
Abstract
Introduction
Lung cancer progression is shaped by vascular dysfunction, hypoxia, desmoplastic remodeling, and immune exclusion. Beyond its endocrine role, the renin–angiotensin–aldosterone system (RAAS) functions locally and may regulate these microenvironmental states. This review evaluates RAAS–mediated stromal and immune regulation in non–small cell lung cancer (NSCLC).
Method
This narrative review synthesized English-language evidence from PubMed/MEDLINE, Web of Science, Scopus, ScienceDirect, and Google Scholar through April 2026. Lung cancer studies were prioritized; evidence from other settings was included to clarify mechanisms.
Results
Angiotensin II signaling through the angiotensin II type 1 receptor (AT1R) in fibroblast-rich niches may promote cancer-associated fibroblast activation, extracellular matrix accumulation, solid stress, impaired perfusion, hypoxia, and CD8+ T-cell exclusion. Accordingly, AT1R blockade may function as a microenvironment-normalizing adjunct rather than a direct tumor-cell-intrinsic cytotoxic strategy. The angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas receptor axis, angiotensin II type 2 receptor signaling, and mineralocorticoid receptor signaling show context- and compartment-dependent effects, while transcript abundance alone cannot establish peptide availability or receptor engagement. Clinical evidence linking RAAS inhibitors to improved outcomes, particularly during immune checkpoint inhibitor therapy, remains retrospective and susceptible to confounding.
Conclusions
Translation requires biomarker-stratified prospective studies demonstrating intratumoral target engagement and linking outcomes to measurable changes in matrix architecture, perfusion, hypoxia, and spatial immune organization.
Keywords
Introduction
Cancer therapy has evolved from surgery, radiotherapy, and cytotoxic chemotherapy toward molecularly targeted therapy, immune checkpoint blockade, antibody–drug conjugates, cellular therapies, and increasingly biomarker-driven multimodality strategies. 1 Lung cancer remains a central challenge within this therapeutic continuum because its clinical course is shaped not only by tumor-intrinsic genomic alterations but also by a complex tumor microenvironment (TME) that constrains drug delivery, immune access, and treatment durability.2–5 These figures underscore the need for therapeutic strategies that address both oncogenic signaling and microenvironmental barriers. Lung cancer remains a leading cause of cancer mortality and is increasingly understood as a disease shaped by reciprocal interactions between tumor-intrinsic oncogenic programs and a multicompartment TME characterized by vascular dysfunction, hypoxia, desmoplastic remodeling, and immune exclusion.6–10 The renin–angiotensin–aldosterone system (RAAS), classically viewed as a circulating endocrine regulator of blood pressure and volume, is now also recognized as a tissue-resident peptide network with paracrine and autocrine functions. 11 In cancer-related settings, angiotensin II (Ang II) signaling through the angiotensin II type 1 receptor (AT1R) can promote pro-angiogenic, inflammatory, and stromal programs, whereas pharmacologic blockade—most commonly with angiotensin receptor blockers (ARBs) such as losartan—can attenuate selected components of this response.12,13
Viewed from a systems perspective, RAAS signaling can be analyzed as a multiscale regulator linking vascular state, stromal mechanics, and immune accessibility to translational opportunities in lung cancer. At the same time, RAAS outputs are highly context dependent and can be distorted by comorbidities, concomitant medications, and model-specific assumptions. This review therefore synthesizes RAAS signaling in lung cancer within an evidence-based framework, highlights mechanistic links between RAAS-mediated stromal remodeling and immune checkpoint responsiveness, and distinguishes what is currently supported from what remains hypothesis-generating.
To maintain a mechanism-first perspective, this review is anchored primarily in lung cancer—especially non-small cell lung cancer (NSCLC)—while drawing on other pulmonary tumor contexts only when they illuminate shared constraints on vascular abnormalization, desmoplastic remodeling, or immune access. Cross-entity comparisons are therefore used cautiously and interpreted according to evidence provenance, including human tissue data, patient-derived models, and immunocompetent versus xenograft-only systems.7,14,15 From a translational standpoint, RAAS modulation is attractive because angiotensin-converting enzyme (ACE) inhibitors, ARBs, mineralocorticoid receptor antagonists, and related agents are clinically available, pharmacologically defined, and readily testable across mechanistic and clinical study designs. However, systemic blood-pressure modulation does not guarantee intratumoral pharmacodynamic activity, particularly in extracellular-matrix-rich, poorly perfused tumors. Interpretation therefore requires pharmacokinetic/pharmacodynamic realism as well as careful handling of indication bias, residual confounding, and exposure misclassification in observational datasets.16,17
The lung is a biologically important RAAS organ: ACE and ACE2 are expressed in pulmonary tissues, and local angiotensin processing is linked to epithelial homeostasis, vascular tone, and injury responses. Lung cancer cohorts have reported reduced ACE2 expression together with relative predominance of the canonical ACE/Ang II/AT1R axis, a pattern often discussed in relation to vascular abnormalization. Some studies also report higher circulating Ang II in advanced disease, but systemic measurements alone cannot establish tumor-specific causation and are heavily influenced by cardiovascular comorbidities and antihypertensive co-medication. Preclinical studies suggest that restoring ACE2 or augmenting Ang-(1–7)/Mas signaling may suppress vascular endothelial growth factor (VEGF)-associated pathways and mitigate vascular dysfunction in selected models.18–20 Nevertheless, the biological directionality of this axis is likely to depend on compartment and state rather than on transcript abundance alone, and it therefore requires validation in immunocompetent models and primary human tissues.
RAAS reconfiguration has also been linked to therapeutic adaptation.21,22 Resistant sublines have shown increased AT1R/ACE expression and higher VEGF output, whereas ACE2 expansion can reduce angiogenesis and tumor vascularization in vivo. At the TME level, Ang II signaling is associated with cancer-associated fibroblast (CAF) activation, collagen deposition, perfusion deficits, and hypoxia—features that may contribute to immune exclusion and treatment resistance. Retrospective cohorts suggest that concurrent RAAS inhibition may be associated with improved outcomes in NSCLC patients receiving immune checkpoint inhibitors (ICIs), but these associations remain vulnerable to immortal-time bias, residual confounding, and heterogeneous exposure definitions,23–25
These observations sharpen the core translational questions addressed in this review: What is the cell- and space-resolved architecture of RAAS ligand–receptor signaling in human lung tumors? Do standard-dose RAAS inhibitors achieve measurable intratumoral target engagement in extracellular matrix (ECM)-rich, poorly perfused niches? Which tumor and TME biomarkers can identify contexts most likely to benefit from RAAS modulation in combination with immunotherapy? In this framework, lung tumors are treated as RAAS-influenced ecosystems in which ACE/Ang II/AT1R signaling may promote vascular dysfunction, fibrotic remodeling, and immune exclusion, while counter-regulatory modules may restrain or redirect these processes in a context-dependent manner.
The central premise of this review is therefore not that renin–angiotensin–aldosterone system activation is a universal oncogenic driver in lung cancer. Rather, we propose that renin–angiotensin–aldosterone system signaling may become translationally relevant in specific tumor states: extracellular matrix-rich, hypoxic, poorly perfused, and immune-excluded tumors in which stromal mechanics limit treatment access and immune surveillance. This cancer-specific framing places renin–angiotensin–aldosterone system biology within contemporary precision oncology: patient selection should depend on tumor phenotype, spatial immune organization, histologic and genomic context, and demonstrable intratumoral pharmacodynamic engagement rather than on antihypertensive exposure alone.
This is a mechanism-focused narrative review rather than a systematic review or meta-analysis. We searched PubMed/MEDLINE, Web of Science, Scopus, ScienceDirect, and Google Scholar for English-language articles published up to April 2026. Search terms included combinations of “renin–angiotensin–aldosterone system,” “angiotensin II,” “angiotensin II type 1 receptor,” “angiotensin-converting enzyme 2,” “angiotensin-(1–7),” “Mas receptor,” “mineralocorticoid receptor,” “lung cancer,” “non-small cell lung cancer,” “tumor microenvironment,” “cancer-associated fibroblast,” “collagen,” “extracellular matrix,” “immune checkpoint inhibitor,” “programmed death-ligand 1,” “myeloid-derived suppressor cell,” and “radiation pneumonitis.” Lung cancer-focused studies were prioritized. Non-lung cancer, cardiovascular, pulmonary fibrosis, or general RAAS studies were included only when they informed mechanisms relevant to stromal remodeling, vascular normalization, immune exclusion, pharmacologic class effects, or translational trial design. Findings derived from indirect models are described as supportive or hypothesis-generating rather than definitive lung cancer evidence.
A three-layer mechanistic framework for RAAS signaling in lung cancer
System-level architecture: Axis–niche–system framework
The RAAS defies single-scale interpretation in lung cancer and operates simultaneously as a peptide–receptor network, a compartmentalized communication circuit among malignant, stromal, vascular, and immune populations, and an endocrine–paracrine system continuously modified by comorbidities and therapy. Single-cell and spatial atlases have improved resolution over the past several years, but they have also highlighted discordance among bulk-tissue signals, circulating biomarkers, and functional phenotypes.7,14,26
We adopt an axis–niche–system paradigm to turn inconsistent readouts into verifiable hypotheses. The axis layer identifies the major ligand–receptor module, the niche layer the sender–receiver linkages between cellular states and geographic proximity, and the system layer defines host and treatment limitations that affect measurability and druggability. At the structural axis, the RAAS balance determines whether the TME predisposes the cell to vascular aberration, inflammatory escalation, fibrotic rigidity, or moderation. Many tumours exhibit an ACE/Ang II/AT1R-predominant state with reduced ACE2/Ang-(1–7)/Mas signalling,27,28 and this result is associated with harmful fibrotic and inflammatory signals; despite this, biased signalling, compartmentalised peptide processing, and non-canonical enzymatic systems may separate the perceived `axis activation’ of the condition from the possible consequences (Figure 1).
