
Abstract
Aims:
To investigate the tumor-intrinsic lymphocyte cytosolic protein 2 (LCP2) in esophageal squamous cell carcinoma (ESCA) and its molecular mechanisms in mediating resistance to programmed death-1 (PD-1) therapy.
Methods:
The expression of LCP2 in ESCA was analyzed using bioinformatics databases and further verified in clinical specimens. Functional studies employed patient-derived organoid models, xenograft models, and molecular assays to assess the impact of LCP2 knockout or overexpression on macrophage polarization, CD8 + T cell exhaustion, and PD-1 therapy response. Mechanistic investigations included nuclear factor-κB (NF-κB) inhibition, signal transducer and activator of transcription 5A (STAT5A) knockdown, chromatin immunoprecipitation, and dual-luciferase reporter assays.
Results:
LCP2 was markedly upregulated in ESCA and correlated with advanced stage, lymph node metastasis, and poor survival. Tumor-intrinsic LCP2 expression positively correlated with M2 macrophage polarization and sorted CD8+ T cell exhaustion. Mechanistically, this association depended on NF-κB pathway activation in EpCAM+ tumor fractions, while STAT5A transcriptionally regulates tumor-intrinsic LCP2 expression as an upstream transcription factor. Knockdown of tumor-intrinsic LCP2 or STAT5A in EpCAM+ tumor fractions suppressed the secretion of immunosuppressive cytokines and restored effector T cell function of sorted CD8+ T cells. In vivo, LCP2 depletion significantly inhibited tumor growth and synergized with PD-1 blockade. This synergistic effect was characterized by reduced tumor volume and increased CD8+ T cell infiltration. Overexpression of LCP2 reversed these effects, confirming its central role in immune escape.
Conclusion:
The STAT5A–LCP2–NF-κB axis remodels the immunosuppressive tumor microenvironment to mediate ESCA immune escape and PD-1 resistance. Targeting this regulatory axis provides a novel strategy to overcome immunotherapy resistance in esophageal cancer. Antioxid. Redox Signal. 00, 000–000.
Introduction
Esophageal squamous cell carcinoma (ESCA) remains a highly aggressive malignancy and a leading cause of gastrointestinal cancer–related mortality worldwide (Siegel et al., 2025). Despite advances in surgery, radiotherapy, and chemotherapy, most patients are diagnosed with locally advanced or metastatic disease and experience high rates of recurrence and poor long-term survival (Peters et al., 2023). Immune checkpoint blockade targeting the programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) axis has introduced a new therapeutic modality for ESCA; however, durable clinical responses are achieved in only a minority of patients (Liu et al., 2022; Whooley et al., 2022). Primary and acquired resistance to PD-1 inhibitors, therefore, constitutes a major unresolved clinical challenge and underscores the need to delineate the molecular mechanisms that govern immunotherapy failure in ESCA.
Innovation
This study offers three key conceptual innovations: (1) We define LCP2 as a tumor-intrinsic immune adaptor co-opted by ESCA cells to suppress antitumor immunity, repositioning it from a passive immune-related expression marker to a central oncogenic immune modulatory hub that drives PD-1 resistance in ESCA. (2) We delineate the complete STAT5A-LCP2-NF-κB regulatory axis as the mechanistic basis for LCP2-mediated immunosuppression, linking oncogenic STAT5A transcriptional programming to NF-κB-dependent remodeling of the TME. (3) We provide preclinical proof-of-concept that targeting this LCP2-centered immune modulatory hub synergizes with PD-1 blockade to restore antitumor immunity, suggesting LCP2 as a candidate prognostic biomarker and a potential therapeutic target for overcoming anti-PD-1 resistance in ESCA, pending large-scale prospective validation.
A growing body of evidence indicates that resistance to immune checkpoint blockade is driven not solely by tumor-intrinsic genetic alterations but by active remodeling of the tumor immune microenvironment (TME). In ESCA, the TME is frequently characterized by enrichment of immunosuppressive myeloid populations and functional impairment of cytotoxic T lymphocytes. Specifically, polarization of tumor-associated macrophages toward an M2-like phenotype promotes immune evasion through the secretion of inhibitory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) (Liu and Cao, 2016). In parallel, sustained antigen exposure and inhibitory signaling result in CD8+ T cell exhaustion, marked by increased expression of immune checkpoint receptors including PD-1, cytotoxic T lymphocyte–associated protein 4 (CTLA-4), and T cell immunoglobulin and mucin-domain-containing protein 3 (TIM-3) (Yan et al., 2024). Together, these processes establish an immunologically suppressive tumor milieu that limits the efficacy of PD-1-based therapies. Nevertheless, the tumor-intrinsic regulatory pathways that coordinate macrophage polarization and T cell exhaustion in ESCA remain incompletely defined.
Lymphocyte cytosolic protein 2 (LCP2), also known as SLP-76, is a canonical adaptor protein required for signal transduction downstream of the T cell receptor (TCR) in hematopoietic cells (Wang et al., 2012). Through its scaffold function, LCP2 orchestrates the activation of multiple downstream pathways, including nuclear factor-κB (NF-κB), which plays a central role in inflammatory signaling and immune regulation. Although LCP2 has classically been regarded as immune-cell restricted, emerging evidence suggests that it may also be aberrantly expressed in solid tumors, where its biological role remains largely unexplored (Sheikh et al., 2023). Preliminary bioinformatic analyses revealed that tumor-intrinsic LCP2 is significantly upregulated in ESCA and exhibits strong positive correlations with immunosuppressive cell populations and immune checkpoint molecules, raising the possibility that tumor-intrinsic LCP2 in EpCAM+ tumor fractions contributes to immune escape rather than merely reflecting immune cell infiltration.
