
Abstract
Hereditary haemochromatosis (HH) is the commonest genetic condition among populations of Northern European ancestry. Mutations to the HFE gene leads to uninhibited intestinal iron absorption followed by excess iron deposition in various organs such as the liver, pituitary gland, pancreas and heart. Due to variable biochemical and clinical penetrance, not all individuals with C282Y homozygosity will develop HH. Early diagnosis is crucial to prevent morbidity and mortality but is challenging with most patients not exhibiting any symptoms. Patients with HH should undergo clinical assessment to evaluate their symptoms, presence of organ damage and hepatic fibrosis using transient elastography. Patients who are negative for the HFE mutations but have significant liver iron loading seen on magnetic resonance imaging should be reviewed by a specialist and considered for genetic tests looking for the rarer non-HFE mutations. HH patients are predominantly treated with venesection which can improve symptoms, hepatic fibrosis and mortality.
Background
Hereditary haemochromatosis (HH) is a genetic condition that causes excess intestinal iron absorption. This leads to iron deposition in various organs and tissues such as the liver, pancreas, joints, heart and pituitary gland. Ongoing iron deposition over years to decades may cause organ dysfunction if left untreated. Among populations of Northern European ancestry, HH is the commonest genetic condition. 1 C282Y and H63D are the most commonly seen variants caused by mutations to the haemostatic iron regulator (HFE) gene on chromosome 6 which was discovered in 1996. 2 Mutations to the HFE gene in the presence of iron overload are also termed HFE haemochromatosis and inherited in an autosomal recessive pattern. With genetic testing now widely available, the historical description of HH, ‘bronze diabetes’ characterised by the presence of liver cirrhosis, diabetes and skin hyperpigmentation, is rarely seen.
Rarer genetic mutations that cause haemochromatosis (non-HFE) include haemojuvelin (HJV), hepcidin antimicrobial peptide (HAMP), transferrin receptor 2 (TFR2) and SLC40A1 (ferroportin disease). 1 These non-HFE mutations are also inherited in an autosomal recessive manner except for ferroportin disease which is autosomal-dominant. In contrast to HFE haemochromatosis, the frequency of non-HFE mutations are very rare and can affect any population. The most severe iron overload is seen with HJV mutations often termed juvenile haemochromatosis, due to the early onset of heart failure and endocrine dysfunction presenting before the age of 30. 3 When to test for these mutations is discussed below. Other causes of genetic iron overload unrelated to hepcidin function are aceruloplasminaemia and atransferrinaemia, and both very rare.