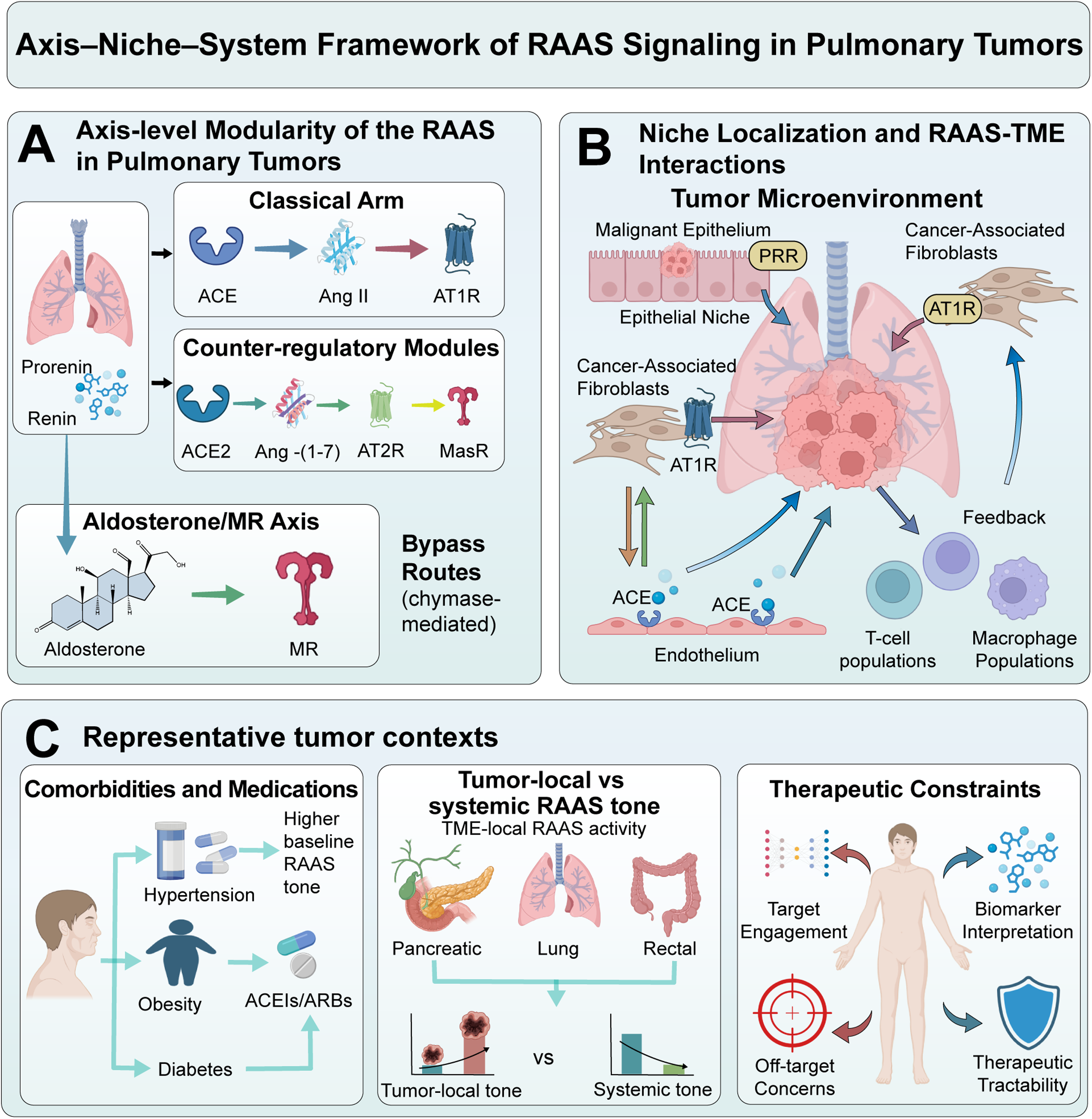
Axis–niche–system framework and modular RAAS architecture in pulmonary tumors (A)Axis-level modularization of the renin–angiotensin–aldosterone system (RAAS), highlighting the classical ACE/Ang II/AT1R arm, counter-regulatory modules (ACE2/Ang-(1–7)/MasR and AT2R), the aldosterone/mineralocorticoid receptor (MR) axis, and representative bypass routes (e.g., chymase-mediated Ang II generation and the (pro)renin receptor). (B) Niche localization of RAAS components across malignant epithelium, cancer-associated fibroblasts (CAFs), endothelium, and immune populations within the tumor microenvironment (TME). Transcript abundance does not equate to peptide availability, receptor occupancy, or signaling flux. (C) System-level context—including comorbidities, concomitant medications, and oncologic therapies—reshapes systemic RAAS tone and tumor-local ligand processing, constraining both biomarker interpretation and therapeutic tractability. Cross-layer coupling and feedback are expected features of RAAS biology. ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; Ang II: angiotensin II; AT1R: angiotensin II type 1 receptor; AT2R: angiotensin II type 2 receptor; CAF: cancer-associated fibroblast; MasR: Mas receptor; MR: mineralocorticoid receptor; PRR: (pro)renin receptor; RAAS: renin-angiotensin-aldosterone system; TME: tumor microenvironment.
Angiotensin II (Ang II) can promote endothelial activation and vascular dysfunction through angiotensin II type 1 receptor (AT1R)-dependent signaling. Clinically, elevated tumor AT1R expression and circulating Ang II have been associated with advanced disease stage and poor outcomes. These associations are difficult to interpret because ACE-independent Ang II formation may alter local ligand availability, whereas cardiovascular comorbidities and antihypertensive co-exposure can obscure observational associations.29,30
RAAS signaling in tumors is compartmentalized and diversified, which limits an ACE-centric interpretation of this system. Proteases such as chymase can generate Ang II independently of ACE and may contribute to matrix remodeling, whereas the (pro)renin receptor can regulate local angiotensin production. These mechanisms may partly explain class-specific differences between ACE inhibitors and ARBs, but they should be treated as candidate effect modifiers until validated in lung cancer-specific cohorts.30,31
The modularisation of the axis alone cannot explain why RAAS inhibitors appear beneficial in some cohorts but not others; niche localisation is the major limitation. Spatial transcriptomics identifies components of the RAAS in specific cellular microenvironments, but transcript abundance does not reliably reflect the availability of secreted peptides, receptor occupancy, or signalling dynamics. As a result, claims about RAAS-specific transcriptomics need peptide/protein anchoring and functional validation. Ultimately, clinical feasibility is dictated by systemic limitations. The endocrine and tissue-local renin–angiotensin–aldosterone system (RAAS) can at times diverge, allowing localized angiotensin activity to act on tumour morphologies whereas systemic levels remain constant. Therefore, circulating Angiotensin II is a poor marker of intratumoral signalling because it primarily reflects systemic physiologic and pharmacologic conditions. In retrospective cohorts of NSCLC, concurrent use of RAAS inhibitors was associated with improved outcomes with ICIs, yet immortal-time bias and poor measurement of dose-timing limit attributions of causality, making mechanism-based prospective studies of NSCLC with specific exposure and biomarker-defined contexts necessary.12,25,32Core modules, dominant niches, state outputs, and interpretability constraints are summarized in Table 1.
RAAS modules in pulmonary tumours: compartments, phenotypes, translational levers, and interpretability constraints.
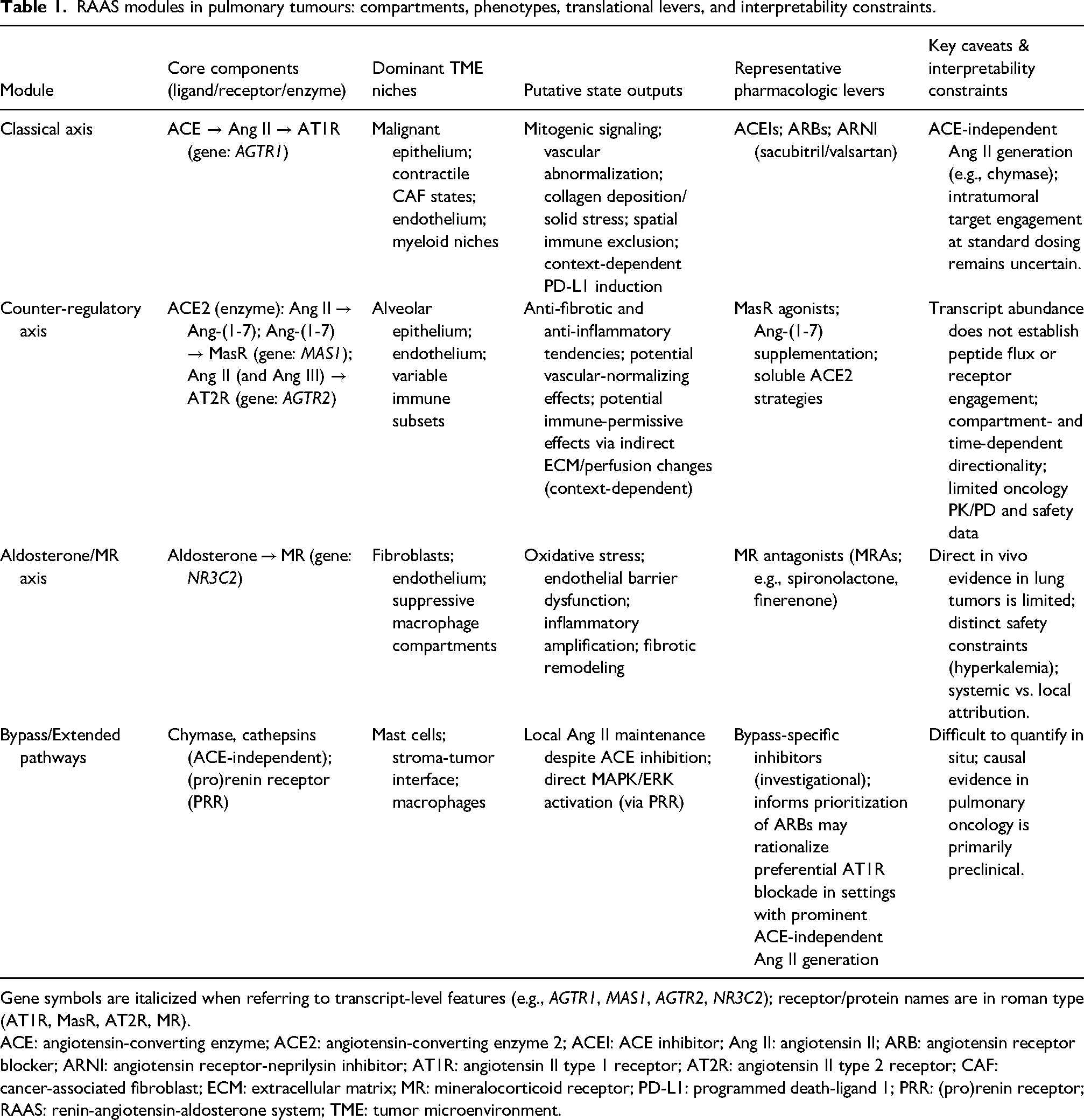
Gene symbols are italicized when referring to transcript-level features (e.g., AGTR1, MAS1, AGTR2, NR3C2); receptor/protein names are in roman type (AT1R, MasR, AT2R, MR).
ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; ACEI: ACE inhibitor; Ang II: angiotensin II; ARB: angiotensin receptor blocker; ARNI: angiotensin receptor-neprilysin inhibitor; AT1R: angiotensin II type 1 receptor; AT2R: angiotensin II type 2 receptor; CAF: cancer-associated fibroblast; ECM: extracellular matrix; MR: mineralocorticoid receptor; PD-L1: programmed death-ligand 1; PRR: (pro)renin receptor; RAAS: renin-angiotensin-aldosterone system; TME: tumor microenvironment.
ACE/Ang II/AT1R: Mapping “pro-tumor programs” of the classical RAAS axis
The canonical ACE → Ang II → AT1R pathway sustains a self-perpetuating, malignant environment by linking tumour-intrinsic signalling to stromal remodelling, vascular irregularities, and immune suppression. AGTR1-enriched states correlate with proliferative pathways, desmoplasia, and diminished effector immune infiltration. Because non-ACE Ang II generation and cellular heterogeneity fracture tumor-homogeneity assumptions, we organize this program map into three interlocked domains stabilized by feed-forward wiring (Figure 2).