The NF-κB signaling pathway represents a key molecular nexus linking inflammation, oncogenesis, and immune suppression (Labib et al., 2019; Takeda et al., 2020). Constitutive NF-κB activation in tumor cells promotes the transcription of cytokines and chemokines that shape the TME, enhances macrophage polarization toward an M2 phenotype, and upregulates immune checkpoint molecules that enforce T cell dysfunction (Cornice et al., 2024). Importantly, NF-κB is also a redox-sensitive transcription factor, integrating inflammatory and oxidative stress signals to regulate immune homeostasis. Prior studies have shown that LCP2 can activate NF-κB signaling in immune cells through protein–protein interactions involving kinases such as ZAP-70 and adaptor complexes including Grb2-associated downstream signaling (GADS) protein (Meri et al., 2025). Whether a similar LCP2-dependent mechanism operates in ESCA tumor cells to modulate the immune microenvironment, however, has not been determined.
Signal transducer and activator of transcription 5A (STAT5A) is a key transcription factor within the JAK-STAT pathway that regulates diverse processes in immunity and cancer (Rani and Murphy, 2016; Smith et al., 2022). Persistent STAT5A activation has been implicated in macrophage polarization and CD8+ T cell exhaustion, as well as in transcriptional reprogramming of tumor cells (Liu et al., 2025; Liu et al., 2021; Oueslati et al., 2022). Notably, transcriptional correlation analyses indicate a positive association between STAT5A and LCP2 expression in ESCA, and bioinformatic prediction identifies putative STAT5A-binding sites within the LCP2 promoter region. These observations suggest that STAT5A may function upstream of LCP2, linking oncogenic transcriptional signaling to immune microenvironment remodeling.
In this study, we systematically investigate the role of a previously unrecognized STAT5A–tumor-intrinsic LCP2–NF-κB regulatory axis in mediating PD1 resistance in ESCA (Fig. 1). Using integrated analyses of public datasets, clinical specimens, patient-derived organoids (PDOs), and immunocompetent in vivo models, we demonstrate that tumor-intrinsic LCP2 in EpCAM+ tumor fractions promotes M2 macrophage polarization and sorted CD8+ T cell exhaustion through NF-κB pathway activation. We further establish STAT5A as a direct transcriptional regulator of LCP2 and show that genetic or functional disruption of this axis suppresses immunosuppression and synergizes with PD-1 blockade to inhibit tumor progression. Collectively, these findings suggest LCP2 as a tumor-intrinsic immune adaptor co-opted by malignant ESCA cells to suppress antitumor immunity, which acts as an oncogenic immune modulatory hub linking STAT5A signaling to NF-κB-dependent immune escape and PD-1 resistance. This work provides a novel conceptual framework for understanding immunotherapy failure in ESCA and a mechanistic rationale for novel combinatorial immunotherapeutic strategies in this disease.
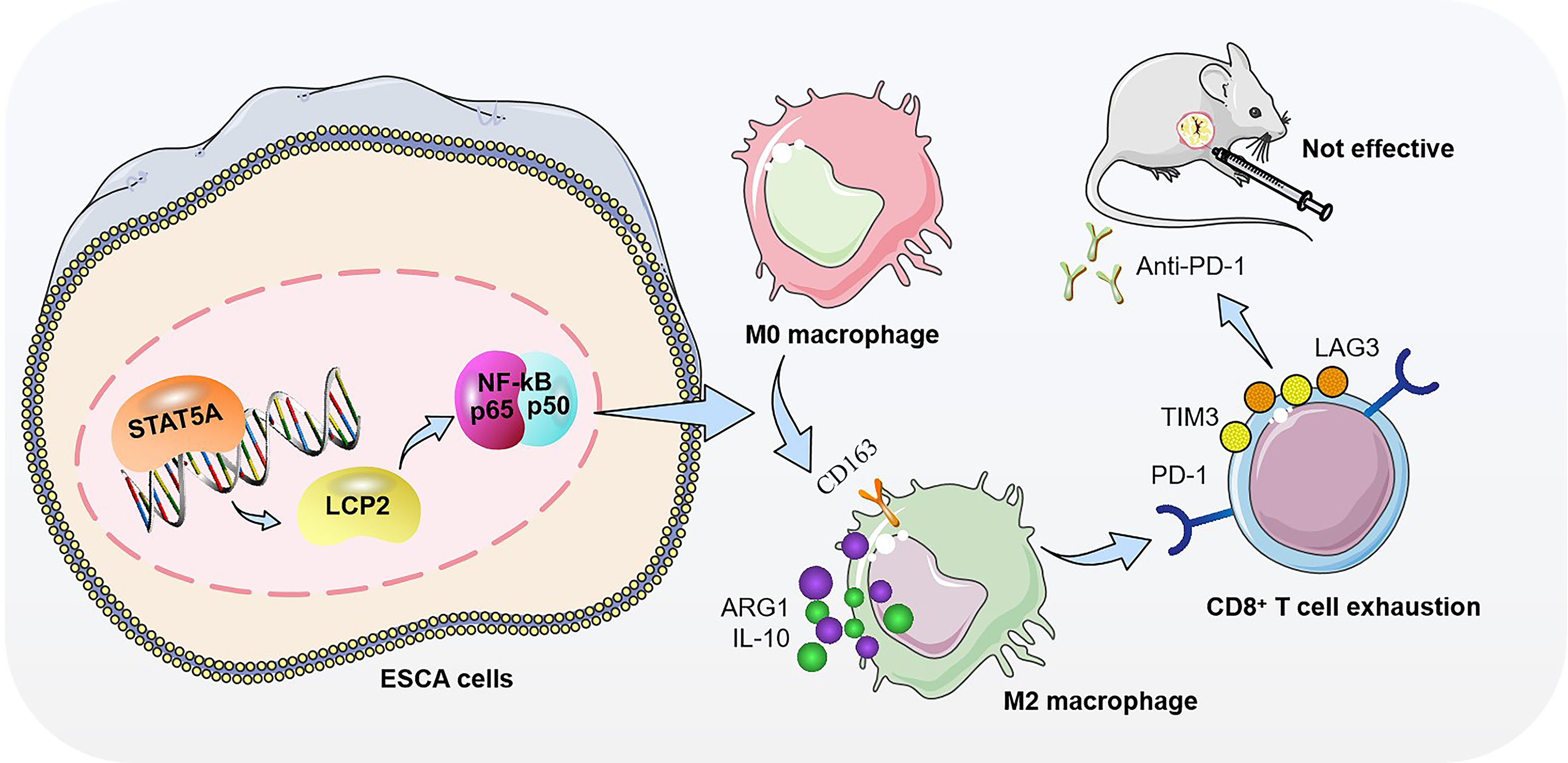
Clinical problem addressed
Only ∼20% of ESCA patients achieve durable responses to anti-PD-1 immunotherapy, with primary/acquired resistance representing a major clinical bottleneck that leads to treatment failure and poor survival. This study addresses this unmet clinical need by identifying the STAT5A–LCP2–NF-κB axis as a driver of PD-1 resistance in ESCA and validating this axis as a therapeutic target to enhance immunotherapy efficacy.
