
Abstract
A 39-year-old woman with a 4-year history of limited cutaneous systemic sclerosis (lcSSc) and no prior exposure to D-penicillamine presented with bilateral fluctuating ptosis of 1 week duration. On evaluation, she was found to have elevated acetylcholine receptor antibody levels and a positive neostigmine test confirming the diagnosis of ocular myasthenia gravis (MG). The patient was treated with oral prednisolone and pyridostigmine resulting in significant improvement within 2 weeks. The co-occurrence of systemic sclerosis (SSc) and MG is rare and when such an association is seen, it is usually seen in the background of D-Penicillamine therapy for SSc. The presence of fluctuating ptosis in SSc is a valuable clinical clue to suspect co-existence of neuromuscular junction disorder like MG.
Introduction
Systemic sclerosis (SSc) or scleroderma is a chronic multisystem autoimmune disease characterised by fibrosis of the skin and internal organs. Limited cutaneous systemic sclerosis (lcSSc) is a subset of SSc with skin thickening seen distal to the elbow and knee. LcSSc may be seen in association with other autoimmune diseases such as Hashimoto thyroiditis, polymyositis, primary biliary cirrhosis and autoimmune hepatitis. 1 The co-occurrence of lcSSc and myasthenia gravis (MG) is rare and when such an association is seen, it is usually seen in the background of D-penicillamine therapy for the treatment of scleroderma. 2 Herein, we describe the development of ocular MG in a patient with lcSSc who had no prior exposure to D-penicillamine. We have also done a brief review of the literature on previously published reports of this rare association and the pathogenic mechanism involved in such an association.
Case presentation
A 39-year-old woman with a 4-year history of lcSSc (diagnosed on the basis of thickening of skin confined to below elbow and face with pigmentation changes, Raynauds phenomenon, auto amputation of right second digit following dry gangrene, multiple digital pits, gastroesophageal reflux disease, severe pulmonary artery hypertension and anti-centromere antibody positivity) presented with bilateral ptosis of one-week duration. She had no history of febrile illness, diplopia, visual disturbances, gait imbalance or generalised muscle weakness. She was on tadalafil 40 mg per day and ambrisentan 5 mg per day for severe pulmonary artery hypertension but had no history of use of D-penicillamine. Examination revealed bilateral fluctuating ptosis with mild eyelid edema. Visual acuity, pupillary responses and extraocular movements were preserved. Systemic examination was notable for pulmonary artery hypertension and thickening of skin confined to face and below elbow bilaterally with a modified Rodnan skin score of 4/51, amputated right second digit, digital pits on fourth and fifth digits of right hand and telangiectasia on the distal aspect of left third and fourth finger and pigmentation changes on dorsal aspect of both hands (Figure 1a and 1b). On admission, her blood investigations including acute phase reactants were normal. She was found to be positive for anti-centromere antibody. The differential diagnosis included mechanical ptosis from localized eyelid edema due to lcSSc, orbital myositis and ocular MG. Contrast-enhanced MRI of the brain and orbits was normal, ruling out orbital myositis or brainstem pathology. An ice pack test was negative, and repetitive nerve stimulation was inconclusive. However, elevated acetylcholine receptor antibody levels (5.06 nmol/L; normal <0.40) and a positive neostigmine test (administration of 1.5 mg neostigmine intramuscularly; Figure 1c and 1d) supported the diagnosis of ocular MG. CT scan of the thorax did not show the presence of thymoma.