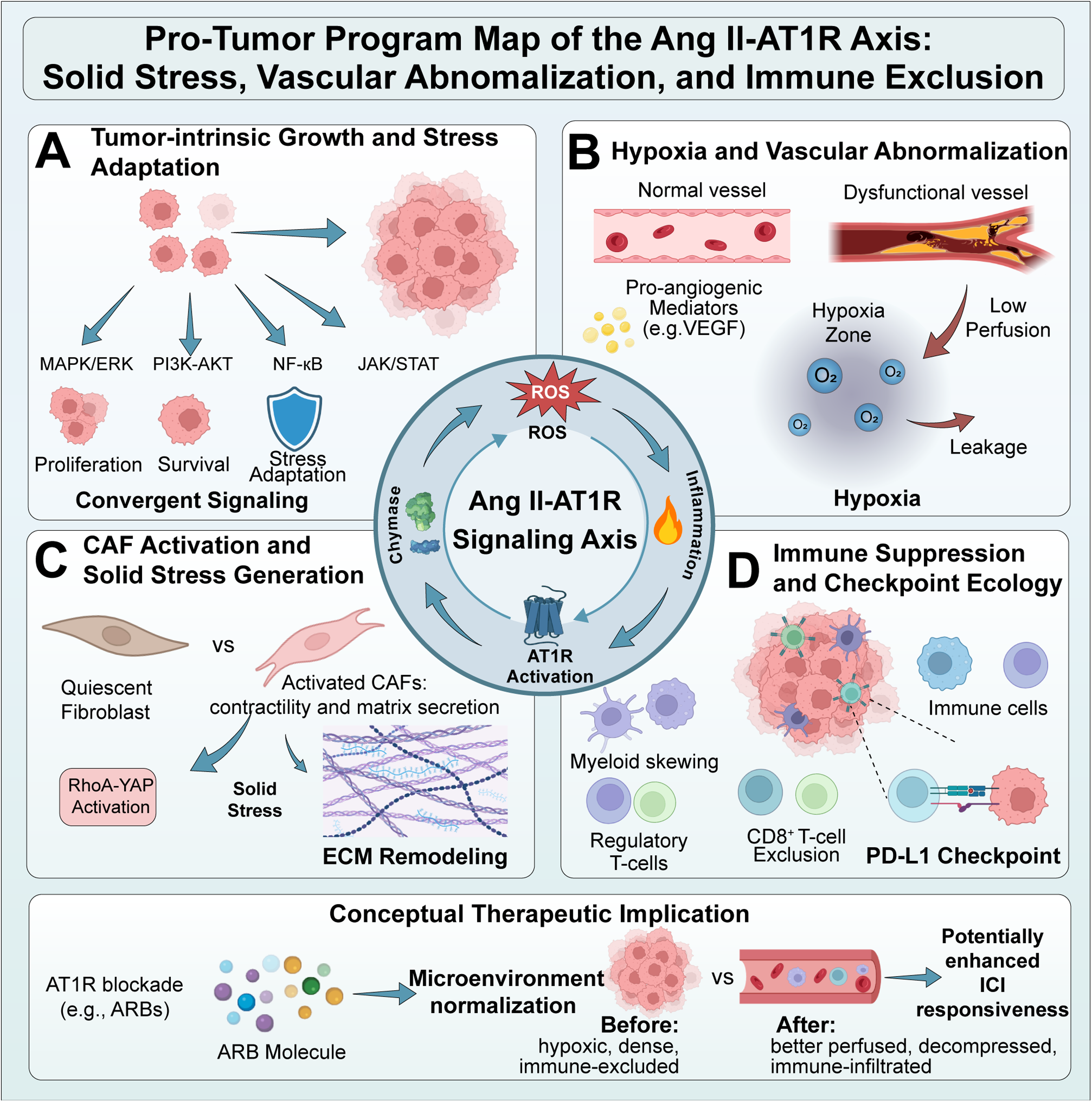
Pro-tumor program map of the Ang II–AT1R axis and its linkage to solid stress, vascular abnormalization, and immune exclusion Conceptual map of coupled domains downstream of Ang II–AT1R signaling in the tumor microenvironment (TME) of pulmonary tumors. (A) Tumor-intrinsic growth and survival amplification through convergent mitogenic and stress-adaptive pathways (e.g., MAPK/ERK, PI3K–AKT, JAK/STAT, NF-κB). (B) Hypoxia and vascular abnormalization, including induction of pro-angiogenic mediators and a dysfunctional, leaky, poorly perfused vasculature that sustains hypoxia and treatment resistance. (C) CAF activation and extracellular matrix (ECM) remodeling, where profibrotic AT1R programs amplify collagen deposition and contractility (e.g., RhoA–YAP–collagen programs), generating solid stress that physically compresses vessels and creates spatial barriers. (D) Immune suppression and checkpoint ecology, including myeloid skewing and CD8+ T-cell exclusion, with context-dependent links to programmed death–ligand 1 (PD-L1) programs. Feed-forward wiring connects hypoxia and inflammation to ACE-independent Ang II generation (e.g., via chymase), reactive oxygen species (ROS), and further AT1R activation. Therapeutic implication (conceptual): Angiotensin receptor blockers (ARBs; e.g., losartan) may act as microenvironment normalizers that reduce solid stress, improve perfusion, mitigate hypoxia, and potentially enhance immune checkpoint inhibitor (ICI) responsiveness, rather than functioning as direct cytotoxics. Ang II: angiotensin II; ARB: angiotensin receptor blocker; AT1R: angiotensin II type 1 receptor; CAF: cancer-associated fibroblast; ECM: extracellular matrix; ICI: immune checkpoint inhibitor; NF-κB: nuclear factor κB; PD-L1: programmed death-ligand 1; PI3K: phosphatidylinositol 3-kinase; ROS: reactive oxygen species; TME: tumor microenvironment.
Tumor-intrinsic coupling of growth and survival signaling
Mechanistically, the classical axis should be interpreted as a ligand–receptor–state program rather than as a nonspecific attachment to familiar oncogenic pathways. Angiotensin II availability in tumors is shaped by ACE-dependent and ACE-independent mechanisms, including chymase-mediated generation in hypoxic or inflammatory niches. Once generated, angiotensin II engages the angiotensin II type 1 receptor on malignant cells, endothelial cells, cancer-associated fibroblasts, and selected immune populations. Receptor activation can converge on mitogen-activated protein kinase/extracellular signal-regulated kinase, phosphoinositide 3-kinase/protein kinase B, Janus kinase/signal transducer and activator of transcription, nuclear factor-kappa B, transforming growth factor-beta/SMAD, and RhoA–YAP signaling, but these pathways should not be treated as equivalent across compartments. In malignant epithelial cells, these signals may support proliferation, survival, epithelial–mesenchymal transition-associated programs, vascular endothelial growth factor production, and matrix metalloproteinase expression. In cancer-associated fibroblasts, angiotensin II type 1 receptor signaling is more directly linked to contractility, collagen synthesis, and extracellular matrix organization. ARB exposure has been suggested to suppress cancer-associated fibroblast collagen programs, potentially through modulation of the RhoA–YAP axis, thereby contributing to a less fibrotic and more immune-permissive tumor state.. These findings support a compartment-specific interpretation: the most therapeutically relevant effect of angiotensin II type 1 receptor blockade may be stromal and immune-access remodeling rather than direct tumor-cell cytotoxicity. 33
Hypoxia–vascular abnormalization: From dysfunctional perfusion to treatment resistance
Ang II induces vascular dysfunction instead of effective angiogenesis, in turn yielding permeable, inadequately perfused networks further contributing to hypoxia. Hypoxia stabilizes hypoxia-inducible factor (HIF) pathways and activates proteases producing ACE-independent Ang II,34,35 while AT1R increases reactive oxygen species (ROS) and inflammatory mediators resulting in endothelial dysfunction;29,30 this feedback cascade reduces radiotherapy oxygenation, limits drug delivery, and inhibits immune trafficking. This wiring supports a conceptual TME-normalization framework in which ARBs may alleviate ECM-associated vascular compression, potentially improving perfusion and enhancing tumor sensitivity to radiotherapy and immune checkpoint inhibitors. Yet, normalization is dependent on dosage, timing, and stromal factors, highlighting the importance of time-resolved sampling with mechanism-proximal assessments.
Immune suppression and checkpoint ecology: Programmed death-ligand 1 (PD-L1) regulation and a T-cell–excluded TME
The link between renin–angiotensin–aldosterone system activity and immune checkpoint inhibitor response should be interpreted through defined immune-oncology mechanisms rather than solely through survival associations. In NSCLC models,12,31,36 angiotensin II signaling via the angiotensin II type 1 receptor has been associated with programmed death-ligand 1 upregulation, supporting a direct link between local angiotensin signaling and the checkpoint ecosystem. 2 In parallel, angiotensin II type 1 receptor activation can promote nuclear factor-kappa B-driven inflammatory transcription, endothelial activation, oxidative stress, and cytokine release, creating conditions that favor suppressive myeloid recruitment and macrophage polarization toward tumor-supportive states. These immune effects interact with stromal mechanics: cancer-associated fibroblast activation and collagen accumulation generate dense extracellular matrix barriers that restrict T-cell trafficking and spatially segregate CD8+ T cells from malignant nests.
This framework provides a mechanistic explanation for retrospective observations linking renin–angiotensin–aldosterone system inhibitor exposure to improved outcomes during immune checkpoint inhibitor therapy, while also explaining why such observations remain insufficient for causal inference. If angiotensin II type 1 receptor blockade enhances immunotherapy, the most plausible mediators are not direct tumor-cell killing but reduced collagen alignment, improved perfusion, reduced hypoxia, altered suppressive myeloid states, and improved CD8+ T-cell access. Prospective studies should therefore measure programmed death-ligand 1 dynamics, macrophage and myeloid-derived suppressor cell composition, CD8+ T-cell exclusion gradients, extracellular matrix topology, and perfusion–hypoxia state before and after treatment. Without these intermediate endpoints, clinical outcome associations remain hypothesis-generating.
Lung cancer heterogeneity and oncogenic context. The relevance of renin–angiotensin–aldosterone system modulation is unlikely to be uniform across NSCLC. Histologic subtype, smoking exposure, oncogenic driver status, stromal architecture, and baseline immune contexture should be considered biological modifiers rather than merely background clinical variables. Lung adenocarcinoma and squamous cell carcinoma differ in stromal composition, vascular organization, smoking association, mutational burden, and immune architecture; therefore, an extracellular matrix-rich, hypoxic, immune-excluded adenocarcinoma may be more plausibly “normalizable” by angiotensin II type 1 receptor blockade than an already inflamed tumor with abundant intratumoral effector T-cell infiltration.
Oncogenic drivers further complicate interpretation. Epidermal growth factor receptor-mutant and ALK-rearranged NSCLC often show lower tumor mutational burden and less favorable responses to immune checkpoint inhibitor monotherapy, making it essential to distinguish targeted-therapy biology from stromal normalization hypotheses. By contrast, KRAS-mutant tumors—particularly those with co-occurring STK11 or KEAP1 alterations—may display hypoxic, metabolically stressed, myeloid-enriched, and immune-cold phenotypes that could theoretically overlap with renin–angiotensin–aldosterone system-driven stromal and vascular constraints. However, these intersections remain insufficiently validated. Future studies should therefore evaluate renin–angiotensin–aldosterone system activity within molecularly annotated cohorts, reporting histology, smoking history, driver alterations, co-mutations, extracellular matrix burden, perfusion–hypoxia status, and spatial immune topology. In this framework, oncogenic context is not a substitute for renin–angiotensin–aldosterone system biomarkers; rather, it defines the biological background in which such biomarkers should be interpreted.
ACE2/Ang-(1–7)/Mas receptor: Tumor-restraint logic and context-dependent liabilities
The ACE2/Ang-(1–7)/Mas receptor pathway can antagonize the classical ACE/Ang II/AT1R axis by reducing Ang II availability and activating Mas receptor-linked signaling. In selected models, this counter-regulatory axis has been associated with anti-proliferative, anti-angiogenic, and anti-fibrotic effects consistent with tumor-suppressive activity. ACE2 expression is usually lower than in normal adjacent lung tissue in patients with pulmonary cancer. ACE2 restoration or Ang-(1–7) delivery has been shown to inhibit tumour growth and vascular anomalies in the orthotopic and xenograft models. The clinical extrapolation process is limited by dynamic remodelling of ACE2 expression by hypoxia and inflammation. This remodelling fails to represent either peptide flux or receptor engagement accurately. Thus, the ACE2/Ang-(1–7)/MasR axis should be analyzed as a probabilistic restriction within the spatial and temporal context of the disease rather than a constant tumour-suppressive factor.