In normal physiology, the duodenal enterocytes of adults absorb approximately 1–2 mg of iron daily from dietary intake. Dietary iron can be broadly classified as haem and non-haem iron. Haem iron which is predominantly from meat sources is directly absorbed. However, most of non-haem iron which is in the ferric state (Fe3+) needs to be reduced to ferrous (Fe2+) iron by the enzyme ferric reductase prior to absorption. 4 In the enterocytes, iron is then bound to ferritin or transported across the enterocyte to the circulation by ferroportin (Figure 1).
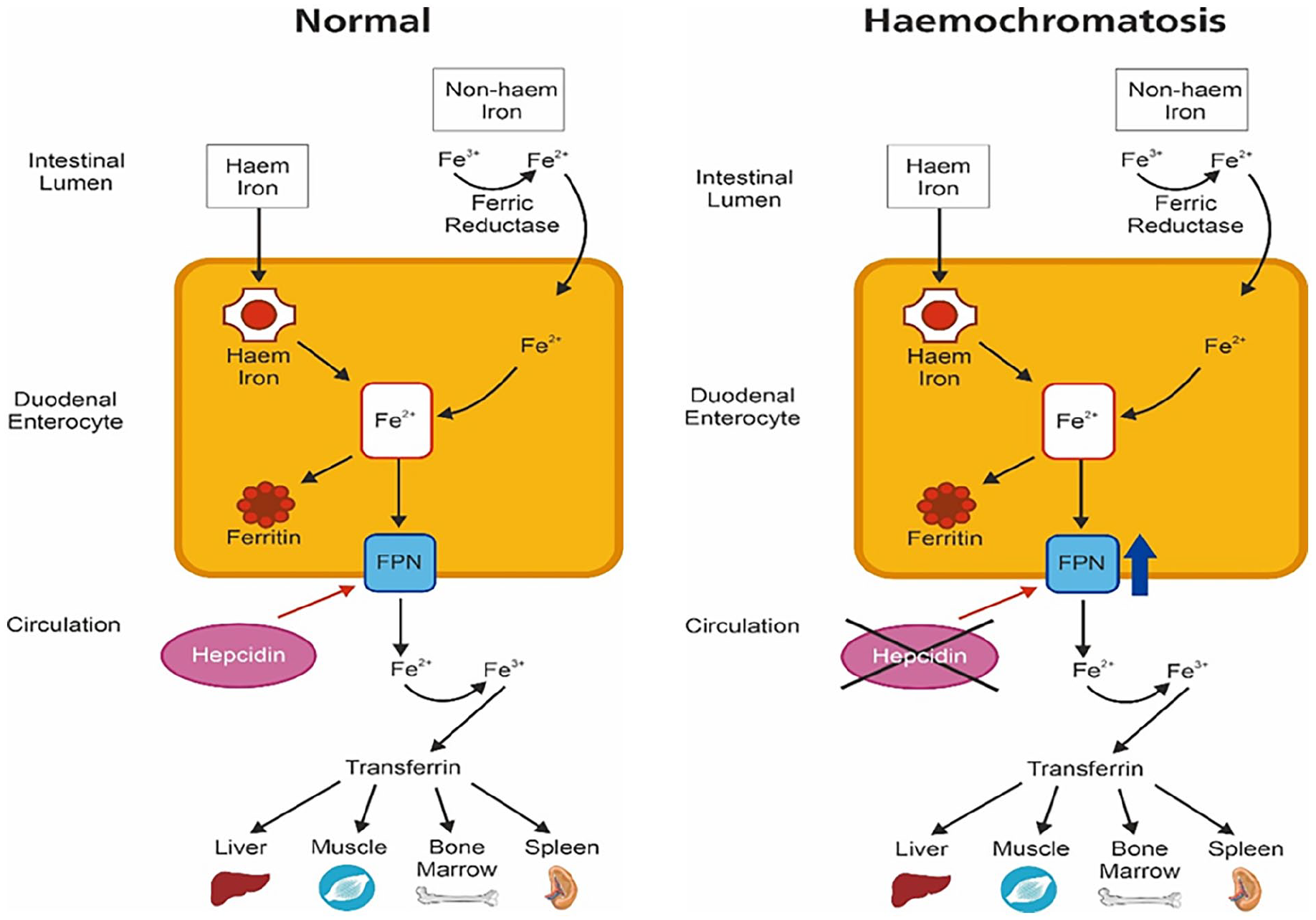
Illustration of intestinal iron absorption occurring via enterocytes in the duodenum. Haem iron is directly transported into the enterocyte. Non-haem iron requires conversion into the ferrous state (Fe2+) by the enzyme ferric reductase prior to being absorbed. Iron in the enterocyte is then either converted to ferritin or transported into the circulation by ferroportin. Once in the circulation, iron is converted to its ferric state (Fe3+) to be bound to transferrin for delivery to the liver, bone marrow and skeletal muscles. Hepcidin influences the amount of iron being transported into the circulation by directly affecting ferroportin activity. In haemochromatosis, HFE mutations causes hepcidin deficiency which prevents degradation of ferroportin. This leads to unopposed iron absorption across the enterocyte through ferroportin.
Hepcidin is a crucial peptide hormone in iron metabolism by regulating the iron transport protein ferroportin. Hepcidin is downregulated in iron deficient states to improve iron availability and upregulated in high iron situations to reduce iron absorption by opposing ferroportin activity by inducing its breakdown. 4 The HFE protein maintains iron haemostasis through its effect on hepcidin. In HFE haemochromatosis, mutations to the HFE gene lead to hepcidin deficiency, thus preventing ferroportin degradation and causing unopposed enterocyte iron absorption and deposition in tissues.
This article is a narrative review focussing on HFE haemochromatosis. We searched the MEDLINE database from inception to June 2024 with the search terms ‘haemochromatosis’ and ‘hemochromatosis’. All publications in English including review articles were eligible for inclusion. Current clinical guidelines on haemochromatosis by the professional liver societies and expert groups were reviewed as well. The reference list of publications identified were also hand searched.
Who has HH?
