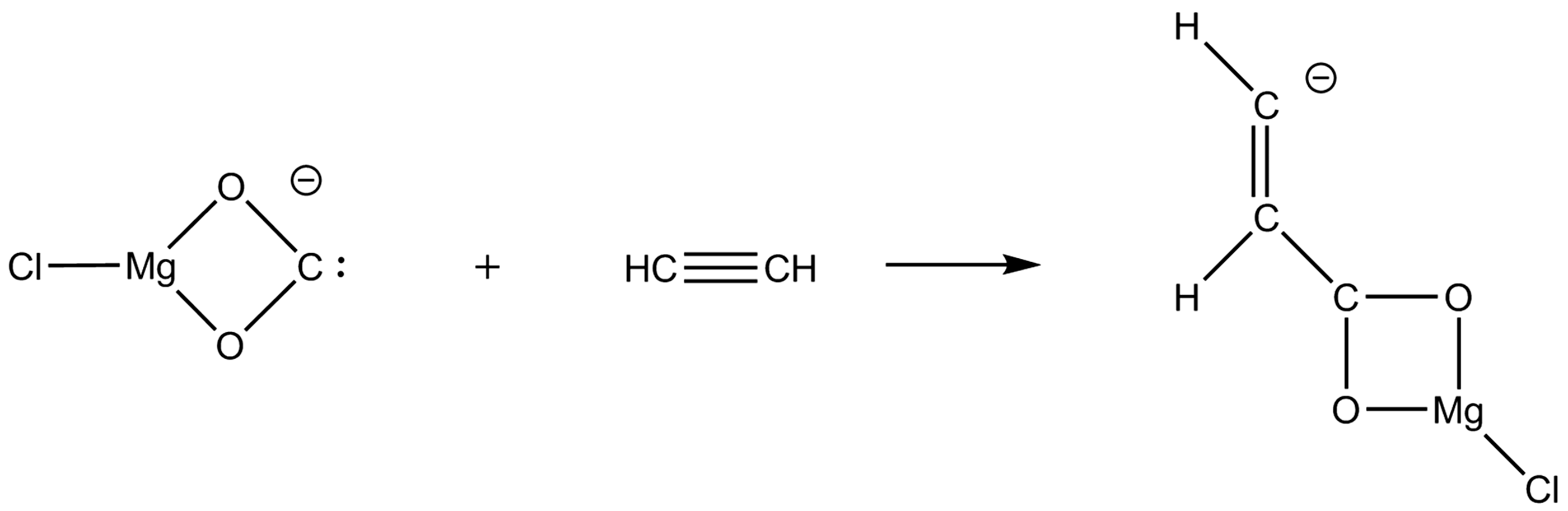
In this paper, we wanted to investigate the closely related succinic acid applying the same experimental strategy. It was of interest to study the effect of replacing the central C=C double bond of maleic acid with the C–C single bond of succinic acid. This may have two consequences. Firstly, a C–C single bond allows for more conformational flexibility. Secondly, the π bond of maleic acid is able to influence the stability by electronic interaction conjugation.
Methods
Experimental details
The experiments were conducted using a modified Micromass/Waters Q-TOF 2 MS with a custom built collision gas inlet system. The instrument has a quadrupole/hexapole/time-of-flight geometry, where the quadrupole serves as the first mass filter, the hexapole as the collision cell, and finally the time-of-flight as the second mass analyzer. The sample was introduced with a 1 mL syringe using an injector flow rate of 2.0 µL min−1. The concentrations of succinic acid and MgCl2 were 1.0 mM and 2.0 mM, respectively, in all experiments. Both compounds were available as crystalline solids (succinic acid: Acros Organics, 99%) dissolved in the given solvent for the experiment at hand (H2O/MeOH in an 80/20 v/v ratio).
The mass spectra were recorded at 3.0 kV time-of-flight (TOF) voltage for the deprotonated acid in order to allow detection of low-mass fragments (cut-off at m/z 14) and at 7.0 kV TOF voltage for the magnesium chloride adduct, as its detection was difficult at lower potentials. The electrospray capillary was operated at 3.0 kV, and the multi-channel plate (MCP) detector had an operational voltage of 2200 V throughout. The cone voltage was kept at 0–15.0 V. For deprotonated succinic acid, HSu–, CID mass spectra were recorded in steps of 1.0 eV collision energy (lab frame) in the range 5.0–10.0 and 25–32.0 eV and in steps of 0.5 eV in the range 10.0–25.0 eV. The collision gas (argon) pressure was kept at 2.0 × 10−4 mbar during these experiments. Similarly, for ClMgSu–, the collision energy was varied 5.0–47.0 eV (lab frame) in increments of 1.0 eV. The collision gas pressure was kept at 3.0 × 10−4 mbar during these experiments. In order to enhance the abundance of ClMg(CH2CH2CO2)– in the reaction experiments with CO2, the cone voltage was increased to 35.0 V. In these experiments, the collision energy was set to 0.4 eV in the center-of-mass frame, while the pressure was kept at a nominal pressure of 8.65 × 10−4 mbar to ensure a sufficient frequency of collisions between the ions and the CO2.
The succinic acid sample was also analyzed with a GC-EI-MS (Bruker Scion 436-GC) and a different ESI-MS instrument (Bruker maXis II ETD) to aid elucidation of the sample solution composition.
Appearance energies were estimated by first fitting a polynomial function to the breakdown curve of a given signal, and subsequently extrapolating the tangent line through the inflection point of the fragment ion curve to the base line. Error estimates were made on the basis of the count statistics. We realize the rather approximate nature of this method but for the present purpose, in comparing appearance energies between competing reaction channels, this semi-quantitative approach was considered appropriate. More sophisticated treatment would involve the use of the methods by the Armentrout or Chen laboratories.19,20 However, the signal-to-noise-ratios of some of our recorded breakdown curves, in particular at very low collision gas pressure, were considered too poor to obtain accurate estimates using these methods.
Quantum chemical calculations
Geometry optimization was done with Gaussian 09 initially using the B3LYP method with aug-cc-pVTZ basis sets to improve the description of polarization in the anionic systems.21,22 Optimized structures were recalculated using Gaussian-4 theory (G4) 23 for increased accuracy, intended to provide energy values within ca. 10 kJ mol−1 of accurate experiments. Intrinsic reaction coordinate calculations were used to connect all transition states to their respective energy minima.