Measuring counter-regulatory activity in situ requires assays that go beyond messenger RNA abundance. Priority approaches include liquid chromatography–tandem mass spectrometry or imaging mass spectrometry for angiotensin II, angiotensin-(1–7), and related peptide ratios; multiplex immunohistochemistry, imaging mass cytometry, co-detection by indexing, multiplexed ion beam imaging, or spatial proteomics to localize ACE2, Mas receptor, angiotensin II type 1 receptor, endothelial markers, fibroblast states, and immune populations within the same tissue architecture; and RNAscope or targeted spatial transcriptomics to support cell-type assignment where protein-level assays are technically limited. Functional anchoring should include ex vivo organotypic tumor-slice or patient-derived model perturbation with angiotensin-(1–7), Mas receptor agonism or antagonism, ACE\2 modulation, and angiotensin II type 1 receptor blockade, coupled to readouts of vascular endothelial growth factor, transforming growth factor-beta/SMAD activity, collagen production, endothelial activation, hypoxia, and immune-cell localization. Such integrated assays would help determine whether the counter-regulatory axis is active, compensatory, suppressed, or potentially maladaptive in a given lung tumor niche.
Functional antagonism of the classical axis and the “ACE/ACE2 ratio” proxy
ACE2 has been shown to alter the flow of RAAS peptides, consequently altering ligand availability from AT1R to MasR. This, in turn, induces a shift in inflammation, fibrosis, and vascular tone. In models of lung cancer, modifications of the ACE2 receptor have been shown to reduce angiotensin II (Ang II)-induced vascular endothelial growth factor (VEGF) formation and angiogenesis-associated characteristics. The ACE/ACE2 ratio acts as a surrogate for RAAS equilibrium within various biological settings. Still, it is important to acknowledge the compartment mixing, therapy-induced alterations in time dynamics, and ACE-independent production of Ang II that may lead to expression ratios independent of AT1R ligand availability in vivo. As a result, compartment-specific estimates must be validated using independent functional data, such as concentrations of Ang II/Ang-(1–7) peptide, and spatial tumour microenvironment parameters (perfusion, ECM density, immunological localization).
Immune remodeling potential: Plausible pathways and lung-specific evidence gaps
The ACE2/Ang-(1–7)/MasR axis can influence immunological trafficking by regulating cytokine networks, endothelial activation, and fibrotic remodelling. In non-pulmonary models, the augmentation of the counter-regulatory renin-angiotensin-aldosterone system (RAAS) has been demonstrated to reduce NF-κB-mediated inflammation, to limit the buildup of suppressive myeloid cells, and to reinstate the permissiveness of effector T-cells.18,19 In the context of pulmonary cancer, the prevailing conceptual framework proposes that augmentation of the ACE2 axis indirectly alleviates immunological exclusion by attenuating the physical barriers associated with hypoxia and fibrosis. 37 However, the direct immune-cell-intrinsic MasR pathways remain to be substantiated. The dependence on immunodeficient xenografts and the absence of spatially detailed human data emphasize substantial deficiencies, underscoring the need for immunocompetent perturbation to substantiate immune remodelling as a valid mechanism. Moreover, the utilisation of ACE2-targeted therapeutics calls for precise infectious disease safety monitoring, given ACE2's role as a receptor for SARS-CoV-2 entry.
AT2R: From presumed anti-proliferative receptor to context-dependent reappraisal
The traditional role of AT2R is to counteract AT1R by facilitating differentiation, apoptosis, and tissue repair. Specific models of lung cancer exhibit growth inhibition upon AT2R activation, thus establishing AT2R as a functional antagonist of AT1R activity. However, forced-expression systems and restricted pharmacologic selectivity have the effect of concealing the physiological significance of these phenomena. Recent evidence indicates that AT2R may have neutral or even pro-tumor effects, likely influenced by the stoichiometry of AT1R/AT2R, cell-type specificity, and state-biased signaling. Determining the directionality of the process necessitates spatial mapping at the protein level, immunocompetent perturbation, and definitive assessments of receptor engagement. Moreover, it is premature to make broad clinical extrapolations until compartmentalised causal data are fully developed.38,39
Aldosterone/MR: An underexplored fibrosis–inflammation–immunity module
The aldosterone/mineralocorticoid receptor (MR) axis has received insufficient attention in the field of pulmonary oncology, despite the body of research in cardiovascular research indicating that MR activation contributes to oxidative stress, endothelial dysfunction, inflammatory enhancement, and fibrotic remodelling. Aldosterone/MR has been demonstrated to mediate ROS production and collagen accumulation in non-neoplastic pulmonary damage via fibroblast activation and TGF-β interaction. Notwithstanding the paucity of lung tumour-specific perturbation data, MR biology provides manageable, compartment-specific hypotheses: Activation of fibroblast mineralocorticoid receptors (MR) may enhance contractility and solid stress, while endothelial MR activity has been shown to exacerbate barrier dysfunction and perfusion heterogeneity. Furthermore, it has been demonstrated that myeloid MR signaling has the capacity to skew macrophage polarization towards immunosuppression. The evaluation of these theories necessitates cell-type–specific MR disruption linked to intermediate goals (e.g., ECM architecture, perfusion metrics, spatial immunological distribution). Consequently, aldosterone/MR should be regarded as a high-priority, testable module rather than an established predictor of tumour growth.
Matrix remodeling and solid stress: RAAS as a bridge from peptide signaling to physical immune exclusion
Solid stress converts extracellular matrix accumulation into a mechanical barrier: proliferative tumor growth and collagen-dense matrices compress blood and lymphatic vessels, restrict perfusion, intensify hypoxia, impair drug delivery, and reinforce immune exclusion. 40 Desmoplastic lung cancers may amplify these constraints, making physical barriers a major contributor to multimodality resistance. Ang II–AT1R signaling in cancer-associated fibroblasts regulates collagen deposition and matrix stiffening, thereby converting peptide signaling into macroscopic stromal barriers. Conversely, AT1R inhibition may alleviate vessel compression and restore perfusion, consistent with a normalization mechanism rather than direct cytotoxicity. 13
CAF-resolved data connects AGTR1 enrichment to ARB-induced disruption of a RhoA–YAP–collagen program, along with enhanced checkpoint responses, so elevating this axis from a biochemical pathway to a physical-oncology mechanism. Translational studies should thus prioritize mechanism-proximal endpoints—collagen architecture (e.g., second-harmonic generation [SHG]), stiffness surrogates (elastography), perfusion/hypoxia metrics, and spatial CD8+ exclusion—and ascertain whether standard antihypertensive dosing and timing yield a consistent human normalization window that facilitates biomarker-enriched prospective testing.12,13,41
Current trends: From epidemiologic controversy to mechanism-guided, biomarker-stratified translation
RAAS-oncology is evolving from unstable incidence connections to concepts that are pertinent to the immunotherapy era. These concepts are both mechanistically analysable and therapeutically stratifiable. This transition is driven by two convergent constraints: the effects of RAAS are determined by the microenvironmental condition rather than consistent class effects, and modern platforms can measure intermediate state endpoints (e.g., ECM architecture/solid stress, perfusion–hypoxia constraints, and immune spatial topology) that enhance causal interpretability and facilitate prospective trial enrichment, instead of depending on post hoc narrative reconciliation.12,42 The delineation of candidate intermediate endpoints and the accompanying diagnostics can be found in Table 2.
Mechanism-aligned intermediate endpoints and companion diagnostics for RAAS-guided translation in pulmonary tumors.
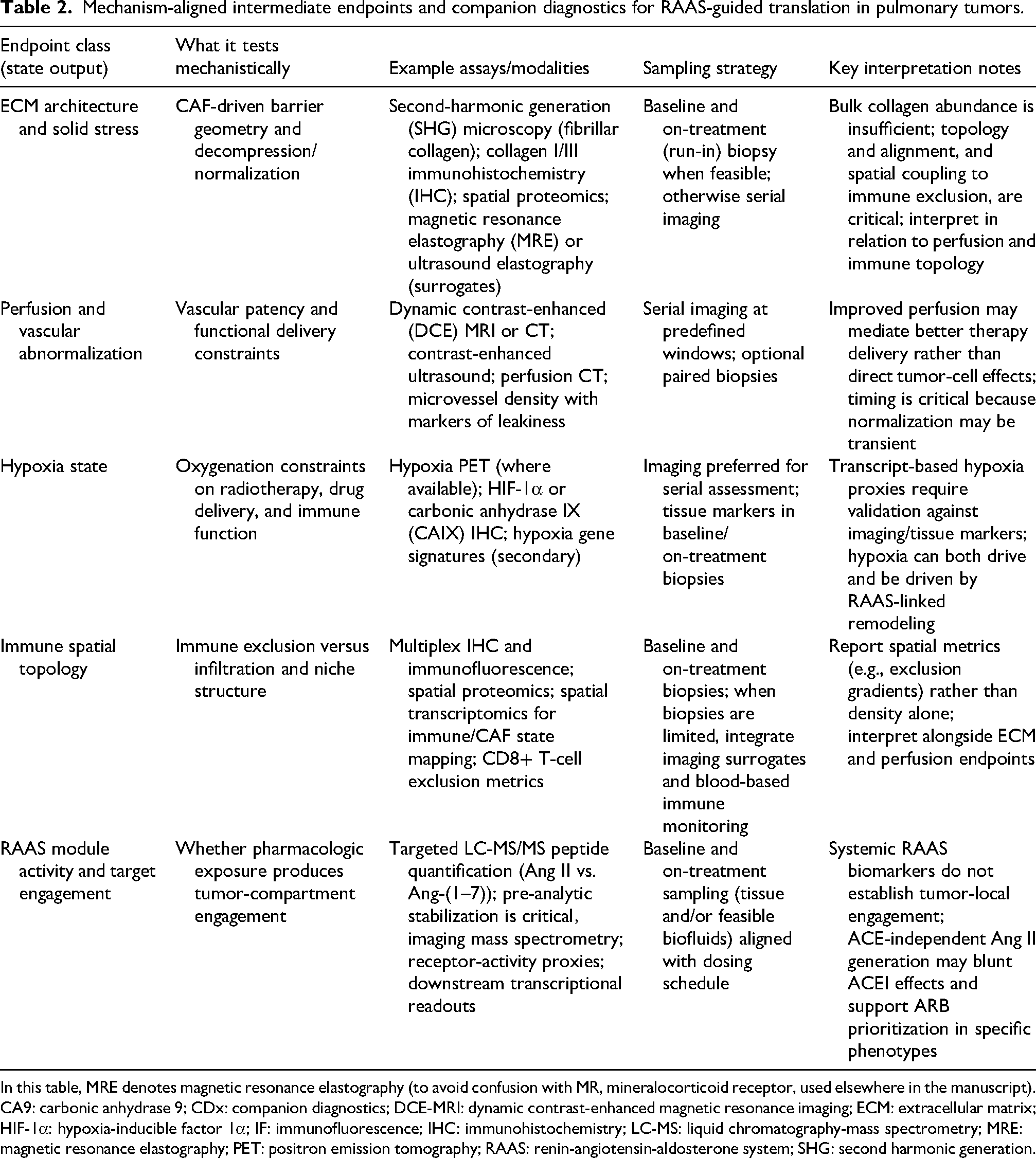
In this table, MRE denotes magnetic resonance elastography (to avoid confusion with MR, mineralocorticoid receptor, used elsewhere in the manuscript).
CA9: carbonic anhydrase 9; CDx: companion diagnostics; DCE-MRI: dynamic contrast-enhanced magnetic resonance imaging; ECM: extracellular matrix; HIF-1α: hypoxia-inducible factor 1α; IF: immunofluorescence; IHC: immunohistochemistry; LC-MS: liquid chromatography-mass spectrometry; MRE: magnetic resonance elastography; PET: positron emission tomography; RAAS: renin-angiotensin-aldosterone system; SHG: second harmonic generation.