C282Y homozygotes (two copies of C282Y) are at the highest risk of morbidity and mortality related to HH. 5 Men are at a higher risk of organ damage especially hepatocellular carcinoma (HCC) and death compared to women due to the protective effect of menstruation.6,7 However, the presence of C282Y homozygosity on its own is insufficient to establish a diagnosis of HH, 8 as not all will develop raised iron parameters (termed biochemical penetrance) or HH-related organ damage (termed clinical penetrance). 9
The variant H63D is increasingly thought to be a common finding in the general population.2,10 Individuals with one copy of C282Y and one copy of H63D variants are termed ‘compound heterozygotes’. Mild elevation of iron indices is seen in compound heterozygotes but are not thought to be pathogenic. 1 Overall, organ dysfunction secondary to iron overload is less likely in compound heterozygotes11,12; however, the risk of liver fibrosis and cirrhosis significantly increases in the presence of cofactors such as alcohol and diabetes. 12
Diagnosis
Early diagnosis and treatment are crucial to prevent the complications associated with iron loading in various tissues, but this can be challenging given the non-specific symptoms of HH. Furthermore, large population studies have not demonstrated the effectiveness of using biochemical measurements such as serum ferritin, transferrin saturation or HFE genetic testing for screening. Even in those with serum ferritin >1,000 µg/L, iron overload is not the commonest cause. 13 Below, we discuss further aspects that influence hyperferritinaemia which needs to be assessed. Currently, population screening is not recommended, 14 but the recent UK BioBank HH mortality and morbidity data suggests this stance may need to be revisited. 15
Presentation
HH patients are initially diagnosed through a variety of presentations including HH-related symptoms, family screening, abnormal liver function tests (LFTs) or incidental finding of raised ferritin on routine blood tests. Patients usually remain asymptomatic especially during the initial phase of the condition. General symptoms seen in patients with HH include fatigue, joint pains and reduced libido. Patients may eventually develop organ-specific signs or symptoms such as decompensated liver cirrhosis, HCC, arthropathy, diabetes, osteoporosis, heart failure and skin hyperpigmentation (Figure 2).15,16 Patients with these conditions should have their iron indices assessed for iron overload.
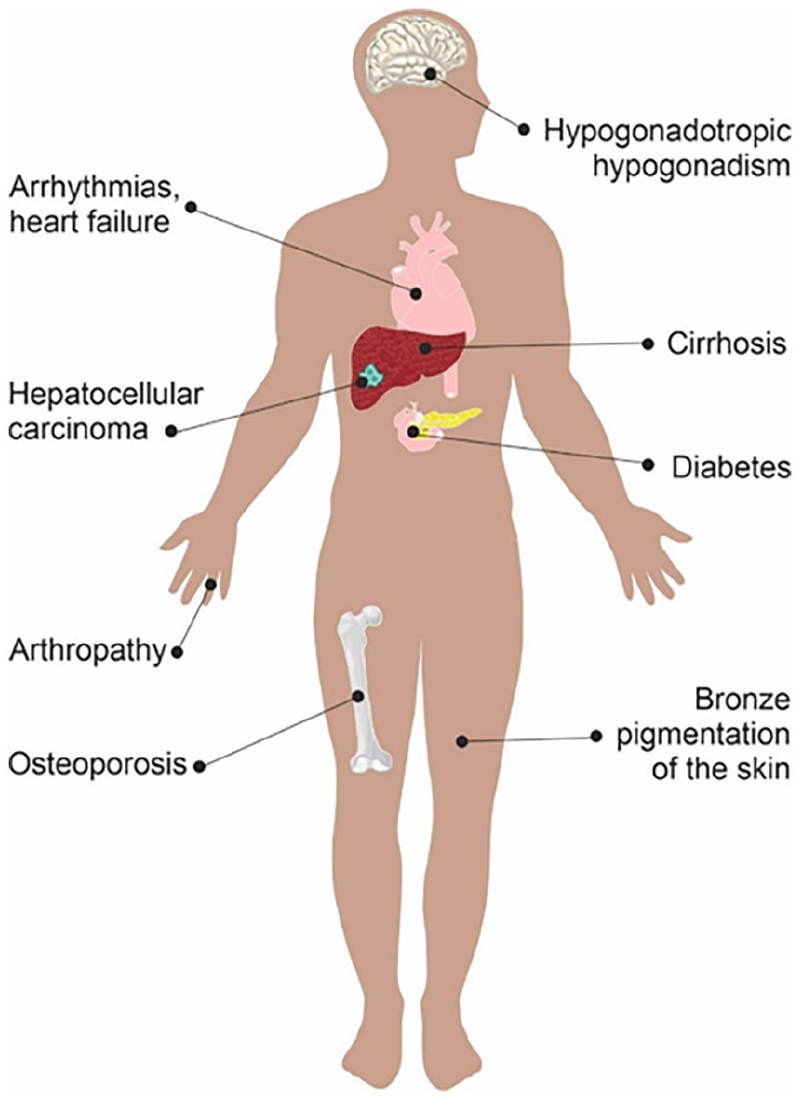
Clinical manifestations in haemochromatosis.
