
Abstract
A method for the analysis of 11 haloacetic acids in water samples has been developed. It involves enrichment of the target analytes from water samples by solid-phase extraction, derivatization to methyl esters, and gas chromatography coupled with tandem mass spectrometry determination. Gas chromatography conditions were optimized for a good separation of all haloacetic acids in a short runtime. Data were acquired in the multiple reaction monitoring mode. Six solid-phase extraction sorbents among the most widely used in environmental analysis were tested. Bakerbond SDB was retained because it has been shown to provide the best results for a large class of targeted haloacetic acids. The performances of the developed method have been assessed according to the French Standard NF T 90-210. The calibration curves for all the studied haloacetic acids had consistent slopes with r2 values > 0.99. Quantification limits between 0.01 and 0.50 µg l−1 were achieved. Satisfactory repeatability (relative standard deviation ≤ 14.3%) and intermediate precision (relative standard deviation ≤ 15.7%) were obtained. Applied to the analysis of 15 untreated water samples collected from three rivers, the method allowed the detection of five haloacetic acids including monochloroacetic acid (in 100% of the samples, <0.5–1.85 µg l−1), dichloroacetic acid (87%, <0.05–0.22 µg l−1), trichloroacetic acid (93%, <0.05–0.52 µg l−1), dibromoacetic acid (53%, <0.01–0.40 µg l−1), tribromoacetic acid (20%, <0.05–0.14 µg l−1), and bromodichloroacetic acid (6%, < 0.05 µg l−1).
Introduction
Haloacetic acids (HAAs) are among the main organohalogen by-products (OXBPs) regularly identified in water. 1 They are primarily formed as a result of disinfection treatments using halogen-based biocides, such as chlorine (Cl2/HOCl/OCl-), chlorine dioxide (ClO2), ozone in presence of bromide (O3/Br−), and monochloramine (NH2Cl). 2 With trihalomethanes, HAAs constitute the largest group of OXBPs by weight in drinking water. 3 The formation mechanism of HAAs is well understood and their concentration levels in drinking water are regulated in various countries. 4 HAAs are also widespread environmental contaminants, their presence in aquatic environments is not only related to human activities but also to natural sources.5–7 Trichloroacetic acid (TCAA) is used as a selective herbicide, an etching or pickling agent in the surface treatment of metals and an auxiliary in textile finishing, while monochloroacetic acid (MCAA) is mainly used as intermediate in the synthesis of a wide variety of chemicals (e.g. drugs, dyes, and pesticides).8–10 HAAs may thus enter aquatic ecosystems through many routes, including discharges of water treated with halogen-based biocides, degradation of halogenated organic compounds, runoff from contaminated soils, atmospheric deposit, as well as natural production.10–15 In vitro and in vivo laboratory studies have shown that HAAs are cytotoxic, genotoxic, mutagenic, and teratogenic.16–20 Several HAAs have been shown to produce developmental and/or reproductive toxicity. 21 Due to their potential adverse effects on human health, the World Health Organization has proposed guideline values for MCAA, dichloroacetic acid (DCAA), and TCAA. 2 In the US, HAAs are regulated by the Environmental Protection Agency (US EPA), which has established a maximum contaminant level for the total concentrations of MCAA, monobromoacetic acid (MBAA), DCAA, dibromoacetic acid (DBAA), and TCAA in drinking water. 2 A guideline value for these same five HAA species also exists in Canada. However, to date, no regulation has been promulgated in the European Union (EU) to control the level of HAA concentrations in drinking water. To protect freshwater aquatic organisms, the European Chemicals Agency included nine brominated and chlorinated HAAs in a selection of relevant disinfection by-products and representative compounds to be addressed in environmental risk assessments under the European biocide law (Regulation (EU) 528/2012). 22 To date, there is a need for a sensitive and specific method permitting the quantitation of these compounds at trace concentrations in river water, in order to evaluate their effects on aquatic ecosystems. Currently, the standard methods more commonly used for HAAs analysis involve liquid–liquid extraction (LLE) followed by gas chromatography using electron capture detection or mass spectrometry (MS) detection (552.2 EPA Method; ISO 23631:2006). However, LLE is coming under increasing criticism because it is time consuming, labor intensive, and it requires large volumes of organic solvents. Solid-phase extraction (SPE) is gradually replacing classical LLE because it generally provides best extraction recoveries with low solvent consumption. Only a few studies using MS operated in the multiple reaction monitoring (MRM) mode rather than selected ion monitoring have been reported, although the potential presence of a large number of interfering OXBPs requires using a very selective mode for unambiguous determination of HAAs.23,24 In the context of trace analysis in complex mixtures, MRM provides unparalleled selectivity, which allows accurate quantitation of HAAs even if they are not fully chromatographically resolved.