RAAS inhibitors and lung cancer incidence: moving from “controversy” to stratified interpretation
The epidemiology links between RAAS inhibitors and lung cancer incidence vary, as estimates are greatly affected by the selectivity of comparator groups (ACEI vs. ARB vs. other antihypertensives), exposure modeling (new-user vs. prevalent-user; time-varying vs. static), and induction and latency assumptions. Studies available on meta-analyses often demonstrate a lower incidence of ARBs, while alternative pooled analyses show a greater incidence with angiotensin-converting enzyme inhibitors (ACEIs). Nonetheless, these are predominantly observational aggregates, which lack meaningful interpretation when relevant confounders are not adequately measured. These confounders may be smoking intensity (pack-years), severity of either chronic obstructive pulmonary disease (COPD) or interstitial lung disease (ILD), socioeconomic status, or healthcare utilization. This makes the comparison of ACEI and ARB especially sensitive to non-exchangeability and contextual prescribing.43,44
A rigorous synthesis should separate distinct sources of heterogeneity and map them to specific bias structures.: active-comparator bias (ACEIs and ARBs are prescribed in different cohorts of clinical severity), protopathic/reverse-causation bias (ACEI-induced cough triggers a switch to ARBs and is associated with airway disease, smoking history, and undetected early lung pathology), latency misclassification (short follow-up leads to detection bias, while longer follow-up compounds cause time-varying confounding as result of switching and discontinuation), and within-class heterogeneity (ARBs have differences in their receptor kinetics, tissue distribution, and extra functionality). Therefore, an approach suitable for incidence-based diagnosis will have to focus on causal inference-tuned approaches to tackle its risk factors. Such approaches must incorporate target-trial emulations with new-user active-comparator initiators, 45 clear induction/latency windows, time-varying exposure models, negative controls, or quantitative bias analyses that consistently assess remaining confounding (specifically, smoking pack-years). Any incidence signal should be contextualized within mechanistically coherent lung/TME phenotypes, rather than as an isolated pharmacoepidemiologic artefact.17,46–50
ARB/RAAS inhibition and immunotherapy: A translational hypothesis from retrospective signals to prospective validation
A major translational hypothesis in the immunotherapy era is that ARBs, and possibly broader RAAS inhibition, may enhance sensitivity to ICIs by modifying stromal architecture and restoring immune accessibility. Retrospective meta-analyses suggest that concurrent RAAS inhibitor exposure may be associated with improved overall survival in immune checkpoint inhibitor–treated cohorts, whereas effects on progression-free survival are less consistent. Within NSCLC, however, these signals remain intrinsically hypothesis-generating because they are highly vulnerable to immortal-time bias, performance-status and comorbidity confounding, and differences in access to care.
Field maturation depends on intervention protocols that pre-specify a TME-normalization rationale, enforce disciplined exposure definitions (molecule, dose, timing, adherence), and embed standardized biomarker acquisition capable of distinguishing tumor-local state change from residual clinical bias.51,52 Mechanistically, CAF-enriched AT1R programs (encoded by AGTR1) can orchestrate collagen deposition and contractility, generating solid stress that constrains perfusion and enforces immune exclusion; 12 AT1R blockade can abrogate these CAF collagen programs, including RhoA–YAP–linked outputs, and enhance checkpoint blockade efficacy in experimental systems. Translation remains uncertain because CAF states are heterogeneous, stromal dominance varies across human lung tumors, and the human “normalization window” likely depends on baseline ECM architecture and demonstrable intratumoral target engagement; prospective studies are therefore most informative when they integrate spatially anchored intermediate endpoints—collagen topology, CAF state programs, perfusion–hypoxia surrogates, and CD8+ T-cell exclusion metrics—and test state change as an explicit mediator within biomarker-stratified analyses.
Angiotensin receptor–neprilysin inhibitor (ARNI) (sacubitril/valsartan) and ICI outcomes: An emerging cardio-oncology interface requiring mechanistic validation
A recently developed cardio-oncology theory argues that ARNI exposure is associated with enhanced ICI outcomes in hypertensive lung cancer cohorts. However, the extant evidence is predominantly derived from single-center, retrospective studies, rendering selection bias and attribution ambiguity inevitable.53,54 The enhancement of heart-failure management and hemodynamic reserve through ARNI can reduce the competing risks of cardiovascular mortality and improve treatment tolerance, consequently extending overall survival without suggesting tumor-local immunomodulation; consequently, ARNI signals should be regarded as explicitly hypothesis-generating until they are prospectively evaluated.54,55 Mechanistically informative studies should predefine dual-domain endpoints (oncologic efficacy and cardiovascular events), utilise competing-risk-aware modelling (e.g., Fine–Gray subdistribution hazards), 56 and integrate tumour-local biomarkers to differentiate cardiovascular stabilisation from genuine transitions in the tumour microenvironment state. 57 In addition to chronic heart failure, the stabilisation of cardiovascular and inflammatory tone through RAAS suppression may provide acute survival gains after severe systemic stress. Preliminary findings show that previous exposure to ARBs is associated with a reduced risk of in-hospital mortality among critically ill patients with solid tumours. This association may be attributable to the ability of ARBs to reduce cytokine storms and prevent multiorgan failure. 58
A definitive evaluation of angiotensin receptor–neprilysin inhibitor exposure in immune checkpoint inhibnon-small cell lung canceritor-treated lung cancer would require a design capable of separating systemic cardiovascular stabilization from tumor-local immunomodulation. A pragmatic framework would be a prospective cardio-oncology study enrolling hypertensive or heart-failure-risk patients with advanced who are initiating immune checkpoint inhibitor-based therapy. Patients could be randomized to sacubitril/valsartan versus an active renin–angiotensin system comparator, such as valsartan, with stratification by baseline heart failure status, blood pressure, renal function, programmed death-ligand 1 expression, histology, and treatment regimen. Cardiovascular endpoints should include blood pressure control, heart-failure events, natriuretic peptides, echocardiographic parameters, immune-related myocarditis, treatment interruption, and non-cancer mortality. Tumor-local endpoints should include paired or early on-treatment assessment of extracellular matrix architecture, perfusion–hypoxia surrogates, myeloid composition, programmed death-ligand 1 expression, and CD8+ T-cell spatial distribution. Statistical analysis should incorporate competing-risk models and mediation analysis to determine whether survival differences are explained by cardiovascular stabilization, improved treatment tolerance, or measurable TME remodeling. Without this dual-domain design, observational angiotensin receptor–neprilysin inhibitor signals should remain hypothesis-generating.
Radiotherapy-related pneumonitis (RP) and RAAS pathways: From normal-tissue protection to combination-enabling translation
Radiation pneumonitis and radiation fibrosis should be distinguished from tumor-local stromal remodeling, even though both may involve renin–angiotensin–aldosterone system-linked inflammatory and fibrotic pathways. In normal lung parenchyma, thoracic irradiation injures epithelial and endothelial compartments, increases oxidative stress, disrupts vascular and alveolar barriers, and promotes the recruitment of inflammatory cells. Angiotensin II type 1 receptor signaling can amplify this injury response through reactive oxygen species, nuclear factor-kappa B activation, transforming growth factor-beta/SMAD signaling, fibroblast activation, myofibroblast differentiation, and collagen deposition. In this context, ACE inhibitors or ARBs may reduce radiation pneumonitis or late fibrosis by attenuating injury-amplifying pathways in non-malignant lung tissue.
In tumor stroma, however, the therapeutic objective is different. Partial suppression of cancer-associated fibroblast contractility and collagen alignment may decompress vessels, improve perfusion, reduce hypoxia, and facilitate immune and drug access. These tumor-local effects could enhance radiotherapy or immunoradiotherapy by improving oxygenation and immune trafficking. Therefore, future trials should not infer tumor reprogramming solely from reduced pneumonitis. They should measure both normal-lung toxicity endpoints and tumor-compartment endpoints, including extracellular matrix topology, perfusion–hypoxia imaging, spatial distribution of CD8+ T cells, and tumor-local transforming growth factor-beta/SMAD activity. This dual assessment is essential to distinguish normal-tissue protection from anticancer microenvironment modulation.
Radiation pneumonitis continues to be a dose-limiting toxicity in thoracic radiotherapy, with retrospective cohorts inconsistently linking ACEI or angiotensin receptor blocker (ARB) exposure to a decreased risk of radiation pneumonitis (RP); however, variability in dose–volume reporting, retrospective grading, confounding factors related to baseline lung function, and co-medication complicate definitive class-specific conclusions. 59 It is evident from the preclinical data that the inflammatory and fibrotic mechanisms associated with RAAS play an important role in the induction of radiation-induced lung injury. However, in the era of immune checkpoint inhibitors (ICI), the most clinically significant perspective is that of combination therapies. The mitigation of toxicity may sustain treatment intensity and continuity, therewith enhancing outcomes through the preservation of dose delivery rather than through direct tumour reprogramming. Prospective studies should pre-specify standardised RP adjudication, include co-primary toxicity and efficacy endpoints, and align normal-lung harm assessments with tumour-compartment state endpoints to differentiate normal-tissue protection from tumour-local regulation.59,60 Table 3 provides a synopsis of the extant clinical evidence landscape, the principal threats to causal inference, and the minimum design principles.
Clinical evidence landscape and minimum design standards for RAAS-pulmonary oncology associations and interventions.
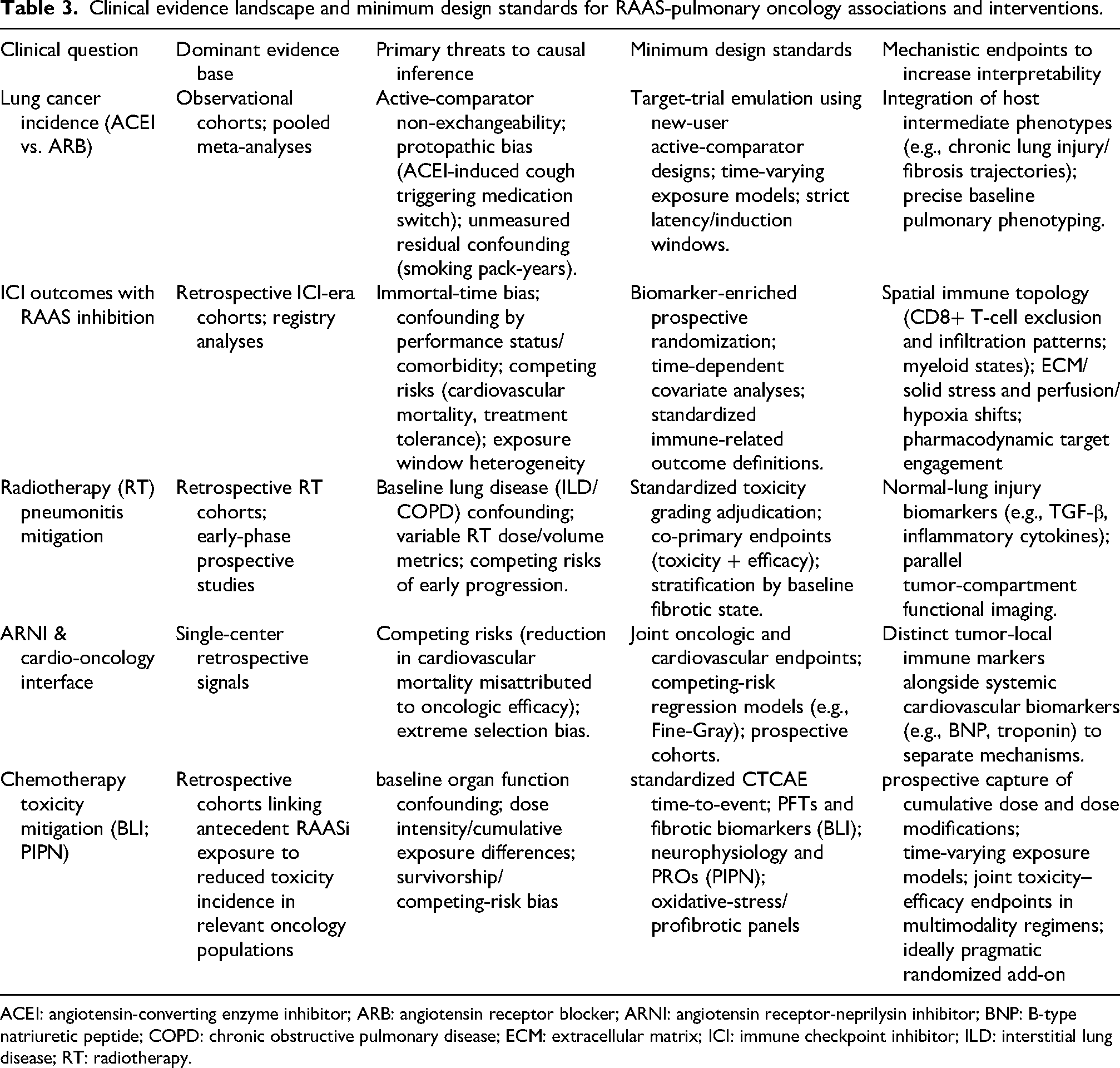
ACEI: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; ARNI: angiotensin receptor-neprilysin inhibitor; BNP: B-type natriuretic peptide; COPD: chronic obstructive pulmonary disease; ECM: extracellular matrix; ICI: immune checkpoint inhibitor; ILD: interstitial lung disease; RT: radiotherapy.