Results
Mass spectra, dissociation reactions and breakdown curves of HSu− (m/z 117)
The results for the hydrogen succinate experiments are illustrated in the spectra of Figure 1. Starting with Figure 1(a), the mass spectrum of succinic acid electrosprayed from a solution of water and methanol, we observe the base peak at m/z 117 as the deprotonated acid, i.e. HSu−. This interpretation is supported by the isotope distribution pattern. The second most intense signal is for m/z 73, corresponding to decarboxylation. Water loss from HSu− is consistent with the fragment ion peak at m/z 99. Both these fragment ions are attributed to in-source fragmentation, although separate GC-EI-MS experiments indicate that a small amount of succinic anhydride is present in the sample solution, which also may contribute to the m/z 99 peak.
(a) Mass spectrum of succinic acid electrosprayed from an 80/20 v/v H2O/MeOH solution. Separate experiments indicate that there is a 2% contamination of acetate (m/z 59) in the succinic acid sample. (b) CID mass spectrum of HSu− (m/z 117) with collision energies averaged over 3.0–5.0 eV (ECOM) at 2.0 × 10−4 mbar Ar pressure, 3.0 kV TOF voltage. Note the different scales on the y-axes.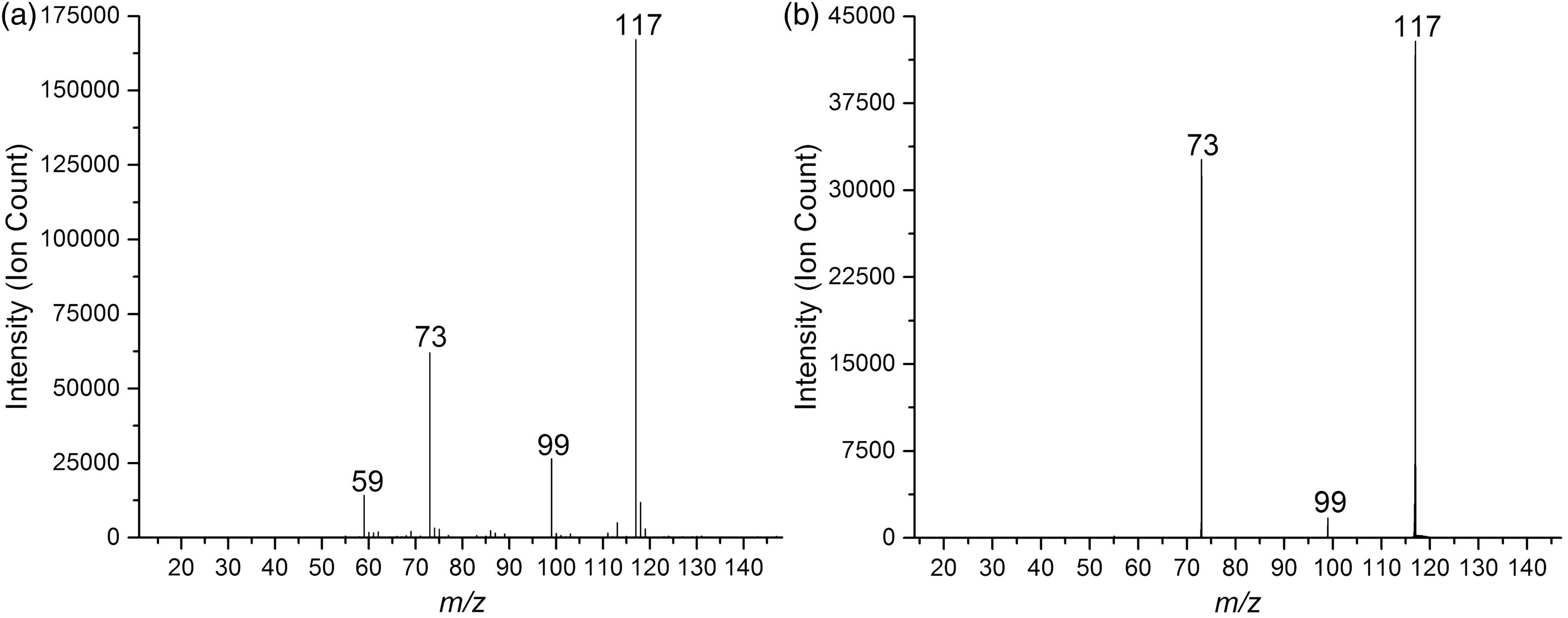
The CID mass spectrum is shown in Figure 1(b). The fragment ion peaks at m/z 99 and 73 appear in both spectra, supporting the assumption that decarboxylation and dehydration are giving rise to these peaks.
Additional peaks were observed in the CID-spectra with higher collision energies at m/z values 55, 45, 27 and 17 (supplementary information). Ions with m/z 55 may be formed by decarboxylation of the dehydration fragment ion at m/z 99, and/or by dehydration of the decarboxylation fragment at m/z 73. The peak at m/z 45 is either due to loss of CO or C2H4 from the fragment ion at m/z 73. The fragment ion at m/z 27 seems to be formed by decarbonylation of the ion with m/z 55. The fragment ion with m/z 17 is most likely the hydroxyl anion, probably formed by decarbonylation of the m/z 45 fragment ion. In addition, there is a peak at m/z 44, which we interpret to be due to carbon dioxide radical anion. Although carbon dioxide has negative electron affinity, EA = −0.60 eV,
24
making it metastable with respect to electron detachment, it is often observed upon CID of carboxylate ions. According to Schröder et al.,
25
formation of

This is consistent with propionate (m/z 73) being the direct precursor in the present experiment, since the ethyl radical (R = C2H5) has negative electron affinity, EA = −0.28 eV 26 ). Separate CID experiments of propionate, not shown here, support this assumption.
The above spectral assignments are summarized in equations (5) to (11) following.