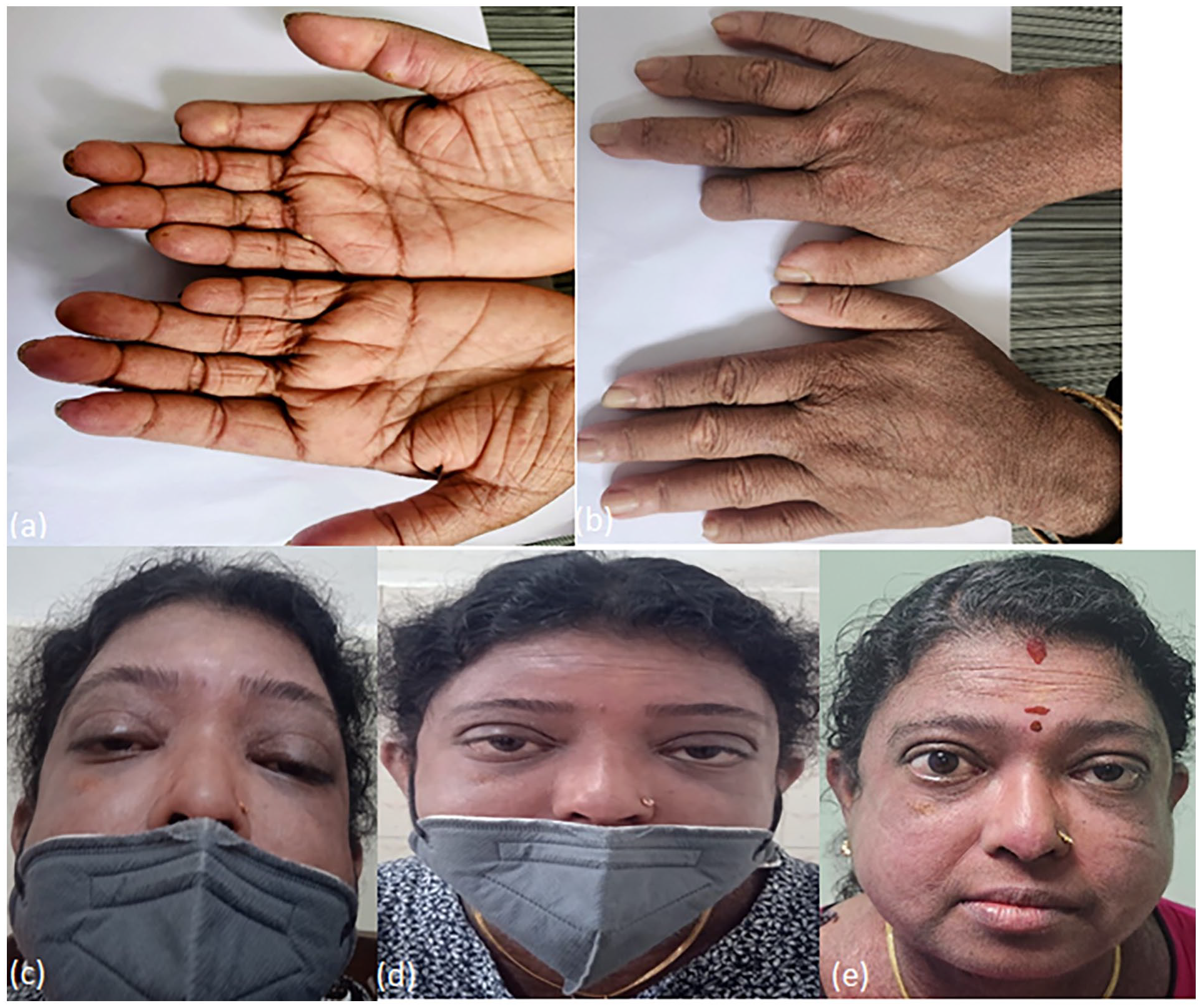
Clinical images of patient: (a) pitting scars on the right ring finger and tip of fifth finger, auto amputated right index finger, telangiectasias on the distal aspect of left middle and ring finger; (b) pigmentary changes in the form of areas of hypo- and hyper-pigmentation on the dorsal aspect of both hands; (c) bilateral ptosis before neostigmine test; (d) improvement of ptosis post neostigmine test; (e) resolution of ptosis with pyridostigmine and prednisolone on follow-up at 2 weeks.
The patient was treated with oral prednisolone 50 mg/day (1 mg/kg) and pyridostigmine (60 mg three times daily), resulting in significant improvement within 2 weeks (Figure 1e). On follow-up, she was initiated on azathioprine as steroid-sparing agent. The dose of prednisolone was tapered by 5 mg every 2 weeks to reach a maintenance dose of 5 mg/day by week 18. The patient is on a maintenance dose of 5 mg oral prednisolone since last 6 weeks and azathioprine 100 mg/day. She has completed 6 months of follow-up at our center after the onset of ocular MG.