The aim of the present work was to develop a fast and efficient alternative analytical method for the simultaneous determination of 11 HAAs including five US EPA-regulated HAAs (MCAA, DCAA, TCAA, MBAA, and DBAA), four unregulated HAAs (tribromoacetic acid (TBAA), bromochloroacetic acid (BCAA), dichlorobromoacetic acid (DCBAA), and dibromochloroacetic acid (DBCAA)), as well as two emerging iodinated HAAs (monoiodoacetic acid (MIAA) and diiodoacetic acid (DIAA)) in river water samples. The analytical approach combines SPE with chemical derivatization and GC–MS/MS analysis in the MRM mode.
Material and methods
Reagents and chemicals
A mixed standard containing MBAA, BCAA, bromodichloroacetic acid (BDCAA), MCAA, DBCAA, DBAA, DCAA, TBAA, and TCAA (EPA 552.2 Haloacetic Acids Mix, 2000 µg ml−1 each component in MTBE, >99%) was obtained from Sigma Aldrich (Saint-Quentin Fallavier, France). Iodoacetic acid (98%), 1,2-dibromopropane (internal standard, 97%) and sulfuric acid were also purchased from Sigma Aldrich, as well as
Pretreatment, extraction, and derivatization procedures
All samples were collected in 2 l amber bottles. Two milliliters of
Instrumentation and GC–MS/MS analytical conditions
GC–MS analysis of the SPE extracts was performed using a Thermo Fisher Scientific “Trace GC Ultra” gas chromatograph equipped with a “TriPlus” autosampler and coupled with a “TSQ Quantum XLS” triple quadrupole mass spectrometer (Thermo Fisher Scientific, Courtaboeuf, France). The chromatographic separation was carried out on a Thermo Scientific “TG–5MS” (5% phenyl, 95% methylpolysiloxane) 30 m capillary column (internal diameter: 0.25 mm, film thickness: 0.25 µm). High purity helium (99.9995%) was used as the carrier gas at a constant flow of 1.4 ml min−1. All experiments were performed using automatic injection of 1.5 µl of sample into a programmed temperature vaporization (PTV) injector in the splitless mode. The PTV conditions were the following: injection temperature, 180℃; cleaning temperature, 270℃; splitless time, 2.00 min; split flow, 30 ml min−1; and cleaning time, 4.00 min. In order to trap the analytes at column head, the oven temperature was maintained at 35℃ for 5.00 min before being raised at 10℃ min−1 to 110℃ and then at 20℃ min−1 to 200℃, for a total duration of 17 min. The solvent delay was set at 4.50 min. The transfer line and ion source temperatures were maintained at 280 and 250℃, respectively. In the first approach, acquisition was carried out in the EI full scan mode from 50 to 450 m/z at 100 ms per scan. The mass spectrometer was operated in the electron ionization mode at 70 eV. The filament emission current was set at 25 µA in the full scan mode and at 50 µA for MS/MS experiments. The electron multiplier voltage was set at 1040 V using automatic tuning (3.105 gain). The tandem MS experiments were performed with argon as collision gas at a nominal pressure of 1 mTorr. The collision-induced dissociation parameters were subjected to optimization and are given below.