Biomarkers and causal inference: From tissue expression to multi-omics integration and Mendelian randomization
The identification of biomarkers is evolving from descriptive correlations to causal frameworks that link RAAS modules to prognosis and immunological contexture. Comprehensive transcriptomic analyses identify AGTR1 as a potential prognostic marker in LUAD and associate its overall abundance with immune contexture. However, confounding factors related to cell composition may misrepresent bulk AGTR1 as a proxy for stromal content rather than an indicator of receptor functionality. 61 Therefore, validation at the protein level and spatial localisation of AT1R within specific CAF, endothelial and immune niches is essential, preferably in conjunction with functional perturbation in patient-derived models.
Genetic instrumental-variable methodologies, such as Mendelian randomization, have been demonstrated to mitigate confounding and reverse causation in RAAS–cancer correlations. However, pleiotropy,20,62 instrument invalidity, ancestry structure, and the disparity between chronic systemic genetic alterations and transient, tissue-specific pharmacologic interventions limit drug-effect inferences. Genetic evidence should emphasise mechanistic pathways rather than replacing oncology pharmacokinetics/pharmacodynamics and safety data. The categorisation of biomarkers for translation is as follows: The following assays are required: (i) tissue/spatial assays that precisely localise ACE/ACE2/AT1R/MasR/mineralocorticoid-receptor modules to specific niches with standardised quantification; (ii) liquid biomarkers that facilitate longitudinal tracking while predominantly reflecting systemic physiology; and (iii) composite signatures that integrate RAAS balance with CAF–collagen programs, perfusion–hypoxia constraints, and immune spatial topology.37,61,63
Single-cell and spatial omics: Making “cell-type–specific RAAS” actionable
Single-cell and spatial transcriptomic platforms now facilitate compartment-resolved mapping of RAAS components across malignant, stromal, vascular, and immune populations, 64 revealing niche-restricted immune-evasion patterns that bulk profiling obscures. However, it should be noted that transcript abundance cannot determine peptide availability, receptor occupancy, or signalling flux in a peptide-based system. This necessitates mechanistic anchoring through orthogonal layers for example spatial proteomics, in situ peptide quantification, and pharmacodynamic readouts. In light of the paucity of RAAS-specific spatial atlases pertaining to human lung cancers, it is imperative to articulate the evidence gap accurately, thereby staving off potential overinterpretation at the transcript level.7,65
It is imperative that an executable workflow is both modular and validation-oriented. In order to do so, it must compute compartment-aware module scores (classical ACE/Ang II/AT1R versus counter-regulatory ACE2/Ang-(1–7)/Mas, along with aldosterone/mineralocorticoid-receptor signalling) instead of single-gene readouts.7,66 Furthermore, these modules must be co-registered with ECM/solid-stress phenotypes (fibrillar collagen topology; contractility/YAP proxies) and immune spatial phenotypes (CD8+ T-cell exclusion gradients and suppressive myeloid niches) to produce falsifiable predictions aligned with normalization hypotheses. In addition, composite signatures must be confirmed across harmonised multi-center cohorts using pre-registered pipelines and stringent exposure definitions prior to patient selection.42,67 In the absence of spatial mapping and functional peptide anchoring, transcriptome abundance remains correlational rather than actionable (Figure 3).

A Spatial-Omics-Enabled Workflow for Mapping Cell-Type-Specific RAAS and Translating Spatial Signatures Stepwise schematic illustrating how single-cell RNA sequencing and spatial transcriptomics can be operationalized to make cell-type-specific RAAS hypotheses actionable. (1) Compute compartment-aware module scores (classical ACE/Ang II/AT1R versus counter-regulatory ACE2/Ang-(1-7)/MasR, plus MR) rather than single-gene readouts. (2) Co-register RAAS modules with ECM/solid-stress programs (fibrillar collagen topology, fibroblast contractility/YAP proxies) and immune spatial phenotypes (CD8+ T-cell exclusion gradients, suppressive myeloid niches) to generate falsifiable spatial predictions aligned with normalization hypotheses. (3) Bridge the transcript-peptide gap using orthogonal functional anchoring (spatial proteomics, in situ peptide quantification, and pharmacodynamic readouts reporting receptor engagement). (4) Externally validate composite signatures across harmonized multicenter cohorts with rigorous exposure definitions before utilizing them for stratification in umbrella/basket trial designs. Without functional peptide anchoring, transcriptomic abundance remains correlative rather than definitively actionable. ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; Ang II: angiotensin II; AT1R: angiotensin II type 1 receptor; CAF: cancer-associated fibroblast; CD8+ T cell: CD8-positive T cell; ECM: extracellular matrix; IHC: immunohistochemistry; MR: mineralocorticoid receptor; RAAS: renin-angiotensin-aldosterone system; YAP: Yes-associated protein.
A further limitation of transcript-based inference is that receptor function is heavily shaped by post-transcriptional and post-translational regulation. Messenger RNA abundance does not determine receptor isoform composition, translation efficiency, membrane localization, ligand accessibility, receptor internalization, post-translational modification, or biased downstream signaling. In complex remodeling diseases, alternative splicing and interconnected pathway regulation can substantially alter protein function and tissue phenotype despite apparently similar transcript-level signals . By analogy, low or moderate expression of angiotensin II type 1 receptor, Mas receptor, or angiotensin II type 2 receptor in bulk or spatial transcriptomic data should not be interpreted as the absence of functional signaling. Conversely, high transcript abundance does not prove receptor engagement. In renin–angiotensin–aldosterone system biology, transcriptomic maps should therefore be paired with spatial proteomics, peptide-level measurements, receptor localization assays, and pharmacodynamic perturbations.
The need for spatially resolved oncology biomarkers aligns with broader trends in complex systems biology. In neurodevelopmental and neuropsychiatric research, cell-specific expression signatures are increasingly integrated with morphometric and structural imaging phenotypes to link molecular programs with organ-scale architecture and functional outcomes. A similar principle applies to lung cancer: renin–angiotensin–aldosterone system transcript or protein signals become biologically interpretable only when co-registered with extracellular matrix architecture, vessel compression, perfusion–hypoxia state, and immune-cell localization. Thus, spatial mapping should not be treated as a mere descriptive visualization but as a method for connecting cell-specific signaling to tissue-scale constraints on therapeutic response.
Current spatial transcriptomic platforms have significant limitations for the study of renin–angiotensin–aldosterone system biology. Many relevant receptors, including the angiotensin II type 1 receptor and the Mas receptor, are relatively low-abundance G-protein-coupled receptors, and their transcripts may be missed due to capture inefficiency, sequencing depth, tissue quality, spot-level averaging, or single-cell dropout. A negative spatial transcriptomic signal, therefore, cannot be assumed to indicate the absence of receptor protein or the absence of signaling. Conversely, detected transcripts do not establish receptor localization at the membrane, ligand exposure, or pathway activation. These limitations are further amplified in a peptide-driven system, where ligand gradients, enzymatic processing, receptor occupancy, and local pharmacodynamics are not captured by transcriptomics. For this reason, spatial transcriptomics should be integrated with spatial proteomics, multiplex immunostaining, in situ hybridization, targeted mass spectrometry, imaging mass spectrometry, and perturbation-based pharmacodynamic assays before renin–angiotensin–aldosterone system modules are used for patient selection.
Challenges and limitations
The RAAS is increasingly recognised as a multi-scale regulator of pulmonary tumour biology; however, its translation is hindered by three interrelated deficiencies: tenuous causal inference in human datasets, ambiguous intratumoral target engagement at clinically administered doses, and underdeveloped biomarker logic for patient selection. These constraints are not peripheral; they govern the interpretation of existing signals and mandate that future experiments incorporate them as design requirements.6,68
Evidence asymmetry: Pathway-level causality without clinical causality
Preclinical perturbation studies substantiate pathway-level causality: Ang II-AT1R signalling has been demonstrated to regulate proliferative and inflammatory programs, enhance outcomes indicative of vascular abnormalities, and intensify stromal remodelling; 13 ACEI/ARBs have been shown to reduce specific readouts under certain experimental conditions. 12 Translational validity is often compromised when models do not succeed to faithfully reproduce the immune-competent, extracellular matrix-dense, immune-excluded microenvironments characteristic of human lung tumours. 69 This can occur when exposures exceed physiological limits or when endpoints predominantly reflect tumour-cell responses while inadequately sampling niche-level state changes. Mechanistic plausibility provides a logical framework; nevertheless, it cannot replace clinical causality.45,70
The preponderance of human evidence comes from observational cohorts, which are often retrospective. In such studies, confounding factors such as indication bias, time-dependent exposure models, and competing risks caused by comorbidities can engender artificial survival differences. 24 Meta-analyses increase precision, but they cannot address design heterogeneity (exposure windows, smoking and pulmonary comorbidity adjustments, endpoint definitions). Furthermore, the commencement of protocols does not constitute evidence of impact. The clinical credibility of the study will depend on the rigor of allocation, the capturing of adherence, and the predefined mechanistic and biomarker endpoints that link any outcome signal to perceptible changes in the tumour-local state.71,72
Why conclusions conflict: Non-exchangeability and exposure modeling, not “right versus wrong”
It is evident that apparent contradictions, particularly between ACEIs and ARBs, frequently arise from non-interchangeable prescribing populations and inconsistent exposure models, rather than from mutually exclusive biological mechanisms. 73 Symptom-driven switching, for example, from ACEIs to ARBs due to cough induced by ACEIs, may occur in conjunction with airway disease, a smoking history, and healthcare utilisation. This may result in biased comparisons of incidence and outcomes due to detection and selection biases. The necessary covariates to accurately assess these biases (pack-years, COPD severity, interstitial lung disease (ILD) subtype, screening frequency) are often lacking or inadequately measured.74,75
Latency assumptions are a major source of instability in incidence analyses. Carcinogenesis unfolds over extended periods, yet many empirical studies rely on short follow-up windows or static ‘ever-use’ definitions that misclassify induction and latency. Short follow-up can amplify detection bias and reverse causation, whereas longer follow-up can accumulate time-varying confounding through switching, discontinuation, and competing mortality. Progress therefore depends on target-trial emulation principles, including new-user active-comparator designs, explicit time zero definition, time-varying exposure modeling, and sensitivity analyses that systematically assess residual confounding. In addition, ACEIs and ARBs should not be treated as homogeneous classes, because they differ in tissue distribution, receptor kinetics, and bradykinin-related biology.