Results
LCP2 is highly expressed in ESCA and associated with poor prognosis
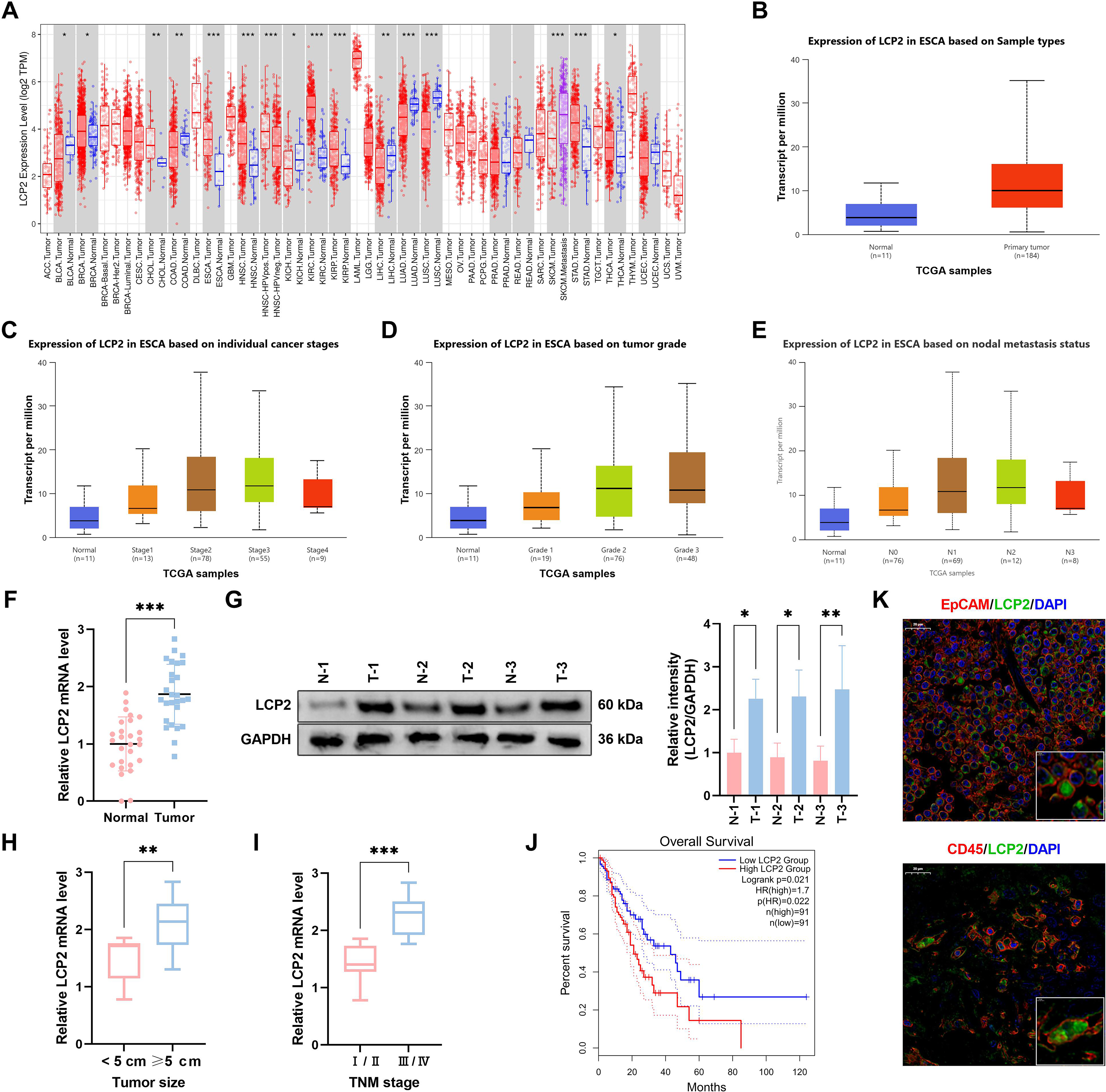
To characterize the clinical relevance of LCP2 in ESCA and establish its potential as a prognostic biomarker, we first performed integrative bioinformatic analyses of public pan-cancer datasets and validated our findings in a single-center clinical cohort of ESCA patients. Analysis of the tumor immune estimation resource (TIMER) database revealed differential expression of tumor-intrinsic LCP2 across the Cancer Genome Atlas (TCGA) pan-cancer dataset (Fig. 2A). Further analysis with the UALCAN database verified significant LCP2 upregulation in ESCA tissues (Fig. 2B). Elevated LCP2 expression was strongly associated with advanced disease, showing increased levels in higher tumor stages (Stage III to IV vs. I to II, Fig. 2C), poorer pathological grades (Grade 2 to 3 [G2 to G3] vs. Grade 1 [G1], Fig. 2D), and lymph node metastasis (positive vs. negative, Fig. 2E). Clinical sample analysis supported these findings: quantitative reverse transcription polymerase chain reaction (qRT-PCR) demonstrated that tumor-intrinsic LCP2 mRNA levels were significantly higher in EpCAM+ tumor fractions compared with adjacent normal tissues (Fig. 2F), and Western blot confirmed significantly elevated tumor-intrinsic LCP2 protein expression in tumor samples (Fig. 2G). This upregulation was particularly pronounced in tumors with large tumor size and advanced clinical stage III/IV (Fig. 2H and I). Notably, GEPIA-based survival analysis of TCGA-ESCA cohorts (n = 182 clinically annotated patients) revealed that high LCP2 expression was significantly associated with reduced overall survival (log-rank test p = 0.021, Fig. 2J). This clinical observation is consistent with our tissue sample validation results (Fig. 2F and G). Immunofluorescence (IF) costaining of tumor-intrinsic LCP2, epithelial marker EpCAM, and pan-immune marker CD45 was performed on clinical ESCA tissues, and the results showed that LCP2 was intrinsically expressed in tumor epithelial cells (Fig. 2K). Collectively, these findings establish tumor-intrinsic LCP2 as a clinically relevant oncogene that is significantly upregulated in ESCA and independently associated with advanced disease and poor overall survival.