Results and discussion
GC–MS/MS characterization
The mass spectrometer and gas chromatograph parameters were optimized by injecting a mixture of all HAAs (methyl ester derivatives) in MTBE each one at 20 µg ml−1. Separation of HAAs was optimized in the gradient mode with the purpose to achieve good chromatographic resolution in a short analysis time. The optimum gradient is described in “Instrumentation and GC–MS/MS analytical conditions” section. The upper part of Figure 1 displays the chromatogram of the mixture of all HAAs (after SPE and derivatization), each at 5 µg l−1, together with the surrogate and internal standards at 4 µg l−1. Most of the target HAAs are well separated with the exception of BCAA, MIAA, DBCAA, and 2,3–dibromopropionic acid (surrogate standard, SA). MS/MS optimization led to the selection of the two most intense transitions per HAA for operation in MRM according to the EU criteria (C.D. 2002/657/EC). The transition providing the most intense signal was used for quantification. MRM transitions and their corresponding collision energies are summarized in Table 1.
Chromatograms of the reference solution of HAAs at 5 µg ml−1 (above) and of a real sample taken on 22 August 2016 (below). 1: MCAA, 2: MBAA, 3: DCAA, IS: 1,2-dibromopropane, 4: TCAA, 5: BCAA, 6: MIAA, 7: DBAA, 8: BDCAA, 9: DBCAA, SA: 2,3-dibromopropionic acid, 10: TBAA, 11: DIAA. Main characteristics of the GC–MS/MS method. LOQ: limits of quantitation.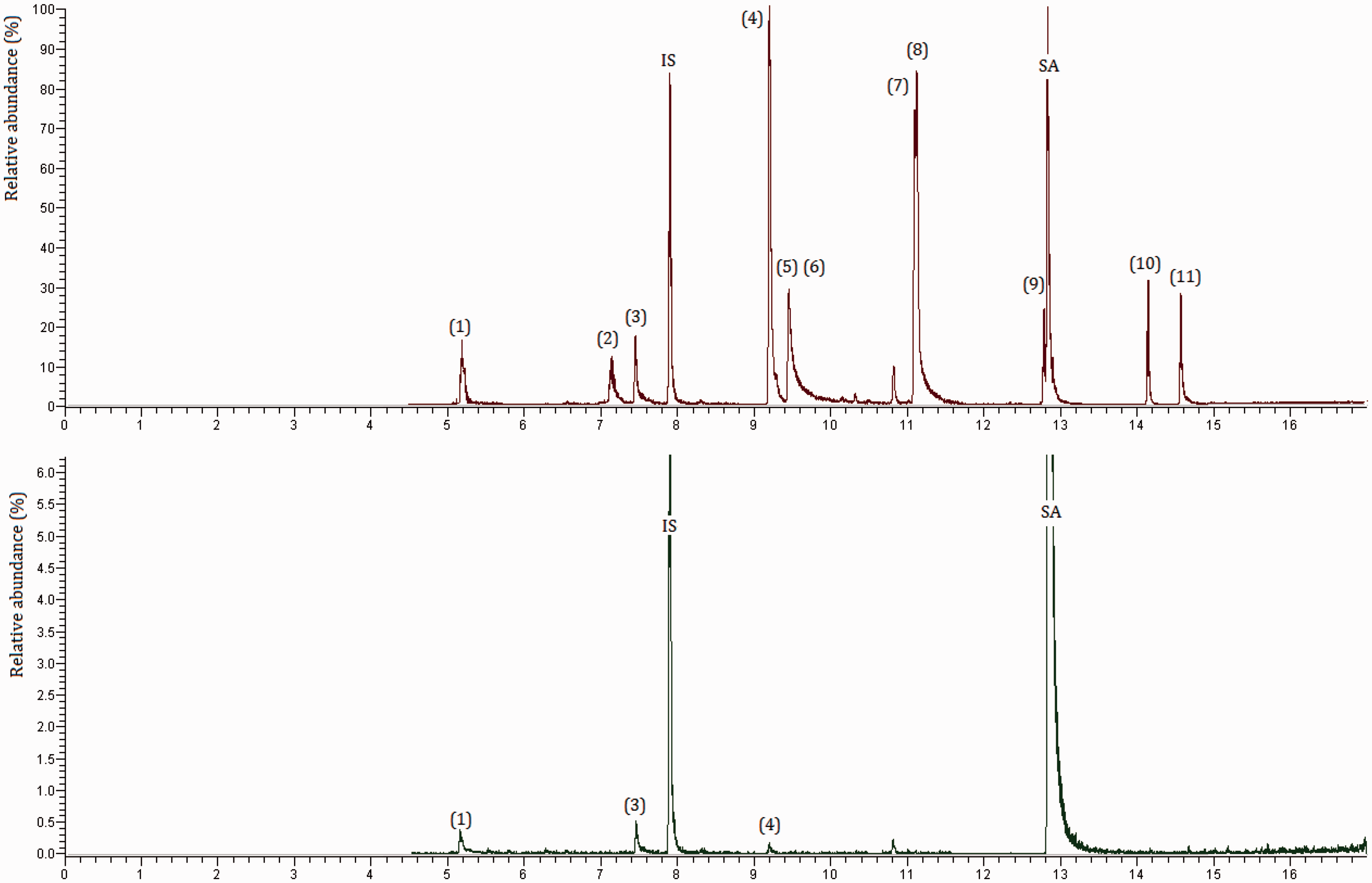
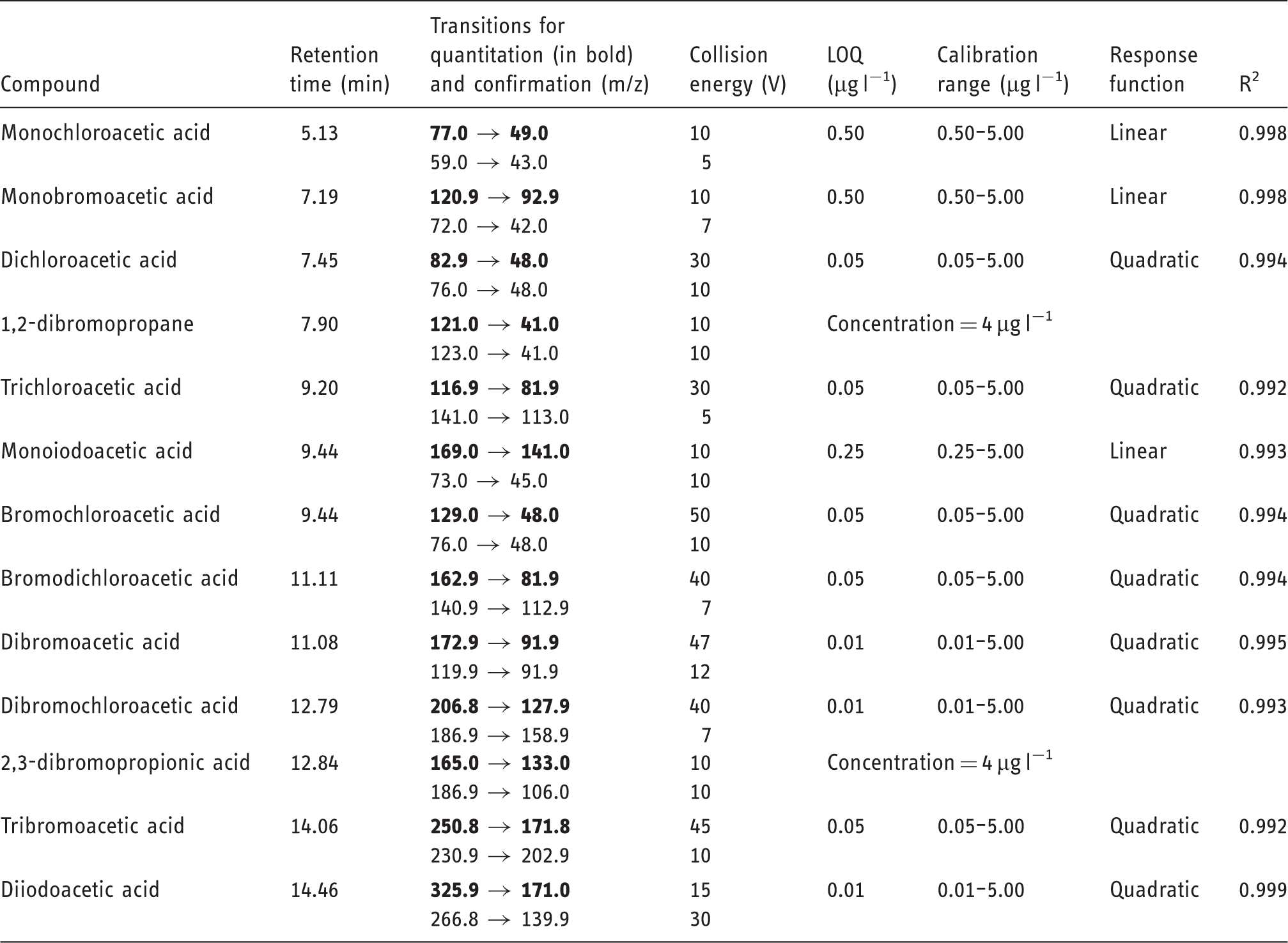
SPE
Relative recovery yields of 11 HAAs from 1 l of river water using six SPE commercial sorbents.
BCAA: bromochloroacetic acid; BDCAA: bromodichloroacetic acid; DBAA: dibromoacetic acid; DBCAA: dibromochloroacetic acid; DCAA: dichloroacetic acid; DIAA: diiodoacetic acid; HAAs: haloacetic acids; MBAA: monobromoacetic acid; MCAA: monochloroacetic acid; MIAA: monoiodoacetic acid; RSD: relative standard deviation (n = 3); TBAA: tribromoacetic acid; TCAA: trichloroacetic acid.
% average recovery of SPE cartridges.
The breakthrough volume, defined as the maximum water sample volume that can be percolated on the SPE cartridge without analyte losses, was evaluated for Bakerbond SDB cartridges. This parameter was tested by analyzing a river water sample spiked with HAAs at 5 µg l−1. For most target analytes, the best results were achieved using a water sample volume of 1 l.
Method performances
Response functions, limits of quantitation (LOQ), trueness (bias), and precision of the analytical method were assessed according to the French method validation standard NF T90-210, as previously described for the validation of a method devoted to monochloramine determination in river water. 30 Table 1 summarizes the method performances achieved for each HAA, in terms of limit of quantitation and correlation coefficient over the calibration range. Various regression functions, including linear, quadratic, and cubic equations, were fitted to the data and compared, in order to determine the best regression model of each HAA. The most suitable mathematical models to describe the relationship between the concentrations of the studied HAAs and their respective responses were the linear model for MCAA, MBAA, and MIAA, and the quadratic model for the other target HAAs. A good correlation was obtained for all the studied compounds with correlation coefficients (r2) values higher than 0.99. The calibration curves were also evaluated with and without using weighting factors. The lower errors were obtained when a 1/x weighting factor was applied, where x represents the concentration in HAAs.