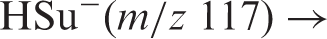
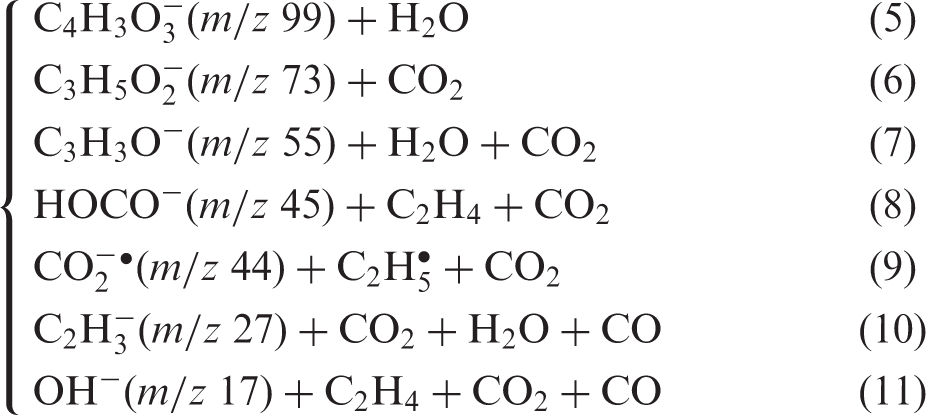
Breakdown graphs for the two fragment ions of lowest appearance energy are presented in Figure 2.
Breakdown curves of hydrogen succinate, HSu– (m/z 117) from a CID experiment recorded at a collision gas pressure of 2.0 × 10−4 mbar, with a TOF voltage of 3.0 kV. (a) Breakdown curves with signals corresponding to dehydration (m/z 99) and decarboxylation (m/z 73) of HSu−; (b) same, but the signal of m/z 99 has been multiplied by a factor of 20 in order to facilitate comparison.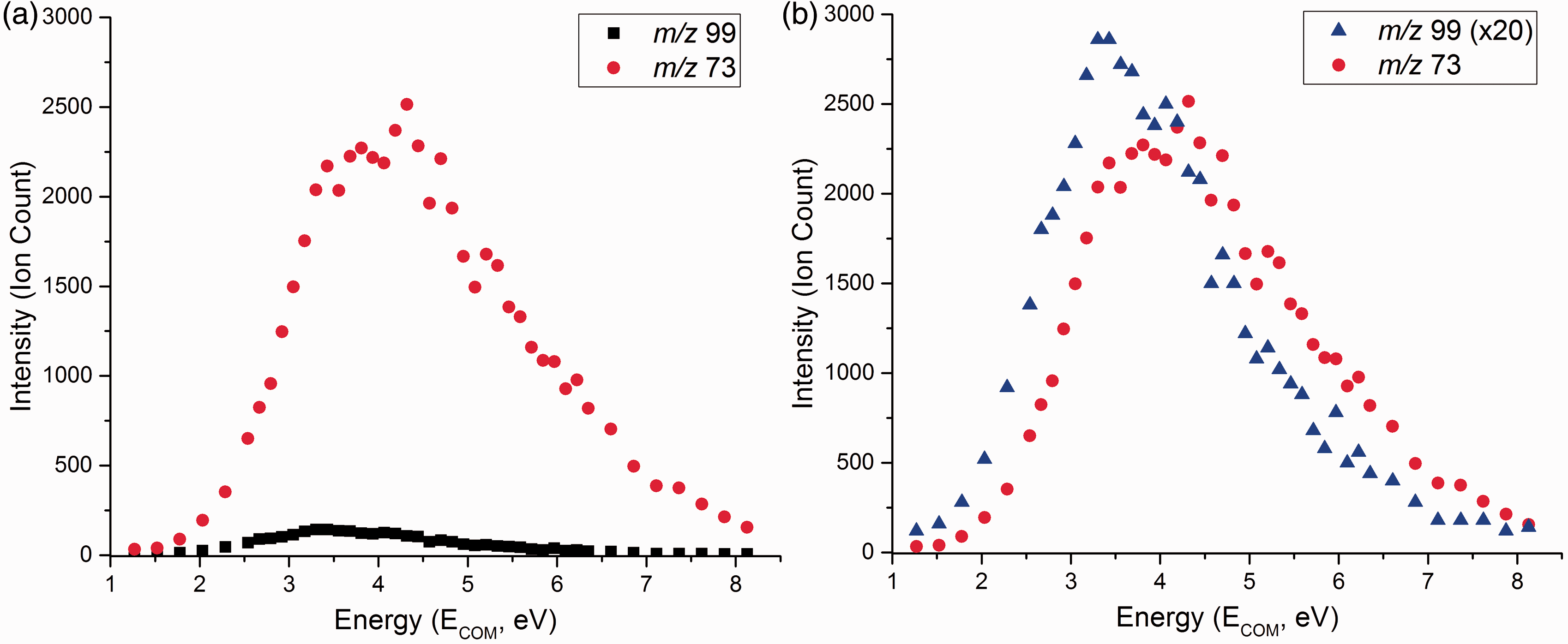
Appearance energies with standard deviations for hydrogen succinate (HSu−) fragment ions.

Mechanisms for dehydration and decarboxylation of HSu−
Next, a survey of the potential energy landscape was conducted. All results from this endeavor can be found in the supplementary information. Here, we will discuss the most prominent features. The potential energy diagram of relevance to decarboxylation and dehydration is shown in Figure 3.