Directionality in a bi-armed system: ACE2 and AT2R are conditional, not universal restraints
RAAS is inherently bi-armed, and therefore directionality should vary according to cell type, niche state, disease stage, and treatment setting. The ACE2/Ang-(1–7)/Mas pathway is frequently characterised as counter-regulatory; however, ACE2 expression in the lung is dynamic and compartmentalised, and transcript-level measurements (e.g., ACE2 mRNA) cannot determine peptide flux, receptor occupancy, or signalling throughput in vivo.18,76–78 In the absence of cell- and compartment-specific evidence, including direct or surrogate assessments of intratumoral Ang II versus Ang-(1–7) activity, assertions of a consistently “protective” axis are conjectural.
AT2R can be demonstrated to display amplified context dependence. Cancer models, which are conventionally linked to anti-proliferative and anti-inflammatory initiatives, have the potential to yield neutral or pro-tumor outcomes when there are alterations in AT1R/AT2R stoichiometry, biased signalling, cell-type localization (such as endothelium, fibroblasts, and tumour epithelium), and ligand microgradients. These are characteristics that are typically overlooked by bulk profiling methods. Translation requires a stratified evidence framework, incorporating spatial protein mapping of receptor location, association with niche-specific downstream state programs, and causal perturbation in immunocompetent or patient-derived systems. 79 In the absence of this stack, clinical extrapolation towards ACE2- or AT2R-targeted therapies is premature.
Pharmacologic reality: Antihypertensive dosing does not guarantee intratumoral target engagement
A prevalent translational failure mode pertains to the assumption that conventional antihypertensive dosages provide adequate intratumoral exposure to replicate preclinical effects. Numerous in vitro studies utilise micromolar drug concentrations, while clinically attainable unbound plasma concentrations (Cu, calculated from total exposure and the unbound fraction, fu) at standard dosing are generally in the nanomolar range. This pharmacokinetic discrepancy is likely intensified in extracellular matrix-dense, poorly perfused areas where delivery is severely limited. It is imperative that translation transitions from “exposure documented” to “engagement demonstrated”. Clinical dosing necessitates tumour-compartment pharmacodynamic assessments that confirm significant AT1R-pathway engagement (or its alleviation) in the niches believed to confer benefit.80,81
The concept of mechanism attribution is predicated on this inherent limitation. RAAS inhibitors are improbable to act as direct cytotoxics; their most credible role in oncology is the modulation of the microenvironment—diminishing solid stress, enhancing perfusion, alleviating hypoxia, and augmenting immune accessibility—thereby sensitizing tumors to immune checkpoint inhibitors, chemotherapy, or radiotherapy. In the context of a hypothesis concerning normalisation, intermediary endpoints (ECM architecture, perfusion–hypoxia measures, spatial CD8+ T-cell exclusion, myeloid states) assume a central role. These endpoints function as the evidentiary link that facilitates a mechanistic interpretation when size-based responses are minimal. Normalization should be considered a transient and schedule-dependent phenomenon. This requires the implementation of time-resolved sampling and biomarker-enriched methodologies, as opposed to indiscriminate continuous-exposure assessments. In essence, the tumour-localised renin-angiotensin-aldosterone system (RAAS) has the capacity to become decoupled from systemic regulation through the production of angiotensin II (Ang II) that is not dependent on the ACE. This process can be facilitated by the action of chymase, a specific enzyme found in the tumour microenvironment. The result of this dissociation is the subversion of upstream inhibitory mechanisms, thereby increasing the dependence on tumour-localised analyses rather than relying on indicators present in the circulation.82,83
Safety and drug–therapy interactions in multimodality regimens
Cardiovascular medicine defines the safety profiles of RAAS-targeting drugs; however, oncology care creates a distinct physiological context in which dehydration, nephrotoxic chemotherapy, infection, and rapid nutritional decline can exacerbate hypotension, renal dysfunction, and hyperkalemia—particularly with MR antagonists or pre-existing renal impairment. During the ICI era, immune-related myocarditis, arrhythmias, and vascular inflammatory events may further reduce hemodynamic reserve. Concurrent RAAS modulation can also influence renal perfusion and blood-pressure trajectories, making toxicity attribution more complex. Prospective studies should therefore predefine monitoring frequency, laboratory surveillance, and stopping criteria in combination ICI/chemotherapy/radiotherapy protocols.
The establishment of a concurrent relationship among safety and efficacy is a key element of thoracic radiation. In certain circumstances, if RAAS modulation mitigates radiation pneumonitis in selected contexts, it may preserve dose intensity and continuity, thereby guaranteeing the maintenance of dose intensity and continuity. This approach is predicated on the premise that it enhances treatment results through the augmentation of delivery effects, as opposed to the reprogramming of tumours. The effective utilisation of this concept necessitates future evaluation through the implementation of standardised assessments and mechanistic outputs. Such evaluations must be capable of distinguishing between normal lung protection and tumour-specific regulation.59,82,84
Overall, the field is limited less by a lack of hypotheses than by the discipline required for translation. Priority gaps include direct or surrogate measurement of intratumoral Ang II/Ang-(1–7) activity, evidence of tumor-compartment target engagement at clinically viable dosing, validated biomarkers that identify ECM-dense and immune-excluded but potentially ‘normalizable’ tumors, mechanistic clarification of ACEI versus ARB heterogeneity, and safety frameworks for RAAS modulation during multimodality treatment.
Future perspectives: An executable roadmap for RAAS-guided pulmonary oncology
RAAS in lung malignancies functions more as a compartmentalised, multi-scale regulator of microenvironmental conditions than as a linear endocrine cascade. Therapeutic leverage should thus be pursued in condition changes, rather than in direct tumor-cell toxicity, by restructuring ECM/solid stress, perfusion–hypoxia limitations, and immune spatial topology. 12 Mechanistic evidence increasingly supports the hypothesis that RAAS-directed stromal mechanics and immunological access are critical factors influencing treatment sensitivity. However, clinical conclusions mainly rely on diverse retrospective relationships. The generation of an executable roadmap is predicated on the generation of three distinct outputs. Firstly, causal models must be produced, defining the operative unit and identifying mechanism-proximal intermediate endpoints. Secondly, biomarker-stratified translation must be employed, replacing empirical co-medication. Thirdly, pharmacologic strategies must be designed for demonstrable intratumoral target engagement in poorly perfused, ECM-dense lesions without intolerable systemic liability. 13
Mechanistic research: Upgrading “pathway diagrams” to a cell–space–system triad
Taken together, the available evidence supports a conditional RAAS–stromal mechanics–immune exclusion model rather than a universal oncogenic model. In this model, tumor-local Ang II–AT1R signaling is most relevant when it occurs within fibroblast-rich and collagen-dense niches. AT1R activation in cancer-associated fibroblasts may enhance RhoA–YAP-associated contractility and type I collagen production, thereby increasing extracellular matrix stiffness and solid stress. These physical changes can compress tumor vessels, reduce perfusion, intensify hypoxia, and spatially exclude CD8+ T cells from malignant epithelial nests. ARB-mediated AT1R blockade would therefore be expected to improve immune checkpoint inhibitor response only in tumors with demonstrable stromal and immune-exclusion phenotypes, and only if intratumoral pharmacodynamic engagement is achieved. This model connects molecular RAAS signaling to tissue mechanics, immune topology, and therapeutic response, while also explaining why unselected retrospective co-medication studies generate inconsistent results.
Thus, a T-cell-excluded environment that diminishes ICI effectiveness is established. The enrichment of CAF-compartment AGTR1 and the ARB-induced inhibition of a RhoA–YAP–collagen I pathway, along with enhanced checkpoint responsiveness, establish a plausible connection over scales that links signalling to ECM mechanics, immunological topology, and response. The main limitation is attributable to the heterogeneity of CAFs: the causative unit is improbable to be CAF abundance and is more likely a collagen-rich, contractile CAF state program geographically associated with CD8 + exclusion gradients.12,85
The concept of executability is predicated on the principle of falsifiability. Disruption of the AT1R, constrained by CAF, or Yap-associated collagen pathways, ought to alter collagen structure towards decompression and significantly reduce spatial exclusion of CD8 + cells. Exposure to ARBs ought to replicate these niche-level effects solely when demonstrable intratumoral pharmacodynamic engagement occurs. Furthermore, if retrospective outcome indicators predominantly reflect systemic confounding, then on-treatment ECM and immune-spatial endpoints should not shift in a mechanism-consistent manner. 86 These predictions require a cell–space–system framework comprising three elements: first, CAF-subtype-specific perturbation in patient-derived systems and immunocompetent models to distinguish fibroblast-intrinsic from tumor-intrinsic effects; second, orthogonal spatial quantification of barrier geometry and immune localization, including second-harmonic generation for fibrillar collagen and multiplex immunohistochemistry or spatial proteomics for CD8+ T cells and tumor-associated macrophages; and third, system-level assessment of ligand sources and peptide flux using direct peptide quantification, such as liquid chromatography–mass spectrometry or imaging mass spectrometry.
A complementary model proposes that compartment-specific activation of ACE2/Ang-(1–7)/Mas can shift peptide balance away from Ang II–AT1R signaling and promote anti-angiogenic and anti-fibrotic states. Genetic prioritization strategies such as Mendelian randomization support continued interest in the ACE2 axis, but they cannot substitute for pharmacologic validation in tumor-specific contexts. Because ACE2/Mas effects are likely to differ across tumor epithelium, endothelium, fibroblast states, and immune compartments, translation should emphasize compartment-resolved perturbation and niche-level intermediate endpoints rather than expression alone.
Biomarker-driven trial design: Transitioning from empiric RAAS modulation to precision stratification
If microenvironment normalization mediates benefit, RAAS-targeted strategies should be evaluated in biomarker-enriched rather than unselected populations. Unselected exposure-based designs risk diluting benefit across biologically heterogeneous cohorts and obscuring tumor-local mechanisms. A minimally viable enrichment gate would prioritize ECM/CAF–collagen–rich tumors together with an immune-excluded topology, while RAAS-related measurements—such as compartment-specific ACE/ACE2 readouts, stromal AGTR1 enrichment, or module-based RAAS scores—should initially function as pre-specified effect modifiers unless they are prospectively validated as inclusion criteria.
These layers inherently facilitate umbrella and basket architectures. An NSCLC umbrella trial has the capacity to randomise an ARB run-in against a control within a predefined ECM/solid-stress–high, immune-excluded stratum. Furthermore, it is possible to employ standardised on-treatment sampling to detect state changes, even when Response Evaluation Criteria in Solid Tumors (RECIST) responses are modest.87,88 A basket design encompassing pulmonary cancers and selectively chosen lung metastases can utilise the same ECM/solid-stress gate to evaluate if normalisation correlates with phenotype instead of histology. In both instances, a status characterised by elevated collagen and high solid stress should function as an enrolment criterion rather than a post hoc subgroup. This assertion is substantiated by scalable assays such as SHG-derived collagen metrics, elastography-based stiffness surrogates, and/or validated radiomics proxies. When serial biopsies are impractical, functional imaging (e.g., dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) / magnetic resonance elastography (MRE)) should act as pragmatic surrogate normalization endpoints.