The LOQ values were first theoretically estimated and then experimentally confirmed by the analysis of blank rivers samples fortified at the estimated levels. For each HAA, the concentration for which the signal-to-noise ratio was found to be equal to or greater than 10 was considered as the presupposed LOQ. The LOQ values were then approved according to the “B test” of the NF T90-210 standard. The LOQ of each HAA was defined as the lowest concentration, which could be accurately and precisely determined with less than 60% total error. As shown in Table 1, LOQ values range from 0.01 to 0.50 µg l−1, depending on the HAA considered. Several research groups have previously developed analytical methods for HAAs analysis. This new method provided lower LOQs for several HAAs in comparison with LOQs reported using methods based on the standard ISO 23631 guidelines (from 0.5 to 10 µg l−1). Li et al. used a method combining LLE and GC–MS/MS analysis; it was equally sensitive with comparable LOQs (from 0.03 to 0.24 µg l−1). 24 Compared with LLE, SPE tends to be relatively more efficient for MCAA and, to a lesser extent, for brominated HAAs, while being slightly less efficient for other species.
The trueness is the closeness of agreement between the average value of a series of measurements and a value considered as true. It estimates the systematic error at each concentration level and is expressed as a relative bias. As no certified reference material is available for HAAs analysis in water samples, the trueness of the developed method was assessed by analyzing river water samples spiked with standard solutions of HAAs. The precision (intra-day