Potential energy diagram (G4 (0K)) for dehydration (right hand side) and decarboxylation (left hand side) of HSu− (H-1), energies in kJ mol−1.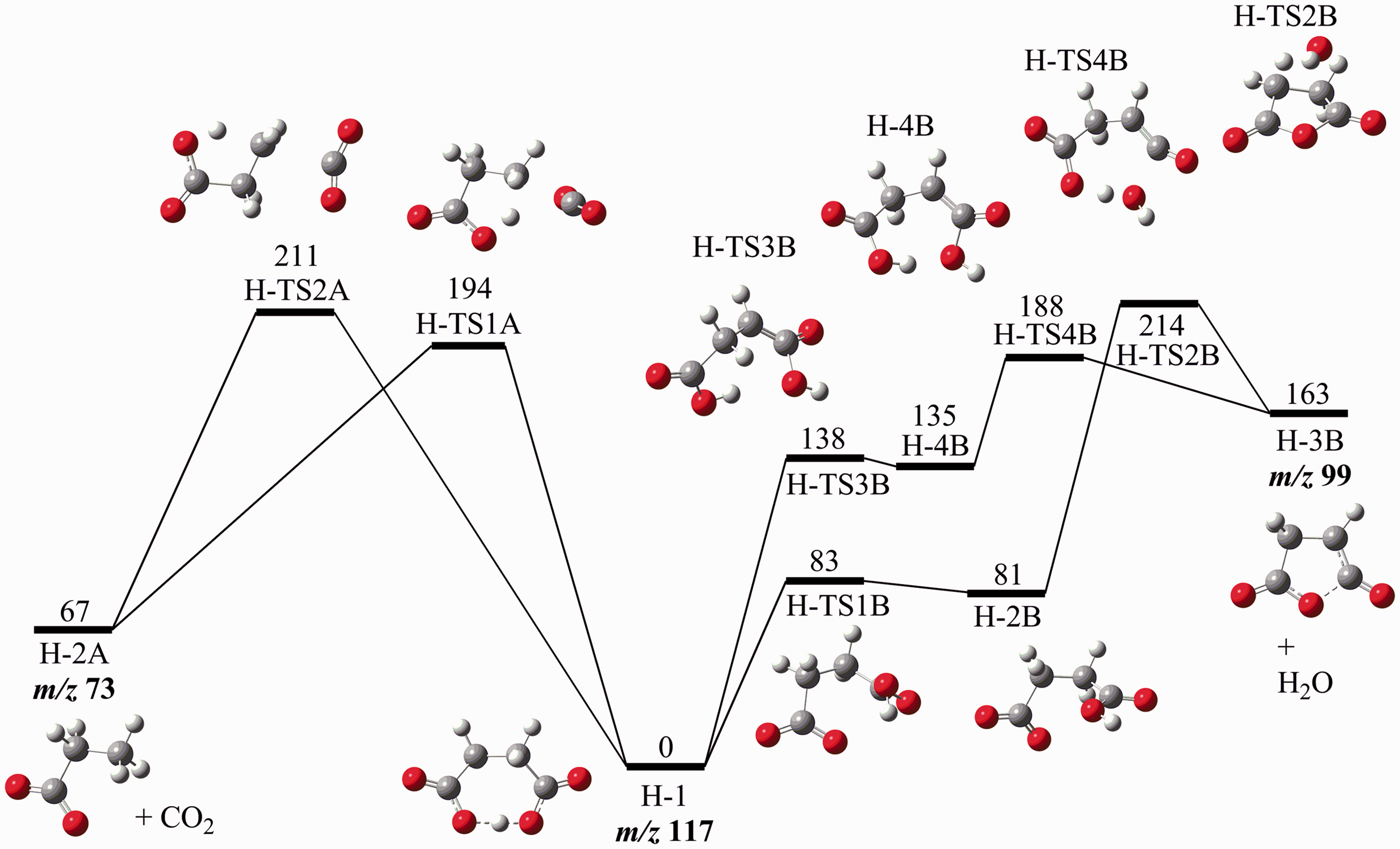
Water loss is identified to occur via the isomeric intermediates
Decarboxylation of HSu− yielding
Both decarboxylation reactions occur in one single reaction step from the most stable conformer of HSu–, indicating that they are entropically more favorable than the dehydration pathways, the latter requiring two reaction steps. This simple fact may explain why decarboxylation is kinetically dominating, although dehydration has lower critical energy. The difference in critical energy is computed to be 6 kJ mol−1.
It should also be mentioned that in the absence of the hidden proton transfer, heterolytic C–C bond dissociation leads directly to the simultaneous formation of three fragments—ethene, carbon dioxide and hydroxycarbonyl anion, HOCO− at 320 kJ mol−1 (not shown in Figure 3, see supplementary information).
Mass spectra, dissociation reactions and breakdown curves of ClMgSu− (m/z 175)
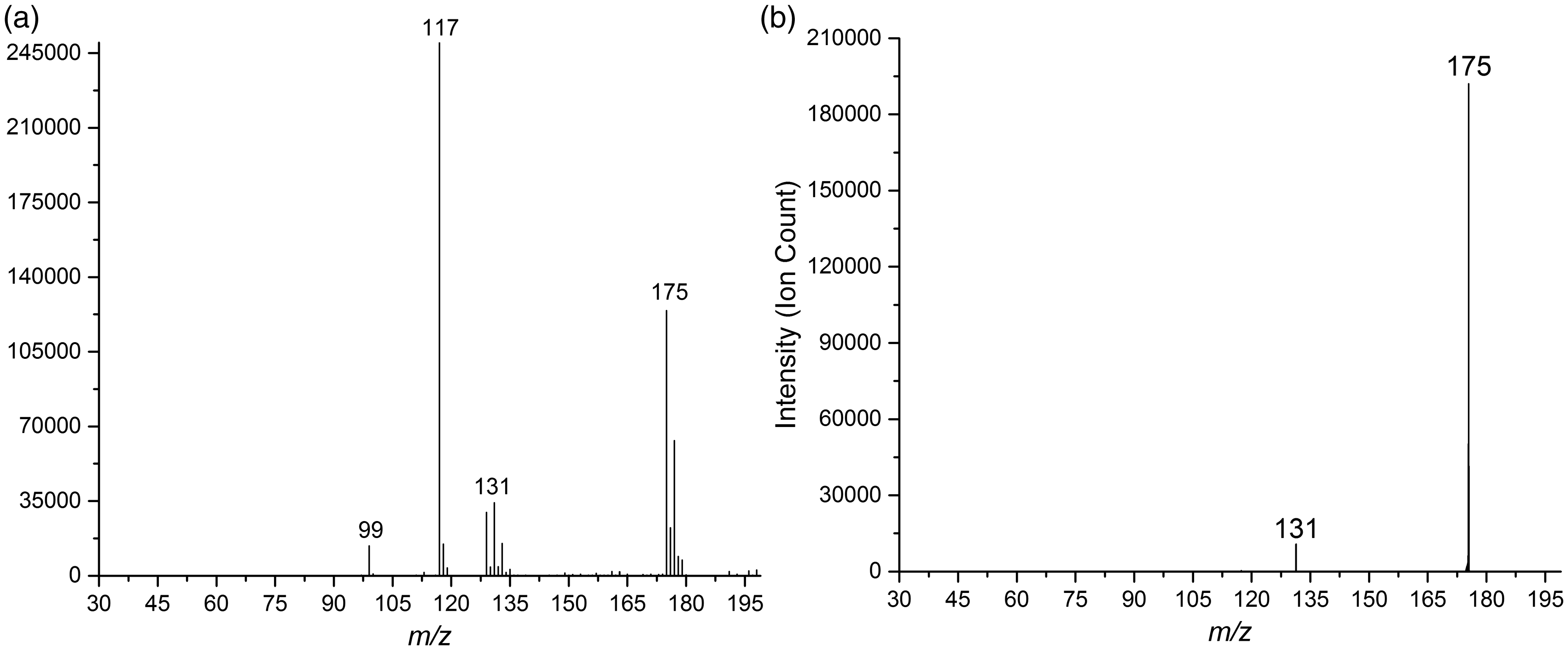
Results from the experiments with the mixture of succinic acid and magnesium chloride are displayed in Figure 4. The relative intensity distribution found for the group of signals in the range m/z 175–179 in the normal mass spectrum of Figure 4(a) is consistent with the theoretical isotopic distribution of ClMgSu−. The group of peaks m/z 129–135 is largely due to MgCl3− anion, but with a contribution (ca. 6 %) from [(ClMgSu) − CO2] −. The CID mass spectrum of ClMgSu− (m/z 175), displayed in Figure 4(b), suggests that the only major fragmentation reaction is decarboxylation. At higher collision energies (supplementary information), a minor peak at m/z 129 indicates loss of H2 from the decarboxylation fragment ion. In addition, a fragment ion with m/z 103 was observed at higher collision energies, most likely due to ClMg(η2-O2C)−,7,16 in analogy with the aforementioned CID experiments with ClMgMa− mentioned in the Introduction.
