
Abstract
Familial hypercholesterolaemia (FH) presents a significant risk for early cardiovascular morbidity and mortality. Several new international guidelines on FH have been published in the last year and similarities and differences between them are described and summarised in this review. New European guidelines discuss other familial dyslipidaemias as well as FH. These guidelines should improve the quality and cost effectiveness of care provided.
Abbreviations and acronyms
ApoB apolipoprotein B
CABG coronary artery bypass grafting
CVD cardiovascular disease
FCH familial combined hyperlipidaemia
FH familial hypercholesterolaemia
HDL high-density lipoprotein
LDL low-density lipoprotein
LDLR low-density lipoprotein receptor
NICE National Institute for Health and Clinical Excellence
PCSK9 proprotein convertase subtilisin/kexin type 9
TC total cholesterol
Introduction
This article compares differences and similarities in the diagnosis and management of familial lipid disorders from several recently published guidelines on familial hyperlipidaemias. The articles reviewed are:
Identification and management of familial hypercholesterolaemia (UK, 2008). 1
Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia (USA, 2011). 2
Familial hypercholesterolaemia: a model of care for Australasia, 2011. 3
European Societies Guidelines for the management of dyslipidaemias, 2011. 4
Consensus on the early management of familial hypercholesterolaemia in Belgium and Luxembourg, 2011. 5
The European guidelines cover several familial dyslipidaemias in addition to FH. 4
Clinical diagnosis
On routine examination and testing, the possibility of FH should be suspected where any of the following are present:
TC > 7.5 mmol/L. 1
Paediatric LDL cholesterol cut-off values vary from > 4.0mmol/L 3 to a lower threshold of > 3.5mmol/L 5 which includes 95% of children with FH, but with more false positives.
Corneal arcus below age 45 years, tendon xanthomata or xanthelasmata.1,2,5
A family history of ischaemic heart disease below age 55 years in males and below 65 years in females.2,5 (However, European guidelines suggest 50 and 60 years respectively. 4 )
Serum lipid profiling should include total, LDL and HDL cholesterol and total triglycerides. The use of non-HDL cholesterol for diagnostic purposes is sporadic. 2 General consensus indicates that two raised fasting lipid profiles are required for a diagnosis of hyperlipidaemia in adults, while for children three raised results are required to confirm the diagnosis with an optimised diet for 2–3 months before the third test.1,2,5 Care must be taken to rule out secondary causes. A clinical diagnosis of homozygous FH is possible where LDL cholesterol > 13 mmol/L (> 11 mmol/L in children). 1 Where potential cases of FH are identified an extensive family history must be obtained (ideally a three-generation pedigree) with particular attention given to relatives with significant vascular incidents, the age of onset of events, cardiovascular risk factors and any formal FH diagnoses.1,4
Use of validated diagnostic criteria for the formal diagnosis of FH is essential. While several internationally recognised criteria are available there are national variations such as the use of the Simon Broome Criteria in the UK (figure 1).1,2 The Dutch Lipid Clinic Network criteria are useful in the diagnosis of paediatric FH due to consideration of age specific LDL levels. 5

Simon Broome criteria as recommended by NICE. 1
Screening and genetic testing
Selective screening using clinical criteria and lipid or genetic testing should be initiated when phenotypic characteristics as previously described are identified, or where a biological relative has been diagnosed with FH or exhibits similar characteristics.1,2,4,5
This is important in paediatric diagnosis, where a family history of FH should prompt the screening process. The earlier the diagnosis and initiation of primary prevention, the more favourable is the outcome for that individual. 5 Opinion as to the optimum time to implement this screening in children ranges from approximately 2 to 10 years of age.1,2 This age range avoids the rapid increase in plasma cholesterol occurring in the weeks after birth and slow increase continuing until approximately 2 years of age, and the pre-pubertal decrease in LDL cholesterol (figure 2).4,5,7-9
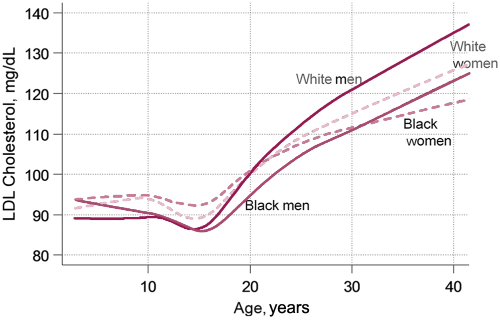
Age specific low-density lipoprotein (LDL) cholesterol levels according to gender and ethnicity.