precision) is the closeness of agreement between the values obtained from repeated measurements. It estimates the random error of the method and is expressed in terms of relative standard deviation (% RSD). The intermediate precision (inter-day precision) was investigated to determine the time-dependent variability of the method.
Trueness and precision of the SPE GC-MS/MS developed method.
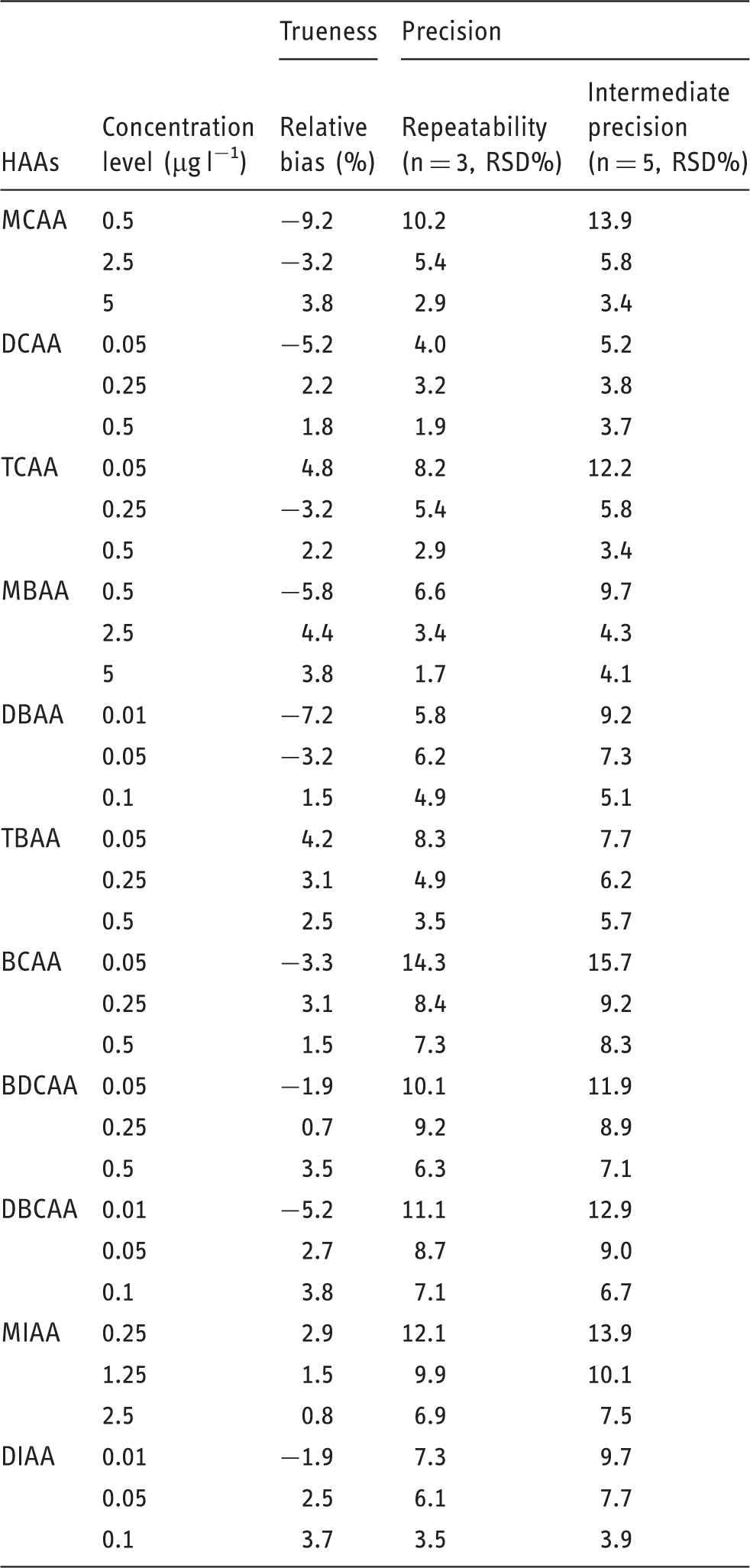
BCAA: bromochloroacetic acid; BDCAA: bromodichloroacetic acid; DBAA: dibromoacetic acid; DBCAA: dibromochloroacetic acid; DCAA: dichloroacetic acid; DIAA: diiodoacetic acid; HAAs: haloacetic acids; MBAA: monobromoacetic acid; MCAA: monochloroacetic acid; MIAA: monoiodoacetic acid; TBAA: tribromoacetic acid; TCAA: trichloroacetic acid.
Accuracy is an important concept in method validation because it represents the global performance of the method (precision and trueness). Based on the obtained trueness and precision values, the accuracy of the method has been evaluated for each HAA. The accuracy profiles obtained respect the maximum acceptable deviation fixed by our laboratory, set at ± 60% for the LOQ and ± 20% in the 5–10 ×LOQ concentration range.
Application to real river water samples
Water quality characteristics of the river waters analyzed.
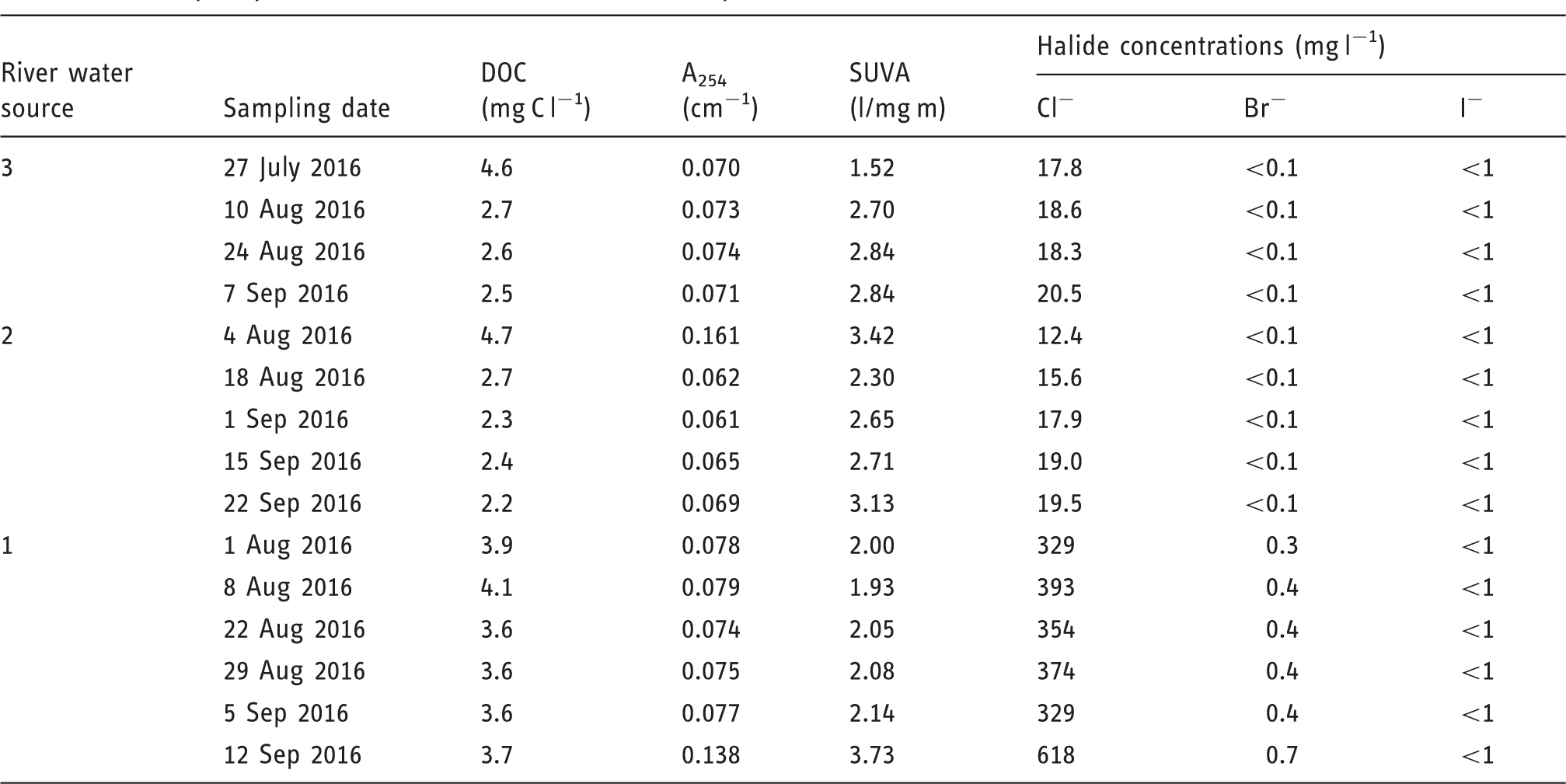
A254: Absorbance at 254 nm; DOC: dissolved organic carbon; SUVA: specific ultraviolet absorbance.
HAAs occurrence (µg l−1) in the three studied rivers.