Discussion
LcSSc is characterised by skin thickening distal to the elbow and knee with features of vasculopathy like Raynaud’s phenomenon, digital ischemia and pulmonary artery hypertension. The natural course of lcSSc can be complicated by the association with other systemic autoimmune diseases such as rheumatoid arthritis, polymyositis, systemic lupus erythematosus, Sjogren disease or by organ-specific autoimmune diseases like Hashimoto’s thyroiditis and primary biliary cirrhosis. The co-occurrence of MG with SSc is rare and when found, it is usually seen with prior therapy with D-penicillamine for SSc. Drosos et al. 3 reported a series of eight patients who developed D-penicillamine-induced MG. Seven of the patients had rheumatoid arthritis and one patient had scleroderma. The authors found that D-penicillamine-induced MG had clinical and immunological features resembling idiopathic MG but was less severe. All patients had complete resolution of myasthenic symptoms after discontinuation of D-penicillamine.
Zivković et al. 4 reported two patients with scleroderma who had no prior exposure to D-penicillamine and developed MG. The authors reviewed literature on the co-occurrence of MG and scleroderma in patients without exposure to D-penicillamine. Among the 12 cases identified, the majority of patients developed seropositive generalized MG. Ocular-limited myasthenia was not reported in this literature review and the duration between onset of scleroderma and myasthenia varied from 6 months to 22 years. The combination of prednisolone and cholinesterase inhibitors led to improvement in myasthenic symptoms in most of the patients, and five patients underwent thymectomy revealing follicular hyperplasia in four and thymoma in one patient. Kokotis et al. 5 reported a 66-year-old with limited scleroderma who developed MG without concomitant exposure to D-penicillamine. The patient improved with a combination of oral prednisolone, pyridostigmine and azathioprine was used as steroid-sparing agent.
Neurological involvement of SSc was found in 40% of patients in a retrospective study of 50 patients by Averbuch-Heller et al. 6 The most common involvement was that of muscle (22%) followed by peripheral nerve (18%) and spinal cord (8%). Neuromuscular junction involvement in the form of MG was seen in only one patient, which shows the rarity of such an association. The authors have not specified the use of D-penicillamine in the patient who developed myasthenia. Ocular abnormalities in scleroderma commonly involve the eyelids, and conjunctiva and may lead to dry eye disease. Eyelid involvement is the most common ocular abnormality and is characterised by eyelid stiffness, tightness and telangiectasia. Eyelid involvement may be seen in 29% to 65% of patients with SSc and occurs secondary to increased deposition of type 1 collagen leading to decreased eyelid motility. Though eyelid involvement may be seen in SSc, ptosis is still a rare symptom.7,8 Patients with orbital myositis may present with diplopia and isolated myositis of levator palpebrae superioris may present with acute onset painful ptosis. Orbital myositis may be seen secondary to autoimmune rheumatic diseases, especially in IgG4 related disease, inflammatory bowel disease and sarcoidosis. In a comprehensive review of orbital myositis, only two cases of scleroderma associated orbital myositis were identified. 9
The association between scleroderma and MG may be explained by a shared HLA B8/DR3 haplotype. 10 Patients with MG have an increased risk for other autoimmune diseases. The frequency of a second autoimmune disease in patients with MG is around 13% to 22%, among which autoimmune thyroid disease (10%), systemic lupus erythematosus (1–8%) and rheumatoid arthritis (4%) are the most common.11,12 The risk of developing of a second autoimmune disease is highest in females with early-onset MG. Hence, when a patient with autoimmune disorder develops a new onset neuromuscular weakness, concomitant MG should be suspected. The overlap between scleroderma and myasthenia may be difficult to diagnose as both diseases may have common features like fatigue and generalized weakness. However, the presence of fluctuating weakness highlighted by diurnal variations and ocular abnormalities in the form of fluctuating ptosis as was seen in our patient should alert the clinician to suspect the development of MG. The prompt diagnosis of this rare clinical overlap also has therapeutic implications. A high dose of corticosteroids is generally avoided in SSc as it can precipitate scleroderma renal crisis. 13 However, in cases of SSc-MG overlap, a higher dose of oral corticosteroids may be required in the initial phase of treatment with cholinesterase inhibitors and a steroid-sparing agents such as azathioprine, mycophenolate mofetil or rituximab may be added. 14 In our patient, we used azathioprine as steroid-sparing agent, and we were able to reduce the dose of oral corticosteroid on follow-up. In our literature review of prior reports of overlap of scleroderma with MG treated with corticosteroids, we could not identify any patient who developed scleroderma renal crisis.4,5,10,15 –20 The majority of the patients reported in literature with SSc-MG overlap had generalised MG which is contrary to our patient who developed ocular limited MG. A comparison of our case with the previously reported cases is shown in Table 1.