It is imperative that endpoints are aligned with normalization as opposed to cytotoxicity. Overall survival (OS) and progression-free survival (PFS) remain definitive; however, mechanism-proximal intermediate endpoints should be incorporated as essential secondary or co-primary outcomes to ascertain target engagement and facilitate early go/no-go determinations. Mechanism-proximal endpoints should include ECM remodeling, assessed by collagen density and topology; perfusion–hypoxia transitions, assessed by imaging or validated hypoxia markers; immune redistribution, assessed by multiplex immunohistochemistry of CD8 + exclusion gradients and myeloid-state composition; and adaptive immune activation, assessed by T-cell receptor dynamics and interferon-γ-associated pathways. The assimilation of these readouts as companion diagnostics is not a superficial undertaking; causal mediation analysis necessitates quantifiable intermediate state shifts in order to ascertain whether reprogramming can feasibly mediate clinical benefit. 89
A practical biomarker-driven trial should begin with phenotype enrichment rather than unselected antihypertensive exposure. Candidate inclusion features could include high extracellular matrix burden, collagen alignment or stiffness, cancer-associated fibroblast enrichment, hypoxia or perfusion defects, and spatial exclusion of CD8+ T cells. These features could be assessed using second-harmonic generation microscopy, multiplex immunohistochemistry, spatial proteomics, elastography, radiomics, dynamic contrast-enhanced imaging, hypoxia imaging, or validated composite signatures. Renin–angiotensin–aldosterone system markers, including stromal angiotensin II type 1 receptor expression, compartment-specific ACE/ACE2 balance, Mas receptor localization, or peptide ratios, should initially serve as prespecified effect modifiers until prospectively validated.
A feasible design would be a randomized window-of-opportunity or run-in study in extracellular matrix-rich, immune-excluded NSCLC before immune checkpoint inhibitor-based therapy. Patients could receive an angiotensin receptor blocker or a control intervention for a predefined interval, followed by repeat tissue or imaging assessment. Go/no-go criteria should include evidence of intratumoral target engagement, reduced cancer-associated fibroblast contractility markers, decreased collagen density or alignment, improved perfusion or reduced hypoxia, altered suppressive myeloid composition, and reduced CD8+ T-cell exclusion. Clinical endpoints such as objective response, progression-free survival, and overall survival remain essential. Still, mechanistic intermediate endpoints are required to determine whether any clinical benefit is mediated by TME normalization.
Pharmacologic innovation: Beyond ACEI/ARB repurposing toward axis engineering and intratumoral target engagement
Repurposing ACEIs and ARBs offers immediate clinical access, but durable progress requires a strategy that addresses RAAS directionality and the pharmacokinetic/pharmacodynamic bottleneck imposed by collagen-rich, poorly perfused tumors. In this context, classical-axis suppression should be treated as microenvironmental modulation whose efficacy depends on local Ang II generation pathways and the presence of ECM/immune phenotypes compatible with ‘normalizable’ states. Counter-regulatory approaches—including Mas agonism, ACE2/Ang-(1–7)-oriented strategies, and AT2R agonism where mechanistically justified—should be advanced only through phased translation grounded in compartment-resolved pharmacodynamic endpoints rather than transcript-level assumptions. Aldosterone/MR modulation remains a testable module for fibrosis- and oxidative-stress-dominant contexts, provided that tumor-specific effects are separated from systemic physiology.
It has been established that the most “normalizable” tumours are frequently the least perfused. Therefore, delivery-enabled target engagement should be regarded as an enabling technique. The evaluation of local or niche-biased delivery, inclusive of pulmonary-local methods when pertinent, should be informed through its potential to augment intratumoral exposure and pharmacodynamic interaction, whilst concomitantly minimising systemic adverse effects (e.g., hypotension, renal impairment, and hyperkalemia). It is imperative that initial studies explicitly measure exposure and engagement, as opposed to solely relying on clinical outcomes, in order to avert the primary translational failure mode in RAAS-oncology: credible biology without verified intratumoral engagement (see Figure 4).
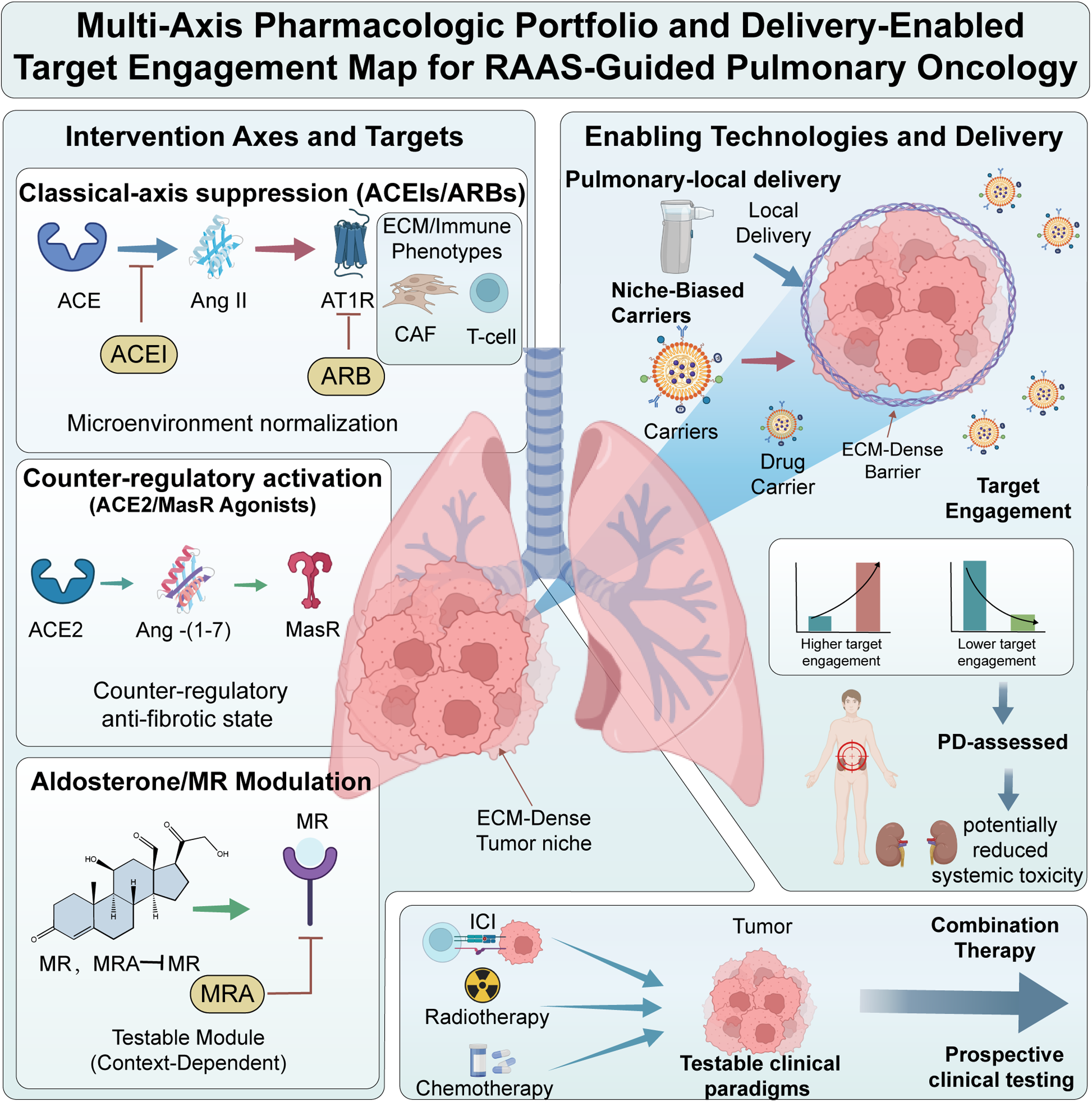
Multi-axis pharmacologic portfolio and delivery-enabled target engagement map for RAAS-guided pulmonary oncology Schematic integrating three intervention families across key tumor compartments and measurable state outputs. (1) Classical-axis suppression (ACE inhibitors and ARBs) is positioned as microenvironmental modulation whose efficacy likely depends on local Ang II generation routes and ECM/immune phenotype enrichment. (2) Counter-regulatory activation (e.g., MasR agonism, ACE2/Ang-(1-7) strategies) is depicted as an orthogonal approach to impose anti-fibrotic and anti-angiogenic states. (3) Aldosterone/MR modulation (MR antagonists) is highlighted as an underexplored, testable module with explicit safety constraints. Delivery-enabled approaches (e.g., niche-biased carriers or pulmonary-local strategies) are included as enabling technologies to achieve intratumoral exposure and pharmacodynamic engagement in ECM-dense, poorly perfused tumors while reducing systemic liabilities (e.g., hypotension, hyperkalemia, renal dysfunction). Combination nodes map clinically testable sensitization paradigms with immune checkpoint inhibitors, radiotherapy, and chemotherapy. ACEI: angiotensin-converting enzyme inhibitor; Ang II: angiotensin II; ARB: angiotensin receptor blocker; CAF: cancer-associated fibroblast; ECM: extracellular matrix; ICI: immune checkpoint inhibitor; MasR: Mas receptor; MR: mineralocorticoid receptor; PD: pharmacodynamic; PK: pharmacokinetic; RAAS: renin-angiotensin-aldosterone system; ACE2: angiotensin-converting enzyme 2.
Integrating RAAS modulation into contemporary lung cancer paradigms: Sensitization, toxicity mitigation, and cardio-oncology co-management
RAAS translation becomes more actionable when integrated into current lung-cancer treatment frameworks rather than treated as an isolated pharmacologic category. The most relevant interfaces are ICI sensitization through microenvironment normalization, radiotherapy enablement via toxicity reduction, chemotherapy-toxicity mitigation, and selected cardio-oncology settings in which competing-risk effects must be distinguished from true tumor-local remodeling.
In order to facilitate the easing of the extracellular matrix (ECM) barriers in ECM-dense, immune-excluded tumours, it is essential to evaluate ARB-mediated AT1R inhibition as an immune checkpoint inhibitor (ICI) sensitiser. This evaluation should incorporate predefined normalization windows and spatial immune-topology objectives in the study design. RAAS modulation should be assessed as a radiation enablement technique using co-primary or hierarchical objectives that measure both pneumonitis risk and tumour control. This should be underpinned by molecular markers that differentiate normal-tissue protection from tumour-local reprogramming. Cardio-oncology signals, including ARNI hypotheses, must be examined through combined endpoints that encompass tumour outcomes as well as cardiovascular and immune-related events. These should be accompanied by mechanistic correlates that distinguish cardiovascular stabilisation from tumour-local immune or vascular remodelling. This will help to avoid misinterpretation of competing-risk effects as anticancer mechanisms. In addition to radiotherapy, RAAS modulation has been shown to be a mechanistically sound strategy for alleviating particular toxicities generated by chemotherapy. Pre-existing RAAS inhibitor medication has been shown to be independently correlated with a markedly decreased occurrence of bleomycin-induced lung damage (BLI) in bleomycin-exposed populations and paclitaxel-induced peripheral neuropathy (PIPN) in lung cancer populations. These clinical data are consistent with the capacity of RAAS inhibition to counteract therapy-induced oxidative stress and profibrotic pathways.90,91
Conclusion
RAAS signaling in lung cancer is best viewed as a compartmentalized regulatory layer that may couple peptide signaling to stromal remodeling, vascular dysfunction, and immune accessibility. The most coherent translational model links Ang II–AT1R activity, particularly within fibroblast-rich niches, to collagen accumulation, solid stress, hypoxia, and CD8+ T-cell exclusion, thereby providing a rationale for testing ARBs as microenvironment-normalizing adjuncts rather than direct anticancer agents. However, ACE2/Ang-(1–7)/Mas, AT2R, and mineralocorticoid receptor signaling remain context dependent, and current clinical evidence is dominated by retrospective co-medication analyses with substantial susceptibility to confounding. Progress will depend on cell- and spatially resolved analyses of RAAS activity in human lung cancer, direct assessment of intratumoral pharmacodynamic engagement, and biomarker-stratified prospective studies that link clinical outcomes to measurable changes in matrix architecture, perfusion, hypoxia, and immune topology. In this framework, RAAS-directed therapy should be considered a testable microenvironmental strategy rather than an established therapeutic principle in lung cancer.
Footnotes
Abbreviation
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (82403804) and Shanghai Municipality’s 2025 High-Quality Development Plan for Science and Technology Industries: “Application Demonstration of Innovative Pharmaceutical and Medical Device Products" (25SF1907403).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
This article is a review and does not report new primary datasets.