BCAA: bromochloroacetic acid; BDCAA: bromodichloroacetic acid; DBAA: dibromoacetic acid; DBCAA: dibromochloroacetic acid; DCAA: dichloroacetic acid; DIAA: diiodoacetic acid; HAAs: haloacetic acids; LOQ: limits of quantitation; MBAA: monobromoacetic acid; MCAA: monochloroacetic acid; MIAA: monoiodoacetic acid; n: number of water samples analyzed for each river water; nd: not detected (concentration < LOD); Si: sampling dates (see table SI 1, in the supporting information file); TBAA: tribromoacetic acid; TCAA: trichloroacetic acid.
Conclusion
This article presents a sensitive, selective, and specific method for the determination of several classes of HAAs in water samples. In terms of LOQs, the developed method provides performances dramatically enhanced in comparison with those reported by standards methods following the ISO 23631 guidelines. These performances are comparable to those achieved by the method reported by Li et al. 24 SPE allows percolation of large sample volumes and is compatible with the principles of sustainable chemistry as it uses small amounts of organic solvents in comparison to LLE processes. This method is currently employed by our research group for the simultaneous extraction and determination of HAAs in river waters. It has been applied to the determination of target HAAs in water samples from three rivers in France; results indicate the quasi-systematic presence of four of the 11 target HAAs: MCAA, DCAA, TCAA, and DBAA.
Footnotes
Acknowledgements
This research was conducted in partnership between Electricité de France (EDF) and Ecole Polytechnique/CNRS (National Center for Scientific Research).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: A part of the financial support for this work was provided by EDF Research & Development to which the authors are thankful.