2
Also, at high collision energies, fragment ion peaks are found for m/z 87, 71 and 59. In short, the assignments are as follows ((12) to (17)).
(a) Mass spectrum of a mixture of succinic acid and MgCl2 in an 80/20 v/v H2O/MeOH solution. (b) CID mass spectrum of ClMgSu− (m/z 175) with collision energies averaged over 3.0–5.0 eV (ECOM) at 3.0 × 10−4 mbar Ar pressure, 7.0 kV TOF voltage.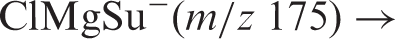
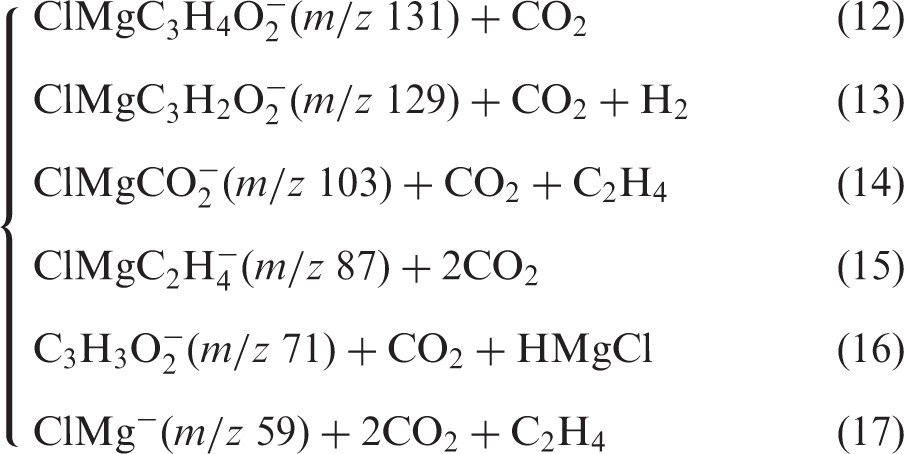
The appearance energies of the fragment ions were estimated from the breakdown curves in Figure 5, summarized in Table 2.
Breakdown curves of ClMgSu– (m/z 175) from a CID experiment recorded at a collision gas pressure of 3.0 × 10−4 mbar, with a TOF voltage of 7.0 kV. The ion count is a function of the centre-of-mass collision energy. Note the different scales: (a) Breakdown curves with measured intensities for all fragment ion signals. (b) Breakdown curves where the decarboxylation signal intensity has been multiplied by 0.03 in order to facilitate comparison with the rest of the signals. Appearance energies with standard deviations for magnesium chloride succinate (ClMgSu−) fragment ions.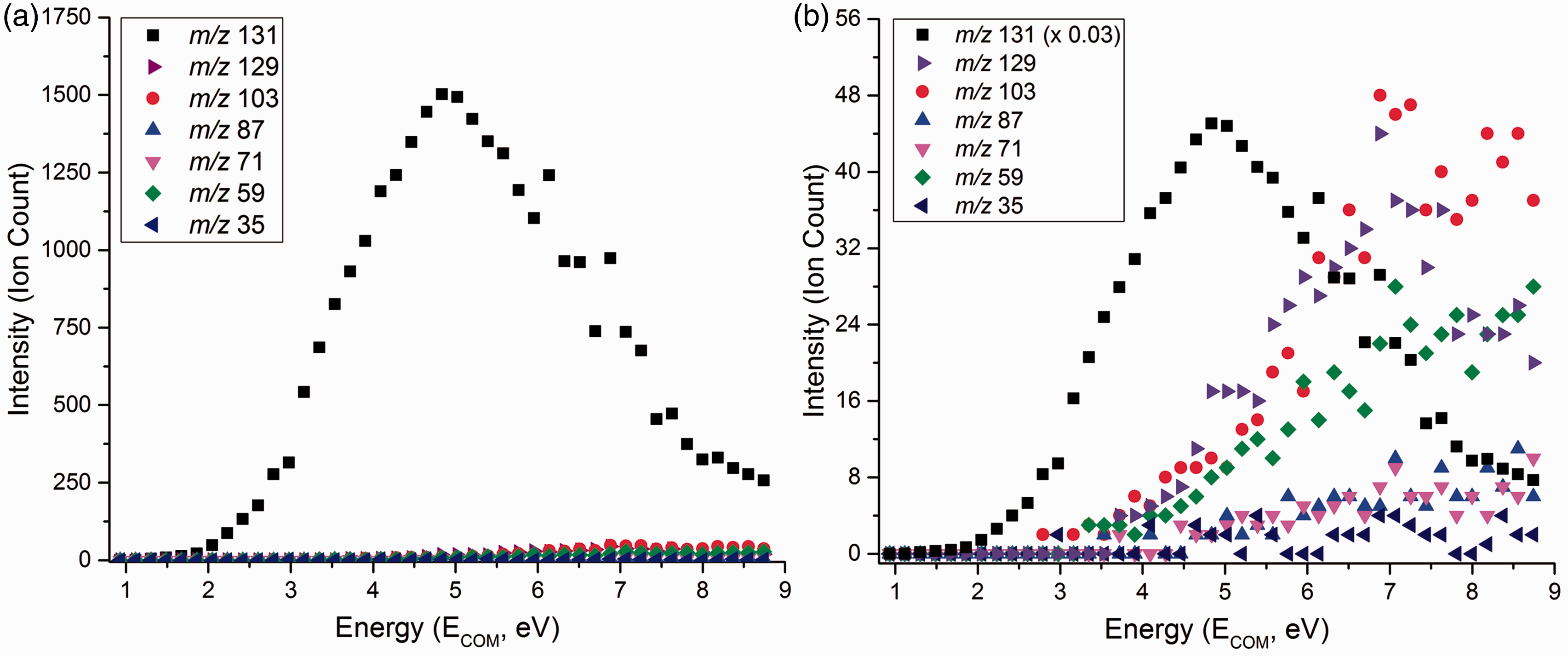

The appearance energy for decarboxylation is measured to 239 kJ mol−1, which within experimental uncertainty is not significantly different from that of the analogous reaction for HSu− at 217 kJ mol−1. No fragment ion peak at m/z 99 was observed that would correspond to the loss of ClMgOH—analogous to water loss from HSu–.