LCP2 regulates immune infiltration in the tumor microenvironment
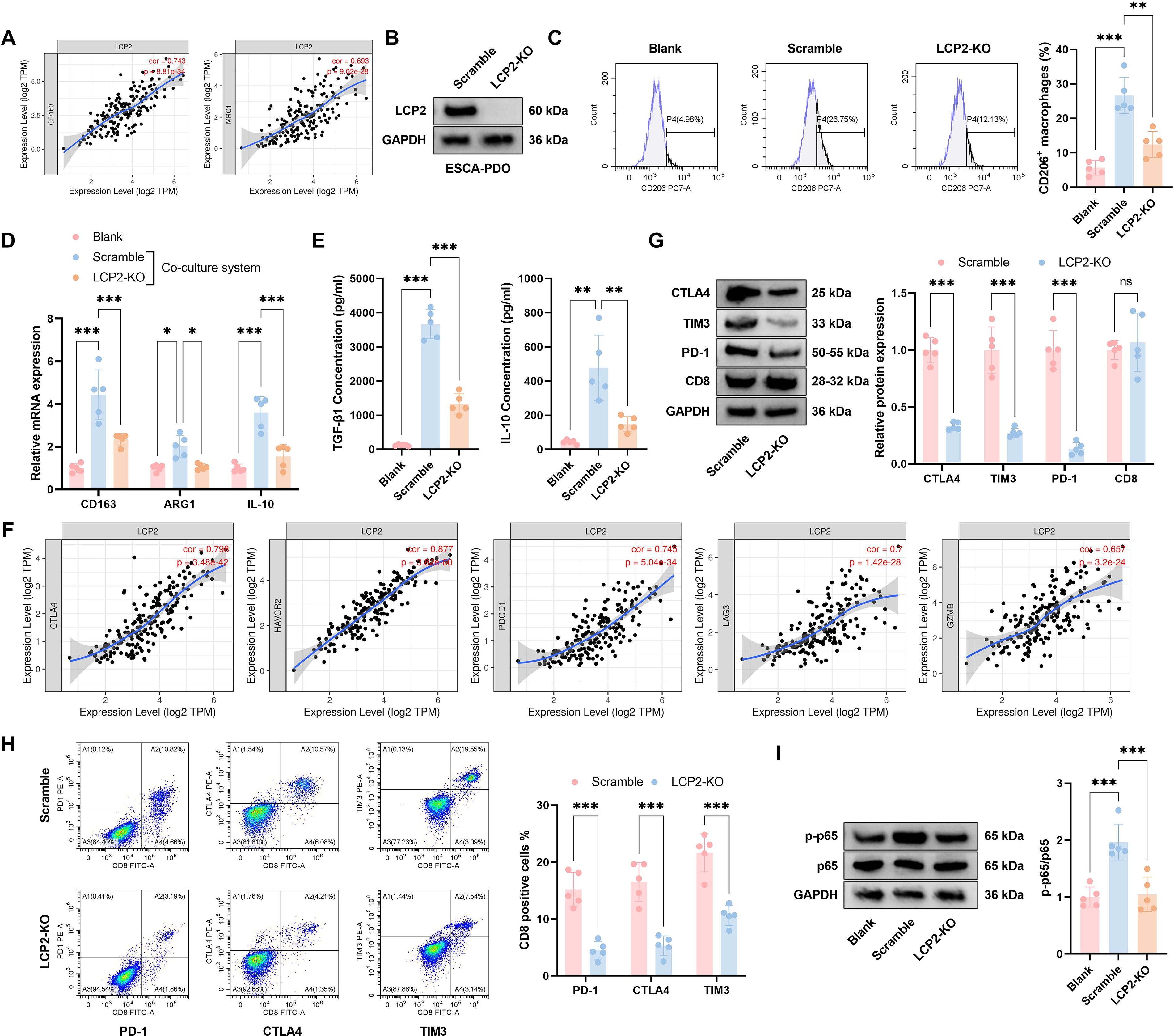
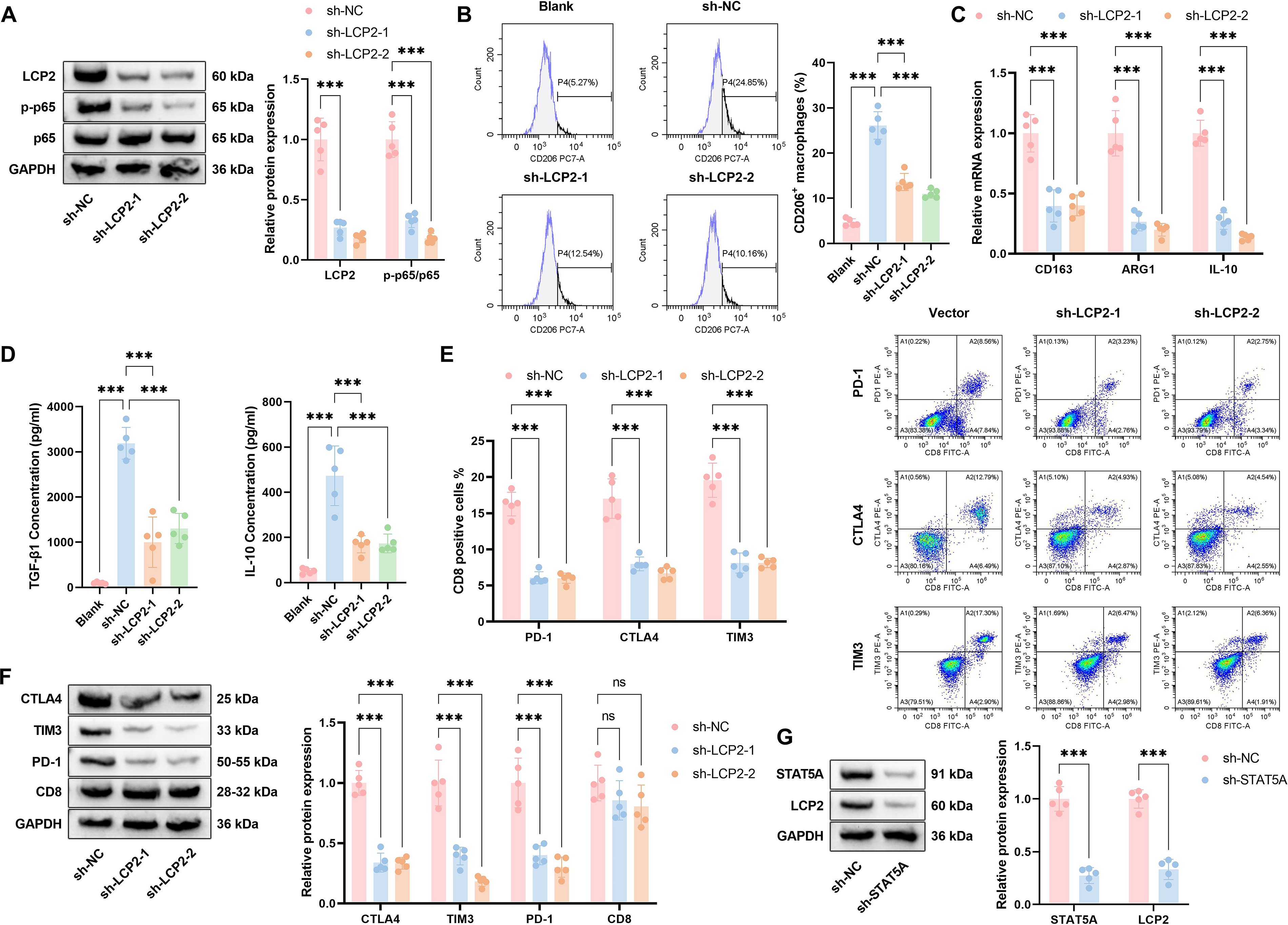
Given the strong clinical association between LCP2 expression and ESCA progression, we next sought to investigate whether LCP2 modulates the immunosuppressive TME—a primary driver of PD-1 therapy resistance in ESCA—using a PDO model that faithfully recapitulates the cellular and molecular features of primary ESCA tumors. Gene expression correlation analysis showed that tumor-intrinsic LCP2 expression in EpCAM+ tumor fractions positively correlated with transcriptional levels of M2 macrophage markers CD163 and mannose receptor C-type 1 (Fig. 3A). To investigate its functional mechanism, we successfully established an LCP2-knockout (LCP2-KO) esophageal cancer PDO (ESCA-PDO) model (efficiency confirmed by Western blot, Fig. 3B). We verified the epithelial purity of our ESCA-PDO model via flow cytometry, confirming that the PDOs were >95% positive for EpCAM and <1% positive for CD45, ruling out immune cell contamination in our functional experiments (Supplementary Fig. S1). Flow cytometry demonstrated a marked reduction in CD206+ macrophages following LCP2 knockout relative to Scramble controls (Fig. 3C). In a coculture model, qPCR revealed substantially decreased expression of immunosuppressive genes (CD163, Arginase 1 [ARG1], IL-10) in LCP2-KO cells, with effects being more evident under coculture conditions (Fig. 3D). Enzyme-Linked Immunosorbent Assay (ELISA) further verified significantly reduced concentrations of immunosuppressive cytokines TGF-β and IL-10 in the supernatant of LCP2-KO cells (Fig. 3E). Western blot analysis of sorted CD8+ T cells from the coculture system demonstrated substantial downregulation of immune checkpoint molecules (CTLA-4, TIM-3, PD-1) in the tumor-intrinsic LCP2-KO coculture group, whereas CD8 expression remained unaltered (Fig. 3G). Correlation analysis revealed a significant positive association between LCP2 expression and multiple immune checkpoints (CTLA-4, hepatitis A virus cellular receptor 2 [HAVCR2], programmed cell death 1 [PDCD1], lymphocyte-activation gene 3 [LAG3], Granzyme B [GZMB]) in ESCA-PDOs (Fig. 3F). Flow cytometric analysis was performed on CD45+CD3+CD8+ T cells gated from the coculture system, and the results significantly reduced proportions of PD-1+, CTLA-4+, and TIM-3+ T cells in LCP2-KO compared with control groups (Fig. 3H). In addition, phosphorylation of p65, a central NF-κB pathway component, was markedly reduced following knockout of tumor-intrinsic LCP2 in EpCAM+ tumor fractions (Fig. 3I). These data indicate an association between LCP2 expression, M2 macrophage polarization, and CD8+ T cell exhaustion in ESCA.