First-degree adult relatives of those diagnosed with HH are advised to undertake genetic screening making this a common pathway to diagnosis. Current guidance does not recommend screening in compound heterozygotes. 14 Abnormal LFTs typically triggers a liver aetiology screen which includes serum ferritin and transferrin saturation to assess for iron overload causes. Raised serum ferritin and/or transferrin saturation may be incidentally identified as part of work-up for other symptoms and should prompt clinical evaluation followed by HFE genetic testing if indicated. 17
Biochemical abnormalities
Confirming a diagnosis of HH usually follows a step-wise process of identifying raised serum ferritin and transferrin saturation followed by HFE genetic testing. Liver biopsy is no longer required to diagnose HH. Despite hepcidin being a key regulator of iron metabolism, its measurement is not widely established and is currently not recommended. HFE genotyping should be undertaken in men/post-menopausal women with serum ferritin >300 µg/L and transferrin saturation >50% and women with serum ferritin >200 µg/L and transferrin saturation >45% regardless of clinical symptoms or signs. 10
Isolated hyperferritinaemia in the absence of elevated transferrin saturation should not lead to immediate genetic testing as it unlikely to be HH. 14 Concomitant C-reactive protein measurement is crucial as ferritin is an acute-phase protein which is elevated during acute infections and inflammatory conditions. In addition, the presence of other conditions associated with hyperferritinaemia such as excess alcohol consumption and metabolic syndrome should be assessed. 17 Rare conditions such as adult-onset Still’s disease and haemophagocytic lymphohistiocytosis can cause significantly raised serum ferritin levels of >10,000 µg/L. If raised ferritin is found during an acute illness, it is recommended to repeat the test in 6 weeks to guide further investigations. 17 Patients who have underlying haematological conditions and who require regular blood transfusion, such as thalassaemia, aplastic anaemia and myelodysplastic syndrome, may also develop secondary iron overload resulting in elevated serum ferritin levels. Figure 3 provides a practical approach to assessing a patient with suspected haemochromatosis and those with elevated serum ferritin.
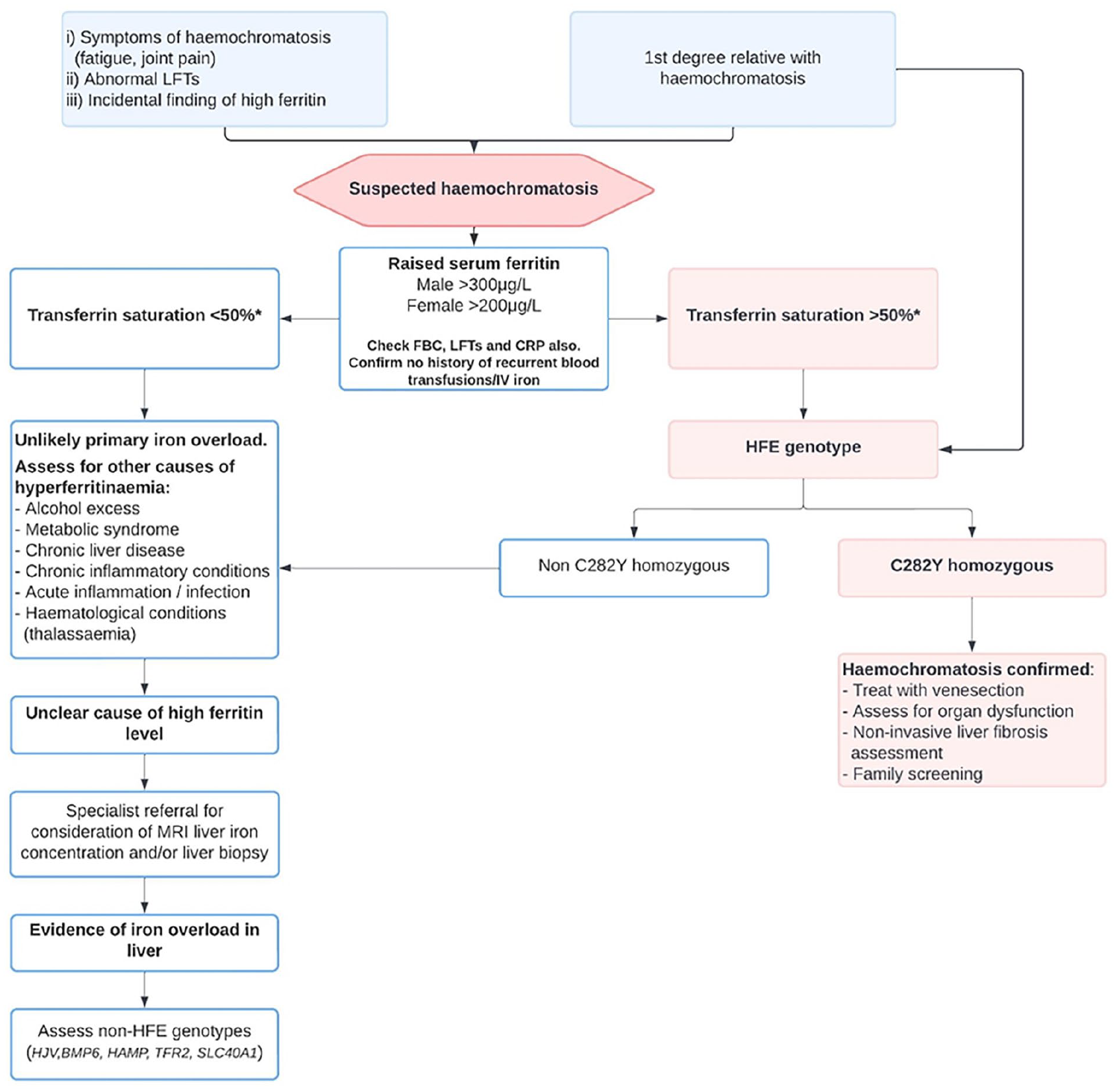
Algorithm to guide the assessment and investigations of patients with suspected haemochromatosis.
In C282Y homozygotes, those with serum ferritin >1,000 µg/L have the highest risk of cirrhosis and mortality. 18 Current UK and European guidelines recommend hepatology review for assessment of liver fibrosis if the serum ferritin is >1,000 µg/L or in the presence of deranged LFTs.10,14