Mechanisms for decarboxylation of ClMgSu−
The computed potential energy diagram describing CO2 loss from ClMgSu− is shown in Figure 6. Two isomeric forms of ClMgSu–, Potential energy diagram (G4 (0 K)) showing the decarboxylation of ClMgSu−, energies in kJ mol−1.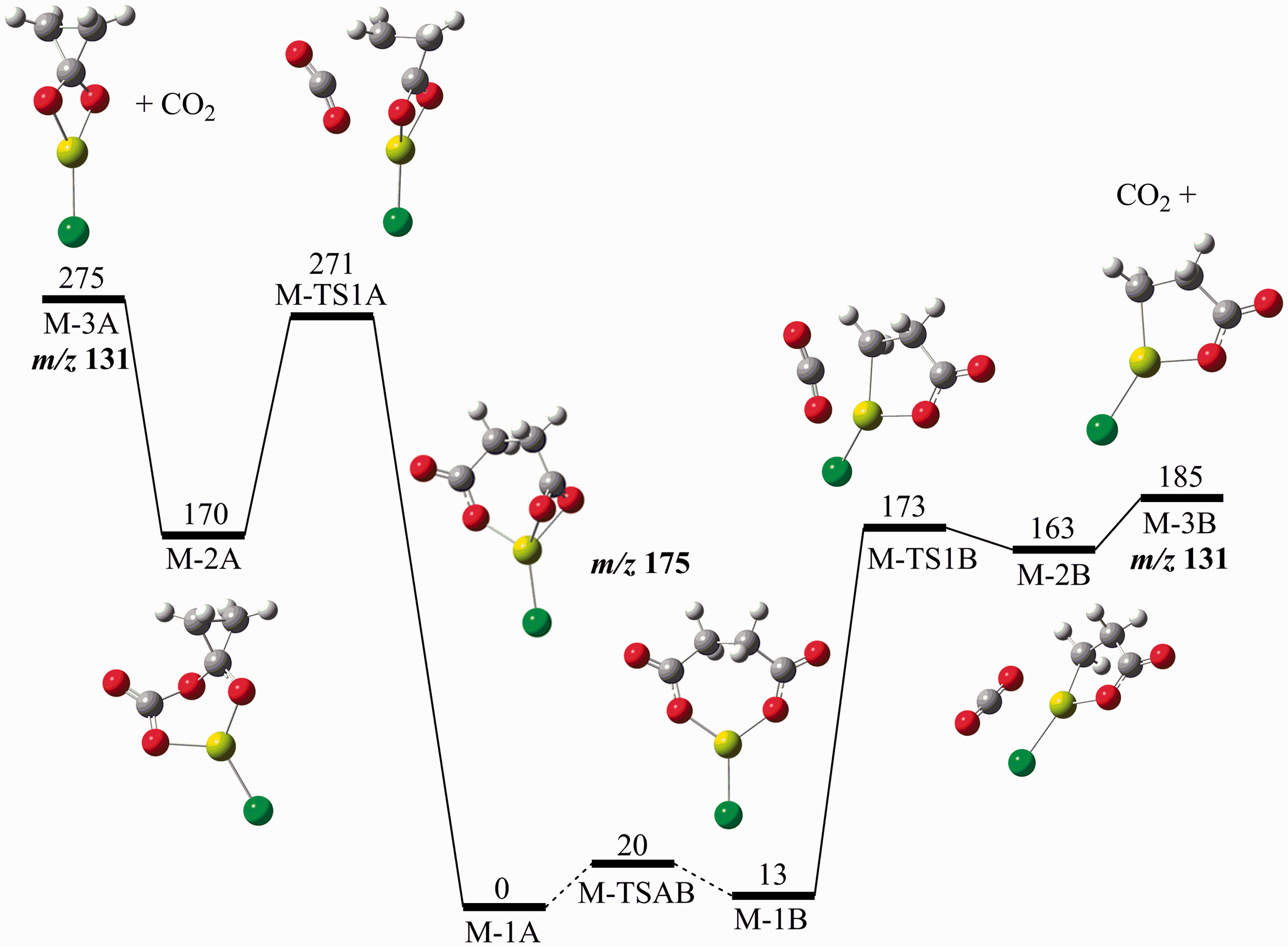
Mechanism for formation of ClMg(η2-O2C)−
Starting from the spiro compound Potential energy diagram (G4 (0 K)) for the formation of ClMg(η2-O2C)−(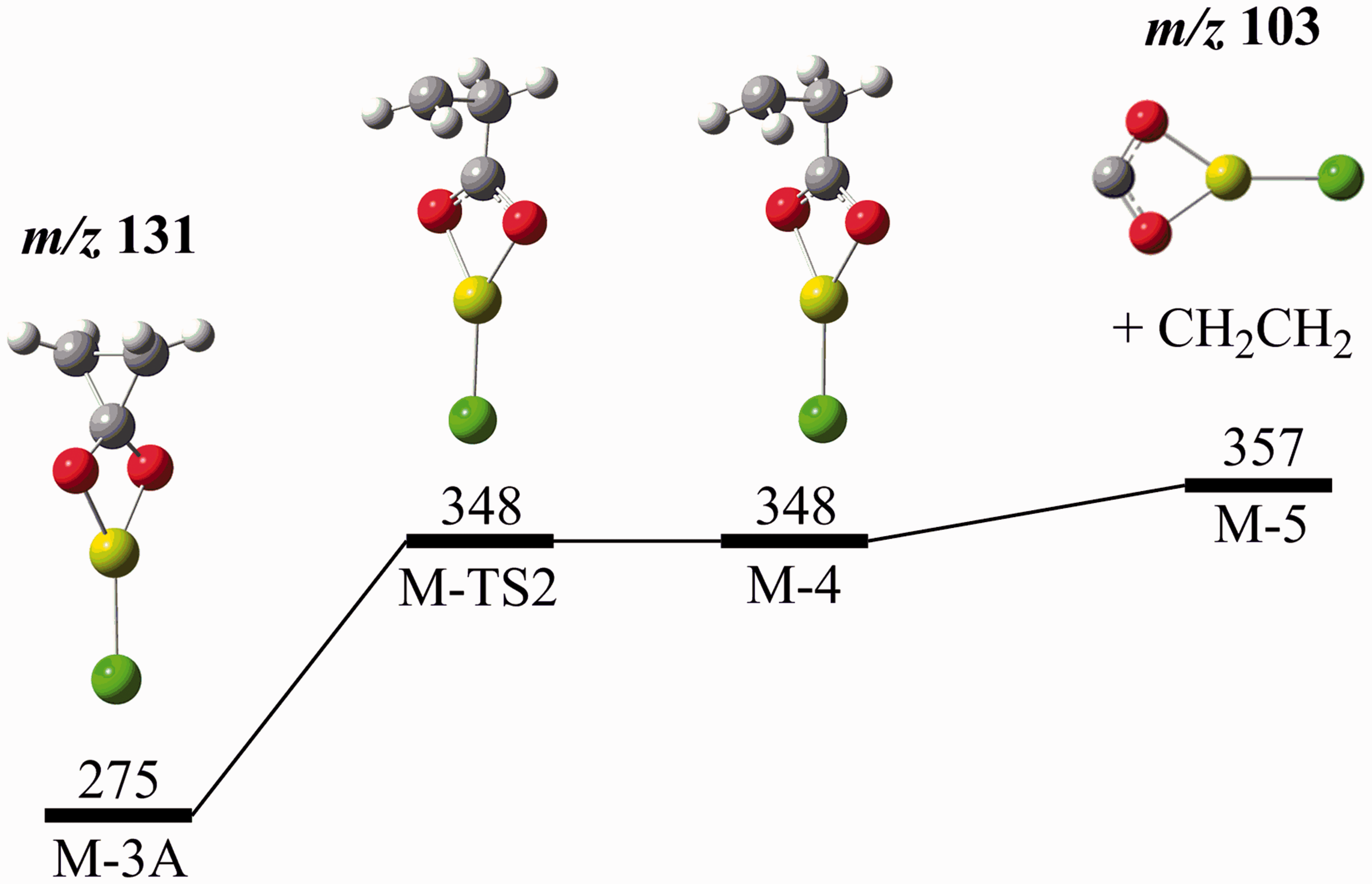
Discussion
G4 (0 K) critical energies in kJ mol−1 for the main pathways of dissociation for succinate and maleate adducts with H+ and MgCl+.
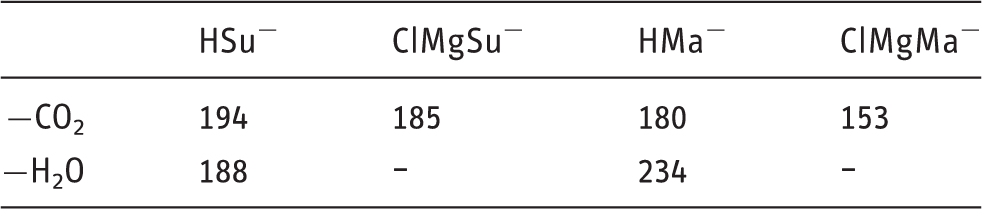
In comparing CO2 loss from HSu− and HMa−, we see that the latter has a slightly lower barrier. The reaction mechanisms are similar in producing the propionate and acrylate anions, respectively, by intramolecular transfer of a proton to the carbanionic site gradually formed as the CO2 molecule is departing.
However, the mechanisms for dehydration are different, and the barrier for dehydration of HMa– is 46 kJ mol−1 higher for than for HSu–. While the products deprotonated succinic anhydride + H2O are 160 kJ mol−1 relative to HSu–, deprotonated maleic anhydride + H2O are 230 kJ mol−1 relative to HMa−. This considerable stability difference is linked to major structural differences between the geometries and electronic structures of deprotonated succinic anhydride and deprotonated maleic anhydride molecules. In the former, the charge can be effectively delocalized to the nearby oxygen atoms, while in the latter, the restrictions imposed by the C=C double bond hampers this. As a result, the energy landscapes for the dehydration reactions are quite different.