Genetic analysis should identify the three most commonly defective genes for LDLR, ApoB and PCSK9. It is stressed that a negative result on genetic testing in the face of significant phenotypic findings does not rule out FH.1-4 Many causal gene variations remain unidentified and more extensive genetic investigations, while possible, are unlikely to be cost effective or of benefit to the patient.3,4 The decision to utilise genetic testing is one that should be routinely offered to patients with a phenotypic FH diagnosis with a careful explanation of the potential impact of the information, including the implications for cascade screening (see below). However, identification of a pathogenic variant is usually not a diagnostic requirement unless lipid profiling is inconclusive. 2 Some guidelines suggest deferring genetic investigations until a child reaches adulthood if there is not an already identified familial trait in a first degree relative. 3
Cascade screening is internationally regarded as a cost-effective means of identifying individuals with FH, initially through contacting first degree relatives of an index case.1-4 However, care must be taken to obtain consent from the index patient in a sensitive manner with awareness of the ethical and legal implications should this be declined. As new cases of FH are identified, so the cascade process should be repeated. Use of the Simon Broome criteria for the process of diagnosing relatives is not recommended according to NICE guidelines, due to the potential for under diagnosis. 1 A recent UK update has advised against the use of some methods to diagnose both an index case of FH and when performing cascade screening. The update recommends comprehensive genetic analysis in the index case, followed by targeted sequencing for relatives undergoing cascade screening; this is deemed to be a more cost-effective approach. 10
In US and European guidelines, cascade screening does not necessarily incorporate genetic testing, although DNA analysis is noted as being useful in some cases or is recommended whenever resources are available.2,5
Management
A broad range of lifestyle modifications are proposed in the international literature. European guidelines, which cover several different dyslipidaemias, provide the most comprehensively reviewed changes, which are summarised in table 1. 4 There is a general consensus that specific nutritional advice should be provided by dieticians to achieve the best quality support, coupled with moderate levels of physical activity for ≥ 30 minutes per day on ≥ 5 days per week. Quantitative dietary fat intake values are rarely quoted but some suggest < 28–30% of energy intake from total fat and < 7–10% from saturates with a cholesterol intake of < 300 mg/day.1,3,4 Guidelines from Belgium suggest a cholesterol intake for children of < 75 mg/1000 kcal with similar total and saturated fat restrictions. 5 USA guidance quotes a slightly higher total fat intake of 25–35%, coupled with a lower cholesterol intake of < 200 mg/day. 2
The significance of various recommended lifestyle interventions on serum lipid levels

Relating to the impact of such intervention on serum lipid levels.
General agreement on the effects on lipid levels.
Less pronounced effects on lipid levels; weight of evidence/opinion is in favour of efficacy
Conflicting evidence; efficacy is less well established by evidence/opinion.
Not effective and/or uncertainties regarding safety.
All international guidelines underline the importance of smoking cessation with appropriate support being available. UK guidance suggests a target alcohol consumption no different to the current guidelines for the general public (2–3 units/day in females, 3–4 units/day in males). 1 Alternatively, Australian guidelines suggest a single universal limit of ≤ 2.5 British units/day. 3
Considerable divergence of opinion arises when considering the use of dietary supplementation in FH. The general consensus is that the use of supplementation, such as with sterols and stanols, is of benefit in the management of FH, with omega-3 having significant triglyceride reducing effects. Soy protein and red yeast are not widely recommended.1-4 Belgian guidelines state there is no clear recommendation to use supplements in the management of children with FH. This may be due to the practical and ethical difficulties in obtaining evidence of their efficacy through paediatric trials. 5 While UK advisors agree that evidence for the paediatric use of omega-3 is lacking, they advocate the use of sterols and stanols, stressing that consistent adherence is required to achieve clinical benefits. 1