LCP2-mediated M2 macrophage polarization and CD8+ T cell exhaustion are associated with NF-κB pathway activation
Given the established role of LCP2 in activating NF-κB signaling (Herndon et al., 2001) and its demonstrated involvement in CD8+ T cell exhaustion (Rong et al., 2022) and M2 macrophage polarization (Yang et al., 2024a), this pathway was selected for further investigation. To determine whether NF-κB signaling is functionally required for LCP2-mediated immunosuppression, we employed both pharmacological inhibition with the selective NF-κB inhibitor BAY 11-7082 and orthogonal genetic silencing of RELA/p65, the core transcriptional effector of the canonical NF-κB cascade, to validate the causal role of this pathway in our experimental system. Western blot analysis showed that overexpression of tumor-intrinsic LCP2 increased both LCP2 protein level and p65 phosphorylation in EpCAM+ ESCA tumor cells. We then treated OE-LCP2 cells with BAY 11-7082, a selective NF-κB inhibitor. BAY 11-7082 treatment markedly reduced p65 phosphorylation, even with sustained high LCP2 expression (Fig. 4A). Functionally, CD206+ macrophage prevalence was markedly increased in OE-LCP2 versus Vector controls, an effect largely abolished by BAY 11-7082 administration (Fig. 4B). qPCR analysis similarly revealed upregulation of M2 macrophage marker mRNA (CD163, ARG1, IL-10) in the OE-LCP2 group, which was attenuated following BAY 11-7082 treatment (Fig. 4C), indicating that LCP2-mediated M2 polarization depends on NF-κB pathway activation. Western blot analysis indicated elevated expression of immune checkpoint molecules (CTLA-4, TIM-3, PD-1)—but not CD8—in the OE-LCP2 group, which was markedly suppressed by BAY 11-7082 co-treatment (Fig. 4D). Flow cytometry further verified elevated proportions of PD-1+, CTLA-4+, and TIM-3+ T cells in the OE-LCP2 group, which were markedly reduced following BAY 11-7082 treatment (Fig. 4E). Collectively, these data show that LCP2-mediated M2 macrophage polarization and CD8+ T cell exhaustion are dependent on NF-κB pathway activation.
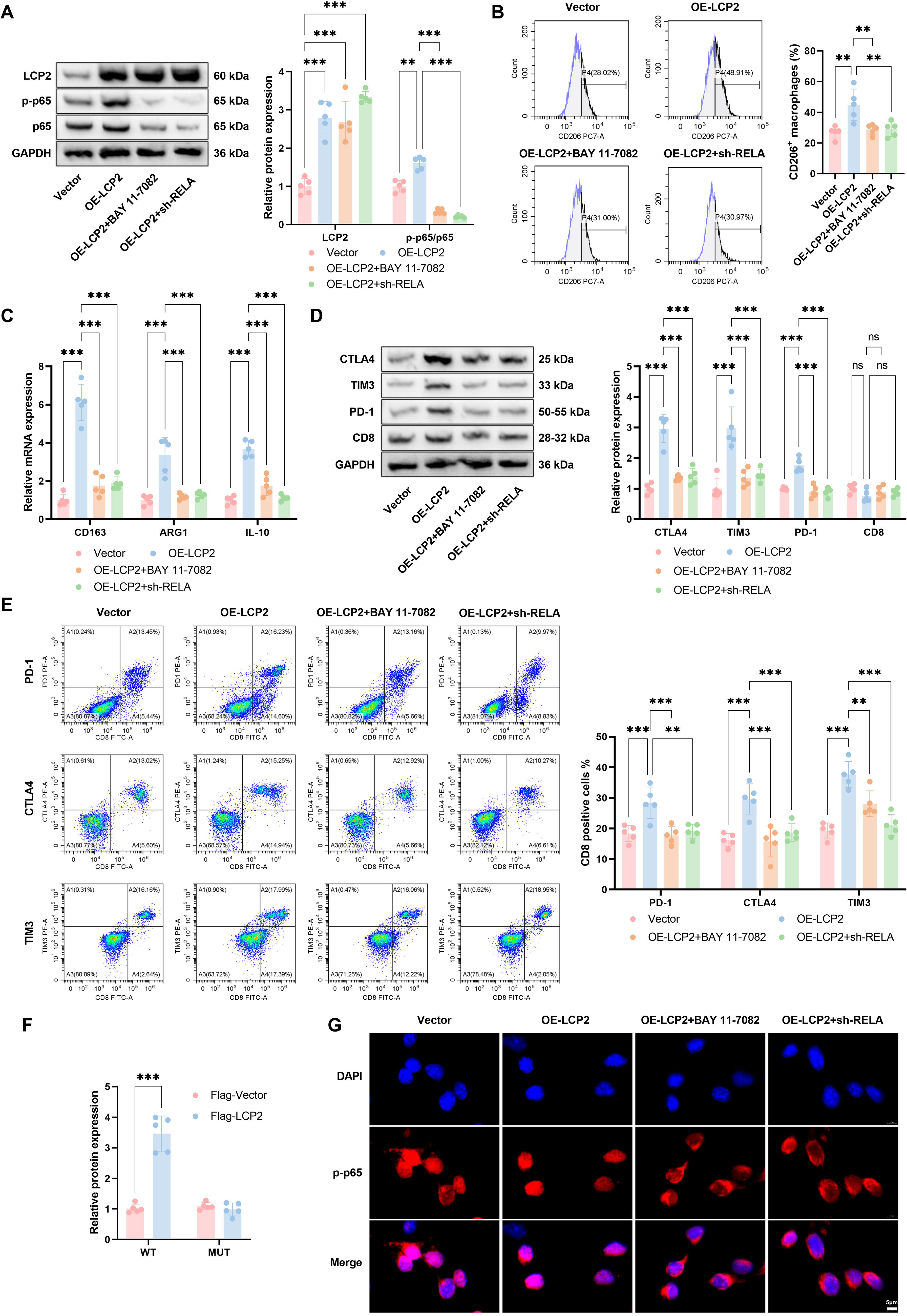
To further validate the causal role of the NF-κB signaling pathway in mediating the biological functions of LCP2, we performed orthogonal genetic validation through targeted knockdown of RELA/p65, the core transcriptional effector of the canonical NF-κB cascade. Western blot analysis confirmed that transduction with short hairpin RNA targeting RELA significantly downregulated total p65 expression and its phosphorylation level in LCP2-overexpressing (OE-LCP2) ESCA cells, without altering exogenous LCP2 expression (Fig. 4A). Functional assays further revealed that RELA knockdown largely abrogated the pro-tumor immunosuppressive phenotype driven by LCP2 overexpression, as evidenced by the abolished promotion of CD206+ M2 macrophage polarization (Fig. 4B), the reversed upregulation of M2 marker genes including CD163, ARG′1, and IL-10 (Fig. 4C), and the eliminated elevation of T cell exhaustion markers PD-1, CTLA-4, and TIM-3 in CD8+ T cells cocultured with OE-LCP2 ESCA cells (Fig. 4D and E). These findings collectively confirmed that the pro-immunosuppressive effects of tumor-intrinsic LCP2 in EpCAM+ tumor fractions are strictly dependent on NF-κB pathway activation, which provides functional evidence for the association between tumor-intrinsic LCP2 and NF-κB signaling, while direct molecular interactions between tumor-intrinsic LCP2 and