Assessment of liver iron concentration
Patients who are not C282Y homozygous but have evidence of ongoing iron overload biochemically, in the absence of other conditions causing hyperferritinaemia, should undergo further assessment of their liver iron concentration with magnetic resonance imaging (MRI).1,10,14 Predating the discovery of the HFE gene, liver biopsy was used to assess hepatic iron to diagnose HH. It also provides iron quantification by assessing hepatic iron grading and the hepatic iron index. However, now, MRI measurement of liver iron concentration is proving to be increasingly valuable in quantifying the presence of excess iron deposition in a non-invasive manner and it acts as a surrogate for total body iron stores. The main MRI techniques that are available to measure liver iron concentration are signal intensity ratio, R2 relaxometry and R2* relaxometry with increasing prevalence of their validity. 19
In non-C282Y homozygous patients who have persistently raised serum ferritin, MRI liver iron concentration is useful to guide hyperferritinaemia assessment and can guide treatment decisions.10,19 A lack of significant iron loading may reflect other processes driving ongoing hyperferritinaemia such as excess alcohol consumption or metabolic syndrome. In addition, Bhuva et al. used MRI liver iron concentration, to demonstrate a lack of significant hepatic iron overload in compound heterozygotes who had a serum ferritin <750 µg/L (100% specificity). 20 In individuals with substantial iron loading on MRI, further genetic testing is suggested for rarer genes associated with non-HFE mutation such as HJV, HAMP, TFR2 and SLC40A1.
Organ dysfunction
Liver
The liver is the main site of iron deposition in HH. Many years of excess iron deposition can lead to hepatic fibrosis and subsequently, cirrhosis. Historically, liver biopsy was carried out to assess for fibrosis and cirrhosis specifically in patients with a serum ferritin ⩾1,000 µg/L and/or elevated liver enzymes. A serum ferritin of <1,000 µg/L with normal serum transaminases at diagnosis provides an excellent negative predictive value for advanced fibrosis and cirrhosis detection. 21 However, this gradual process of iron deposition may not necessarily be reflected by rising liver enzymes. The EASL 2022 guidelines mentions that all patients with HH are assessed for liver fibrosis using non-invasive methods, with transient elastography (FibroScan) of ⩽6.4 kPa ruling out advanced fibrosis. 10 Liver biopsy may still be considered in those whom cirrhosis cannot be ruled out.