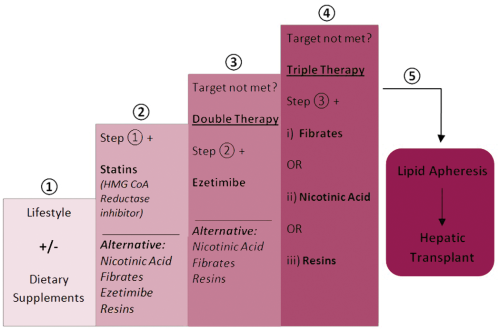
Current guidelines for pharmacological interventions are relatively homogenous and can be summarised in terms of a stepwise approach. Figure 3 consolidates the various guidelines in to a common, widely applicable framework. Opinions suggest that the lowest effective dose of pharmacological therapies should be used to achieve target lipid levels, whilst minimising side effects.1-5 Laropiprant or aspirin can be co-prescribed with nicotinic acid to reduce the associated side effects of facial flushing. 4 It is agreed that baseline muscle and liver enzymes should be measured, before initiating medication which may cause myositis or hepatitis.1,3,4 For children, most guidelines suggest initiating pharmacological intervention after the age of 10 years, using clinical judgement for what age exactly to start.1,3-5 American guidance states that consideration should be given to start at 8 years of age or possibly earlier in special cases, such as for homozygous FH. 2
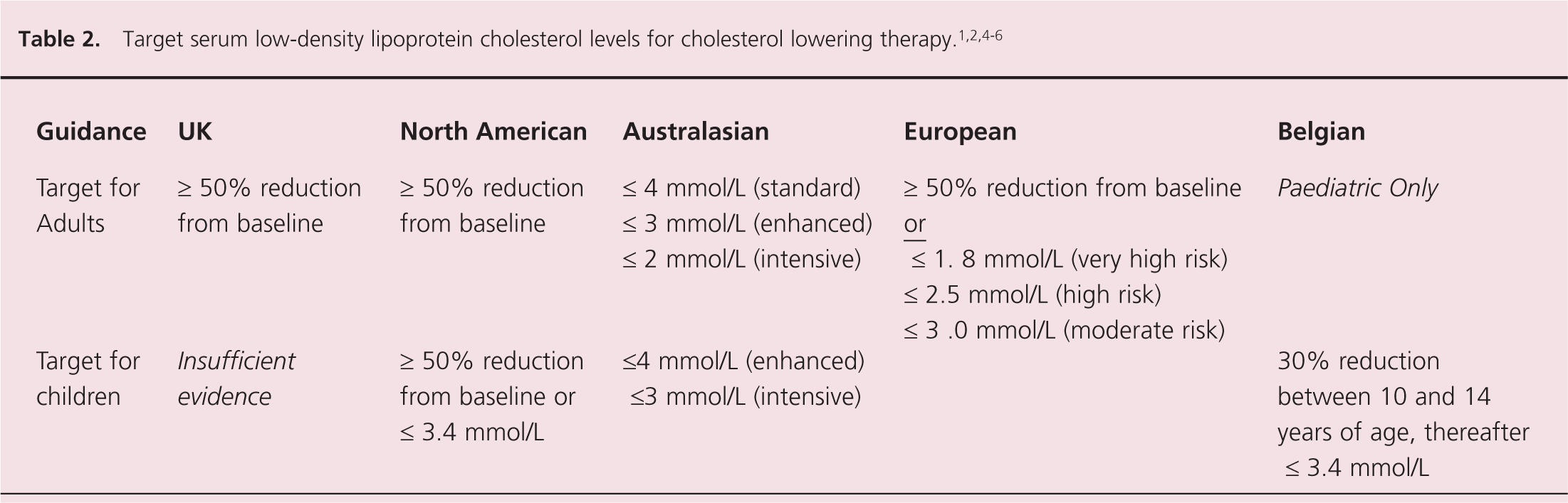
Alternatives to LDL cholesterol as a therapeutic target are non-HDL cholesterol and ApoB but use of these targets are not as commonly advocated. Catapano et al. state: ‘Epidemiological studies show that non-HDL cholesterol and ApoB measurement may correlate modestly better with outcomes, but there are no data on the use in routine clinical settings.’ 4 Opinion on the appropriate target LDL levels varies considerably and is summarised in table 2. Ideal serum triglycerides are widely regarded as being below 1.7–2.0 mmol/L.1-5
Of the guidelines that review the use of lipid apheresis all agree that the primary indication is for patients with homozygous or compound heterozygous FH.1-4 Clinical judgement must be exercised when initiating apheresis in heterozygotes when pharmacological therapy is not tolerated or severely elevated lipids are refractory to medication.1,3,4 UK and European guidance agree that ultimately hepatic transplantation may be considered in homozygous FH to provide functional LDL receptors. Additionally, angioplasty, CABG and cardiac transplantation serve in the secondary prevention of future coronary events.1,4
Patient support and advice
The patient with FH requires careful advice and support to understand their condition and its various implications, e.g. ischaemic heart disease. In addition the introduction to validated online resources and support groups may be of benefit. Where appropriate this should extend to the patient’s family (especially for children) and discussion on cascade screening should be initiated. 1 Pregnancy and contraception in women require discussion because of the potential teratogenic properties of some lipid modifying therapies and potential for hormonal contraception to increase the relative risk of CVD. 1 All guidelines consistently advise discontinuing lipid modifying drugs 3 months before conception and during pregnancy and breast feeding.1,4 In the event of conception without withdrawal of therapy an urgent obstetric referral is required.1,2
Review and follow-up
Patient follow-up should ideally occur in a specialist lipid clinic at a frequency which ranges from every 6 to 12 months for review of lifestyle, lipid profiles and medication adherence. UK, Australian and European guidelines all emphasise the importance of monitoring cardiovascular disease during such reviews.1,3,4 Australian guidance questions the evidence base for using carotid ultrasound. 3 In the UK the use of routine baseline electrocardiograms are recommended and where cardiovascular disease issues arise, a cardiology review should be sought. Patients with homozygous FH or a strong family history of early ischaemic heart disease should be referred for a cardiological assessment. 1 Australasian guidelines indicate the need for monitoring physical and sexual development in children when lipid modifying therapy is used. 3
Key messages
Familial disorders of lipoprotein metabolism, including hypercholesterolaemia, combined hyperlipidaemia and dyslipidaemias, are associated with an increased risk of premature cardiovascular disease
These conditions are both under diagnosed and under treated
Several international guidelines, published in the last year, should increase awareness and understanding of these conditions by healthcare professionals and lead to better prevention of cardiovascular disease
Footnotes
Funding
This reesearch received no specific grant from any funding agency in the public, commercial, or not for-profit sectors.
Conflict of interest statement
There is no conflict of interest.