Demographic and clinical characteristics of reported cases with co-existence of scleroderma and myasthenia gravis.
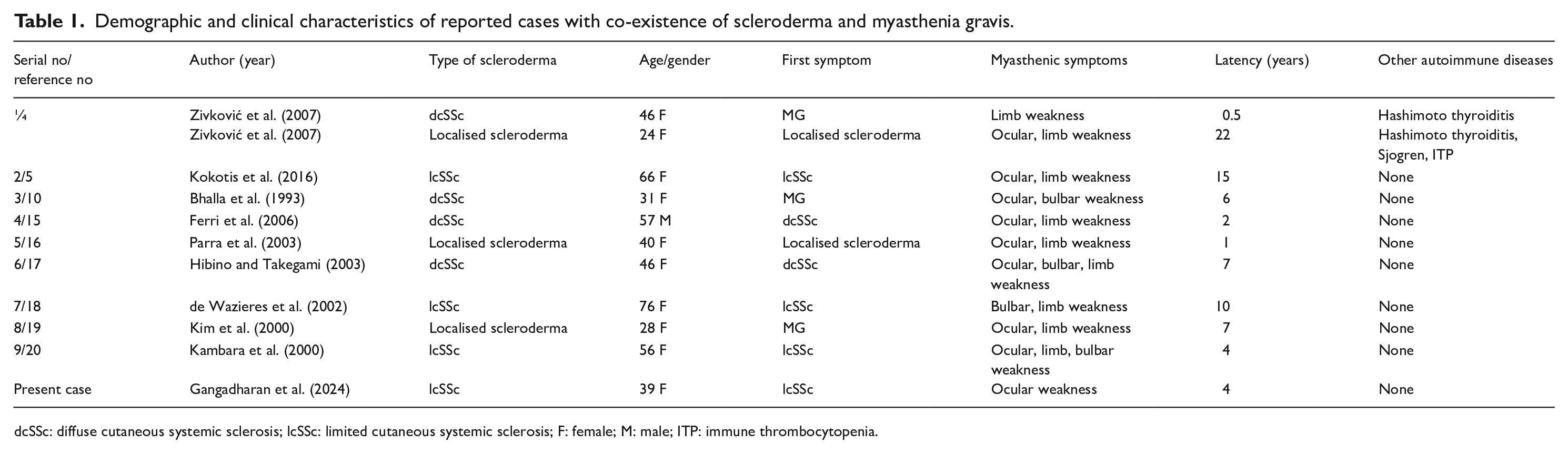
dcSSc: diffuse cutaneous systemic sclerosis; lcSSc: limited cutaneous systemic sclerosis; F: female; M: male; ITP: immune thrombocytopenia.
Conclusion
The development of a new onset neuromuscular weakness in a patient with SSc should prompt the clinician to suspect the co-occurrence of MG. The presence of fluctuating ptosis is a valuable clinical clue to consider an association with neuromuscular junction disorder. Our case study emphasises the need to consider ocular MG in limited scleroderma patients presenting with fluctuating ptosis for a timely diagnosis and treatment.
Footnotes
Acknowledgements
None
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Informed consent
Written informed consent for the paper to be published was obtained from the patient.