Several serum-based non-invasive biomarkers such aspartate aminotransferase to platelet ratio index (APRI) and fibrosis-4 (FIB-4) are now widely used for hepatic fibrosis detection in many chronic liver diseases. Their utility has also been demonstrated to rule out advanced fibrosis in HH. 22 However, they are currently not recommended due to the limited studies on their validity. 10
Patients with HH who develop liver cirrhosis are at risk of several complications including ascites, jaundice, hepatic encephalopathy, variceal bleeding and HCC. A study examining four population-based cohort studies, estimated the prevalence of ‘severe liver disease’ to be 9% amongst C282Y male homozygotes who were untreated for haemochromatosis. 23 Men with C282Y homozygosity are at severalfold risk of developing HCC. 6 Patients who develop cirrhosis complications and/or HCC that meet specific criteria are eligible for liver transplantation. 1 Surveillance should be undertaken in cirrhotic HH patients owing to the significantly increased risk of HCC with six monthly ultrasonography with or without alpha-fetoprotein levels. 10
Arthralgia and arthropathy
Joint pain is one of the commonest symptoms experienced by HH patients and significantly affects quality of life. 24 Also, HH can lead to arthritis and joint damage particularly affecting the second and third metacarpophalangeal joints and ankles. 16 The pathogenesis mimics that of osteoarthritis but in an accelerated form with larger osteophytes and subchondral cysts. Given its aggressive nature, an increase in joint replacements are seen in C282Y homozygotes compared to individuals without these mutations. 25 Arthropathy and liver disease seem to be interlinked with the absence of arthropathy indicating advanced liver fibrosis to be unlikely. 26 ‘De-ironing’ treatments have not shown to improve symptoms or joint damage caused by HH-related arthropathy and can even worsen despite improvement in serum ferritin. 16 Current management involves analgesia and physiotherapy to alleviate symptoms. Overall, there remains a paucity of evidence in the assessment and management of arthropathy among HH patients.
Endocrinopathies
Diabetes and hypogonadotropic hypogonadism are the main manifestations of HH affecting the endocrine system.27,28 The prevalence of diabetes has been demonstrated to be higher in C282Y homozygotes especially men compared to the rest of the population. 15 Diabetes in HH occurs from excess iron deposition in pancreatic islet cells as well as insulin resistance from hepatic damage. 1 Clinicians are advised to assess for diabetes as part of the evaluation for HH patients. 10 The effect of venesection on diabetes remains unclear, 29 although current studies favour improved glucose control if treatment is commenced early. 27
Hypogonadotropic hypogonadism results from iron deposition into the pituitary gland leading to symptoms of reduced libido, erectile dysfunction and amenorrhoea. Presence of these symptoms should prompt assessment of pituitary and sex hormones (follicle-stimulating hormone, luteinising hormone, sex hormone-binding globulin, free testosterone and oestradiol) with input from an endocrinologist for abnormal results.10,17 The reported prevalence of hypogonadotropic hypogonadism in HH patients is approximately 6.4% with the authors acknowledging that this may be decreasing due to earlier diagnosis. 28 Thyroid and adrenal dysfunction are rarely reported due to the predilection for iron deposition in the gonadotrophs within the pituitary gland. 27
Cardiovascular
Excess iron deposition in the heart can lead to cardiomyopathy causing symptoms of heart failure, conduction abnormalities affecting the sinus and atrioventricular nodes and palpitations from various arrhythmias including atrial fibrillation and ventricular arrhythmias. 30 Early in the disease, cardiac iron deposition causes diastolic dysfunction which may eventually lead to dilated cardiomyopathy and reduced ventricular ejection fraction. 30 Over a quarter of HH patients presenting to hospital were found to have cardiovascular complications, with arrhythmias and heart failure being the commonest. 31 Individuals with the HJV genetic mutation are more likely to develop cardiac complications due to the rapid and excessive iron loading in the myocardium. Cardiovascular complications of iron loading in the heart are more commonly seen in transfusion-related iron overload conditions such as thalassaemia. 31
Baseline investigations such as electrocardiography assist the assessment of arrythmias and conduction abnormalities, whilst echocardiography provides information on cardiac contractility. In addition, cardiac MRI can be utilised to objectively quantify the degree of myocardial iron overload and guide treatment strategies. 32 Cardiology input is essential in the presence of cardiac haemochromatosis. 17 Treatment with venesection and/or iron chelating agents prevents further iron loading and improves cardiac dysfunction. 30
Others
Osteoporosis, dementia and psychological effects are other frequently described associations with HH.15,33 Up to a third of HH patients are thought to have osteoporosis, although the pathogenesis of this is uncertain. 27 Data from the UK BioBank cohort highlighted a positive association between male C282Y homozygotes and dementia. 33 From a psychological perspective, patients also report feeling stigmatised as a result of their HH diagnosis and are more likely to suffer from depression. 34 These areas require further research to improve our current understanding of their pathogenesis in HH alongside the influence of other genetic and environmental factors.