Decarboxylation is the major fragmentation reaction for both ClMgSu– and ClMgMa–. Both have two isomers that differ by Mg coordination to two and three oxygens, respectively, of which the 3-coordinated isomers are lowest in energy. For ClMgSu–, the 2-coordinated is 13 kJ mol−1 higher in energy, while for ClMgMa–, it is only 1 kJ mol−1 higher, which may account for some of the difference in that the most facile decarboxylation reaction mechanism for ClMgMa− has a critical energy of 153 kJ mol−1, but for ClMgSu− at 185 kJ mol−1. The other contributing factor lies in the relevant product stabilities. The Mg–C bond distance is 0.02 Å shorter in the Grignard species for Ma than for Su, indicating a stronger interaction. Separate G4 calculations show that the following isodesmic reaction is exothermic by 20 kJ mol−1
The formation of ClMg(η2-O2C)− from both ClMgMa− and ClMgSu− uncovered an interesting opportunity to describe the differences in the reactivity of this carbenoid species with ethene and ethyne. Addition of ClMg(η2-O2C)− ( Nucleophilic addition of ClMg(η2-O2C)− to ethyne (top) and ethene (bottom).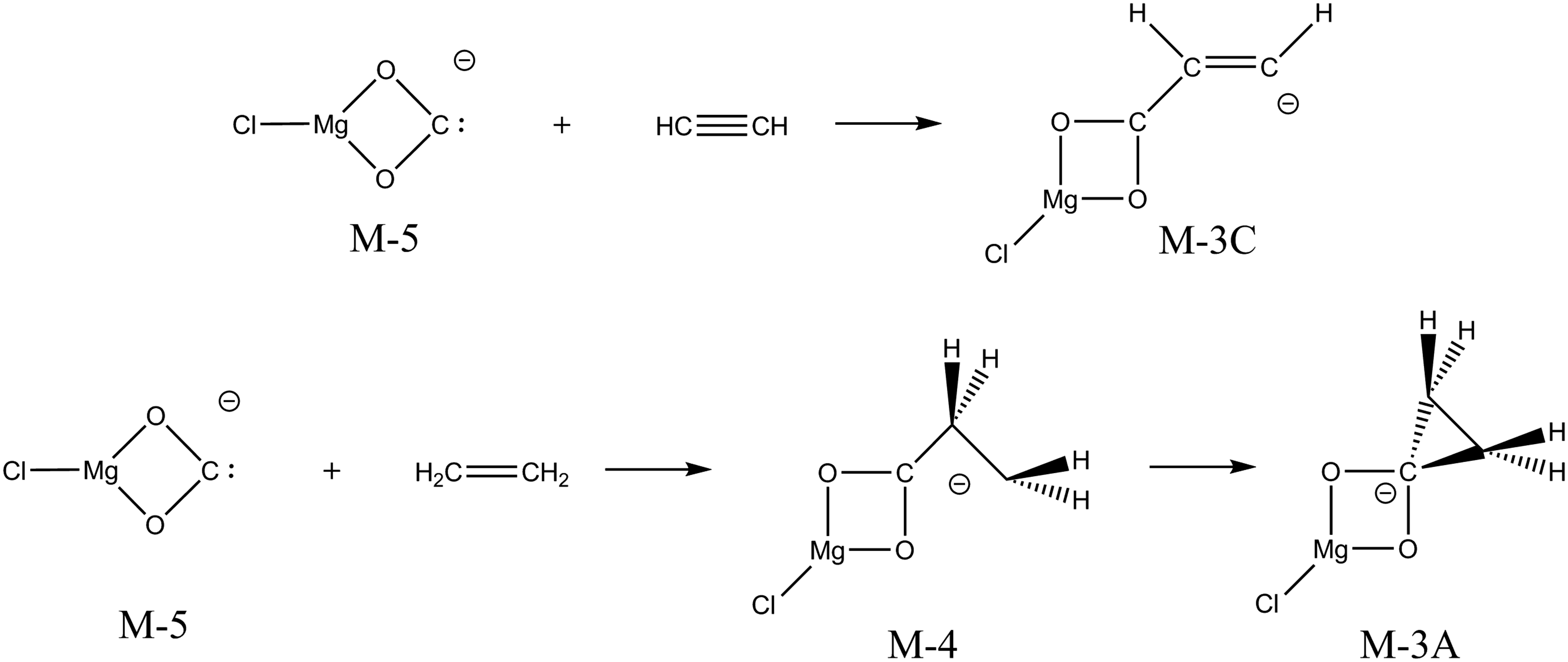
Polino et al.
30
performed quantum chemical calculations (RQCISD(T)/CBS// B3LYP/6-311++G(d,p)) of reactions of singlet methylene (1CH2) with ethyne and ethene, that form cyclopropene and cyclopropane, respectively, with reaction energies of 364 and 416 kJ mol−1. The ethene and ethyne affinity of ClMg(η2-O2C)−, which is an electronic singlet in its lowest energy state, is therefore seen to be significantly lower than that of singlet methylene. In addition, the ethyne and ethene affinities are inverted. However, it should be taken into account that the adduct between ClMg(η2-O2C)− and ethyne was found to be non-cyclic. Separate calculations show that the binding energy of
Conclusion
We have investigated the dissociative patterns of hydrogen succinate and magnesium chloride succinate anions and compared them with the analogous maleate systems. The main fragmentation reactions of hydrogen succinate and maleate were decarboxylation and dehydration. The demands for decarboxylation of the two were found to be quite similar; however, dehydration was more facile for the succinate system. This is attributed to the increase in conformational freedom going from double to single bonds in the carbon backbone, emphasizing the connection between structure and dissociative mechanism as has been reported in the past.2,17,18 The dissociation of magnesium chloride succinate and maleate adduct anions occurs mainly through decarboxylation, which is most demanding for the former. The increased conformational freedom of hydrogen succinate compared to hydrogen maleate facilitates water loss, yet makes decarboxylation more difficult as the stabilization by the intramolecular bond between the carboxylate oxygen atoms and the H+ and MgCl+ ligands is stronger than in the latter, structurally more rigid anion. The nucleophilic metal-CO2 adduct anion is observed as a fragmentation product in both systems, the survey of which has provided interesting details in relation to its reactivity with ethyne and ethene. The ClMg(η2-O2C)− adduct anion adds to these hydrocarbons in one and two steps, respectively. Addition to the former yields a structure with a single bond between ethyne and ClMg(η2-O2C)−, while the equivalent reaction with the latter forms a structure with a double bidentate bonding moiety.
Footnotes
Acknowledgements
The authors are grateful to Glenn B.S. Miller for valuable suggestions.
Authors’ note
This article is dedicated to the memory of Peter Derrick, a great friend and colleague.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Norwegian Research Council by the Grant No. 179568/V30, to the Centre of Theoretical and Computational Chemistry through their Centre of Excellence program, by the grant No. 249788 to the project “The chemistry of CO2 activation and fixation”, and the Norwegian Supercomputing Program (NOTUR) through a grant of computer time (Grant No. NN4654K).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.