NF-κB pathway components have not been verified in this study. In parallel, we performed multiple complementary assays to consolidate the direct regulatory effect of LCP2 on NF-κB signaling activation. Luciferase reporter assays demonstrated a significant increase in RELA promoter activity upon overexpression of LCP2 (Fig. 4F). IF staining further showed that LCP2 overexpression significantly promoted p65 nuclear translocation in ESCA cells, an effect that was inhibited by RELA knockdown (Fig. 4G). These results confirm that the pro-immunosuppressive effects of tumor-intrinsic LCP2 are strictly dependent on NF-κB pathway activation and that both pharmacological and genetic blockade of this pathway abolish LCP2-driven immune dysfunction.
LCP2 promotes ESCA progression by shaping an immunosuppressive microenvironment
To establish whether tumor-intrinsic LCP2 drives tumor progression in a CD8+ T cell-dependent manner and validate the functional consequences of tumor-intrinsic LCP2-mediated immunosuppression, we performed functional assays in ESCA-PDOs and an immunocompetent in vivo xenograft model, with targeted CD8+ T cell depletion to confirm the immune dependence of tumor-intrinsic LCP2’s protumor effects. Organoid experiments revealed significantly reduced organoid formation (Fig. 5A), smaller size (Fig. 5B), and decreased viability (Fig. 5C) in tumor-intrinsic LCP2-KO EpCAM+ tumor fractions compared with control groups. However, upon addition of anti-CD8 antibody (tumor-intrinsic LCP2-KO+ anti-CD8), the number, area, and viability of organoids significantly increased compared with the tumor-intrinsic LCP2-KO group alone. Western blot analysis showed markedly elevated cleaved-caspase3/caspase3 and cleaved-caspase7/caspase7 ratios—established apoptosis markers—in tumor-intrinsic LCP2-KO cells, effects that were substantially reduced following anti-CD8 antibody treatment (Fig. 5D). The xenograft mouse model further corroborated these results, demonstrating significantly reduced tumor weight in the LCP2-KO group compared with controls, an effect that was reversed by anti-CD8 antibody treatment (Fig. 5E). Histological analysis indicated increased Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling (TUNEL)-positive (apoptotic) cells in LCP2-KO specimens, accompanied by reduced Ki67-positive (proliferating) and increased cleaved-caspase3-positive cell proportions; these indicators shifted in the opposite direction after anti-CD8 treatment (Fig. 5F). Mechanistically, p65 phosphorylation was markedly suppressed in tumor-intrinsic LCP2-KO tumors but restored following anti-CD8 antibody administration (Fig. 5G). Flow cytometry indicated a substantial increase in CD8+ T cell infiltration within the TME of LCP2-KO mice, which was significantly attenuated by anti-CD8 treatment (Fig. 5H). Western blot (Fig. 5I) and ELISA (Fig. 5J) analyses demonstrated reduced expression of immune checkpoint molecules (PD-1, PD-L1, CTLA-4) in tumor-intrinsic LCP2-KO tumors, alongside elevated levels of sorted CD8+ T cell-derived effector cytokines (GZMB, IL-2, tumor necrosis factor-α [TNF-α], interferon-γ [IFN-γ]). After anti-CD8 treatment, expression of immune checkpoint molecules partially recovered (though still lower than control), and effector cytokine levels significantly decreased (though still higher than control). These results show that the antitumor effect of tumor-intrinsic LCP2 knockout is required for functional CD8+ T cells.