Management
General principles
Not all individuals who are homozygous for the C282Y variant develop symptoms related to HH, have a raised serum ferritin or suffer from organ dysfunction, indicating variable biochemical and clinical penetrance. However, some individuals develop significant morbidity such as liver cirrhosis and HCC despite treatment with venesection. Why this variation exists remains unknown yet, but the role of cofactors in the form of alcohol, diabetes and metabolic syndrome have been implicated. 35 Clinicians should assess and address cofactors to prevent liver fibrosis progression especially from excess alcohol consumption. 10
Proton pump inhibitor therapy has been associated with reduced intestinal iron absorption requiring fewer venesections, but current guidelines do not recommend its routine use as treatment for HH.10,14 Current guidance recommends vitamin C and iron supplementation should be avoided as well as limiting red meat and citrus fruit consumption. In addition, alcohol consumption should be limited and patients with established cirrhosis should refrain from any alcohol intake. 10 These measures are not a replacement for iron removal therapies.
Venesection
Venesection which leads to ‘de-ironing’ of tissues and organs has remained the mainstay of HH treatment since the mid-20th century. It has shown to improve mortality if commenced before the development of liver cirrhosis. 36 Improvement in liver fibrosis have also been demonstrated as a result of venesection. 37 From a symptom perspective, improvement in fatigue is described following venesection, 29 although the evidence remains inconclusive regarding joint pains which have shown contradictory results. The evidence base concerning improvement in quality of life, diabetes and cardiac disease from venesection is limited but does show some benefit. 29
Treatment with venesection is commenced once a diagnosis of haemochromatosis is confirmed following genetic testing and in the setting of raised iron parameters (men/postmenopausal women and premenopausal women >300 and >200 µg/L, respectively). 10 However, biochemical parameters should be considered alongside comorbidities and frailty to ensure any risks from regular venesection is clearly outweighed by its benefit. The presence of C282Y homozygosity without biochemical iron overload does not merit treatment but should be monitored yearly and commenced on venesection once the above parameters are met. Venesection treatment consists of the induction and maintenance phases to initially remove excess iron and subsequently prevent reaccumulation.
During each venesection treatment session, 450–500 ml of blood is removed. This equates to approximately 200–250 mg of iron. The volume of blood removed or frequency of venesections should be reduced once the haemoglobin levels are <120 g/L. Development of anaemia (haemoglobin <110 g/L) during venesection should prompt a pause in treatment and further clinical evaluation. 10 Patients that have persistently low serum ferritin, even after stopping venesections should be assessed for iron deficiency in accordance with established pathways. Common challenges encountered during venesection are tolerability of frequent bloodletting, difficult venous access and frequency of appointments especially for work and caring commitments. 38
Induction phase
Upon diagnosis, patients undergo weekly to biweekly venesections aiming for a ferritin of 50 µg/L. 10 Whilst a target ferritin of around 50 µg/L reflects the majority of national and consensus guidelines, there have not been clinical trials to ascertain the appropriate target serum ferritin to prevent organ dysfunction and improve symptoms. Patients should have their haemoglobin measured before every venesection and serum ferritin every 4 weeks. The induction phase may take more than a year before achieving the target ferritin. There remain differing opinions within the scientific community regarding the utilisation of transferrin saturation as a treatment target, some guidelines recommending a target of <50%, whilst others do not. Joint symptoms are reported to be worse in those who have transferrin saturation >50% which may have influenced the treatment targets. 39
Maintenance phase
Following successful induction, patients undergo the maintenance phase whereby venesection frequency can vary from every 2–6 months. The primary aim is to maintain a ferritin of 50–100 µg/L and prevent further iron loading, 10 although there is no strong evidence base to support the maintenance phase serum ferritin targets either. Haemoglobin monitoring should continue to be undertaken before each venesection with six monthly serum ferritin measurement to ensure iron deficiency is not developing. Patients in the United Kingdom are encouraged to donate blood regularly via the NHS Blood & Transplant transfusion services during their maintenance phase which provides the added benefit of the venesected blood being used for a good cause. The overall HH care and monitoring of iron parameters remains with the clinical team even if patients donate blood as part of their maintenance phase.
Iron chelating agents
Excess iron stored in the tissues can also be removed with iron chelation therapy which includes desferrioxamine, deferasirox and deferiprone, but may be limited by its side effects. 40 Deferasirox is the most commonly used chelating agent, but is not currently licensed for HH treatment. 10 Evidence of their effectiveness are largely limited to studies related to iron overload secondary to regular blood transfusion, 40 but there is accumulating evidence of their efficacy and safety in HH in smaller studies. 41 Iron chelation is a useful second-line option in patients with HH who are unable to tolerate venesection, have poor venous excess or become anaemic following careful assessment. Iron chelation therapies can be used on top of venesection when cardiac failure occurs especially in juvenile HH. 3
Erythrocytapheresis
Erythrocytapheresis is an alternative to venesection for HH treatment involving removal of erythrocytes only compared to whole blood removal in venesection. Erythrocytapheresis has also been used effectively in other conditions treated with venesection such as polycythaemia 42 and secondary iron overload due to haemoglobinopathies. 43 Specialist equipment is required for this treatment which is not widely available and is costly. 42 However, erythrocytapheresis has been shown to require fewer procedures to ‘de-iron’ HH patients compared to venesection in both the induction and maintenance phases.44,45 A multicentre randomised controlled trial comparing erythrocytapheresis against sham intervention with plasmapheresis showed improvement in patient-reported fatigue as well. 46 Larger-scale prospective studies are required to evaluate its cost-effectiveness including productivity loss from patients having to attend regular treatments.
The future
Rusfertide, a hepicidin mimetic given subcutaneously weekly, has been shown to reduce requirement for phlebotomy in early trials. 47 Another target for future therapy is the enzyme transmembrane serine protease 6 (TMPRSS6) which inhibits hepcidin. 48 In preclinical studies using mouse models with HH, targeting TMPRSS6 is effective in reducing tissue and serum iron accumulation. 49 Early clinical trials using therapeutic agents that inhibit TMPRSS6 activity in polycythaemia are ongoing. 50
Conclusion
Early diagnosis of HH is crucial to identify patients with biochemical and clinical penetrance who will benefit from treatment. Greater awareness of symptoms, family screening, widespread availability of HFE genetic testing and increasing availability of MRI liver iron concentration makes this process more attainable. There remains a lack of understanding as to why some patients develop liver cirrhosis and HCC in middle age, but others remain asymptomatic without organ dysfunction and not requiring venesection. The presence of normal transferrin saturation should prompt assessment of other causes of hyperferritinaemia rather than primary iron-overload disorder. When working up patients with suspected HH, physicians need to be mindful of other conditions that impact upon serum ferritin levels particularly excess alcohol consumption and metabolic dysfunction associated with steatotic liver disease, thus, preventing unnecessary venesection in those that do not require it. Assessment of liver fibrosis with transient elastography should form a core aspect of clinical assessment to identify those at the highest risk of liver disease. Venesection improves mortality, hepatic fibrosis and fatigue but is unlikely to alleviate joint pain. Cofactors for liver disease need to be addressed alongside HH treatment to prevent acceleration of liver fibrosis. Ongoing clinical trials targeting hepcidin show promise in preventing iron accumulation which may mitigate the requirement for venesection during the maintenance phase in the near future.
Footnotes
Acknowledgements
Professor John Macfie and Dr Chao Huang reviewed the article and provided feedback. The authors would like to thank Robyn Sotheran, Medical Artist at York and Scarborough Teaching Hospitals NHS Foundation Trust for creating Figures 1 and ![]() .
.
Author contributions
PS, CM and RD were involved in study concept and design. PS drafted manuscripts. CM and RD critically revised manuscript. All authors approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.