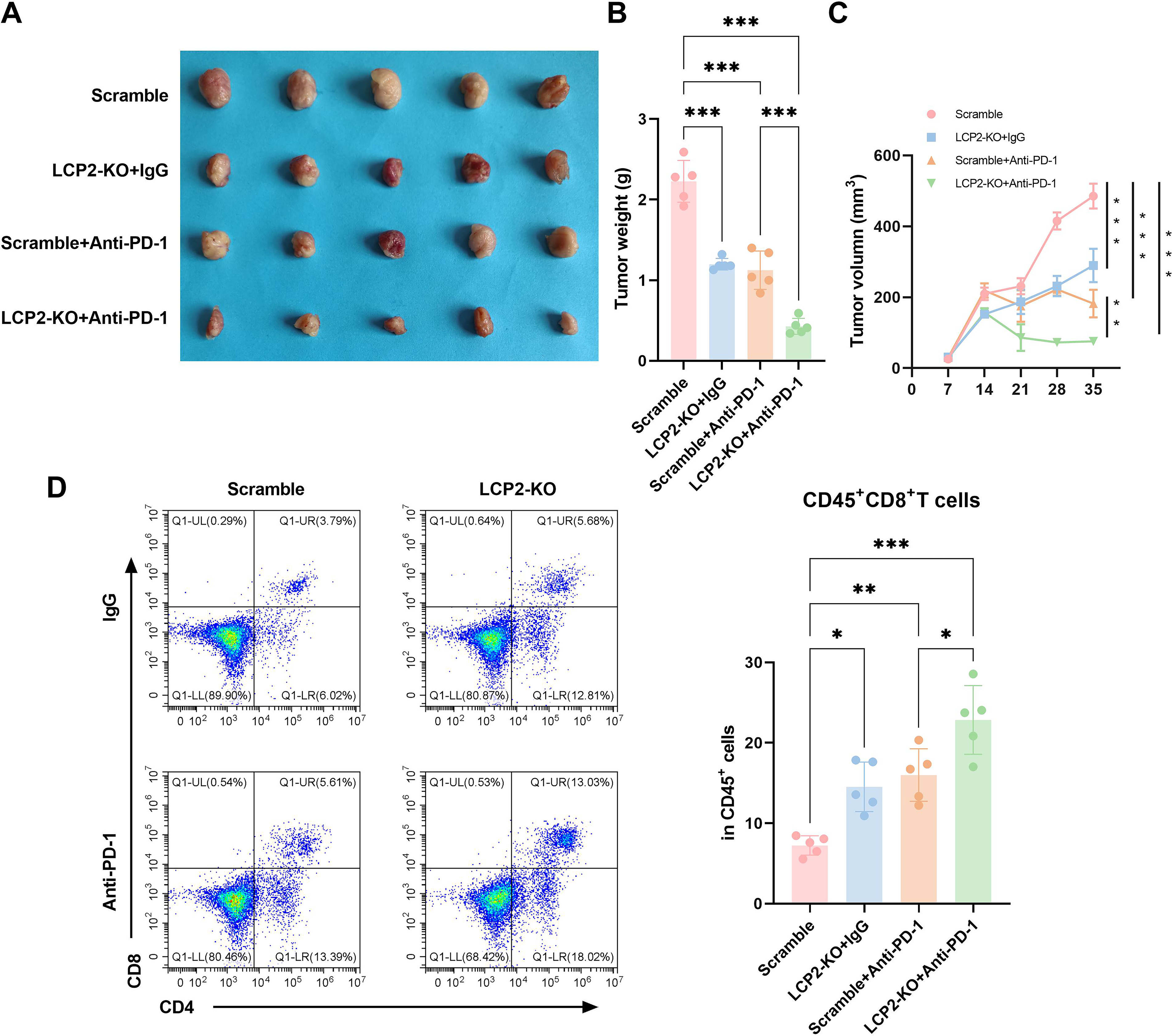
Knockdown of LCP2 enhances the sensitivity of ESCA xenografts to anti-PD-1 therapy
To test our central hypothesis that targeting tumor-intrinsic LCP2 can overcome PD-1 resistance in ESCA, we evaluated the combinatorial efficacy of tumor-intrinsic LCP2 knockout and anti-PD-1 immunotherapy in an immunocompetent syngeneic mouse model, which retains a fully functional adaptive immune system required to assess immunotherapy response. In the immunocompetent C57BL/6J mouse syngeneic xenograft models (Fig. 6A), tumor volume was significantly smaller in the tumor-intrinsic LCP2-KO + IgG group compared with Scramble controls. Both Scramble + Anti-PD-1 and tumor-intrinsic LCP2-KO + Anti-PD-1 groups showed further reduction, with the most pronounced effect observed in the tumor-intrinsic LCP2-KO + Anti-PD-1 cohort. Quantitative analysis of tumor weight (Fig. 6B) demonstrated the highest mass in Scramble controls, followed by tumor-intrinsic LCP2-KO + IgG, a significant decrease in Scramble + Anti-PD-1, and the lowest weight in tumor-intrinsic LCP2-KO + Anti-PD-1 groups, confirming synergistic antitumor effects between tumor-intrinsic LCP2 knockout and PD-1 inhibition. Tumor growth curves (Fig. 6C) indicated consistently smaller volumes in LCP2-KO + IgG versus Scramble, with Scramble + Anti-PD-1 and tumor-intrinsic LCP2-KO + Anti-PD-1 groups showing further suppression, particularly pronounced in the tumor-intrinsic LCP2-KO + Anti-PD-1 cohort during later phases. Flow cytometric analysis (Fig. 6D) indicated a significantly higher proportion of CD45+CD8+ T cells within tumors in the tumor-intrinsic LCP2-KO + IgG group compared with Scramble controls. This infiltration was further enhanced in both Scramble + Anti-PD-1 and tumor-intrinsic LCP2-KO + Anti-PD-1 groups, reaching maximal levels in the LCP2-KO + Anti-PD-1 cohort. In summary, knockdown of tumor-intrinsic LCP2 significantly enhances the sensitivity of ESCA xenografts to anti-PD-1 immunotherapy.
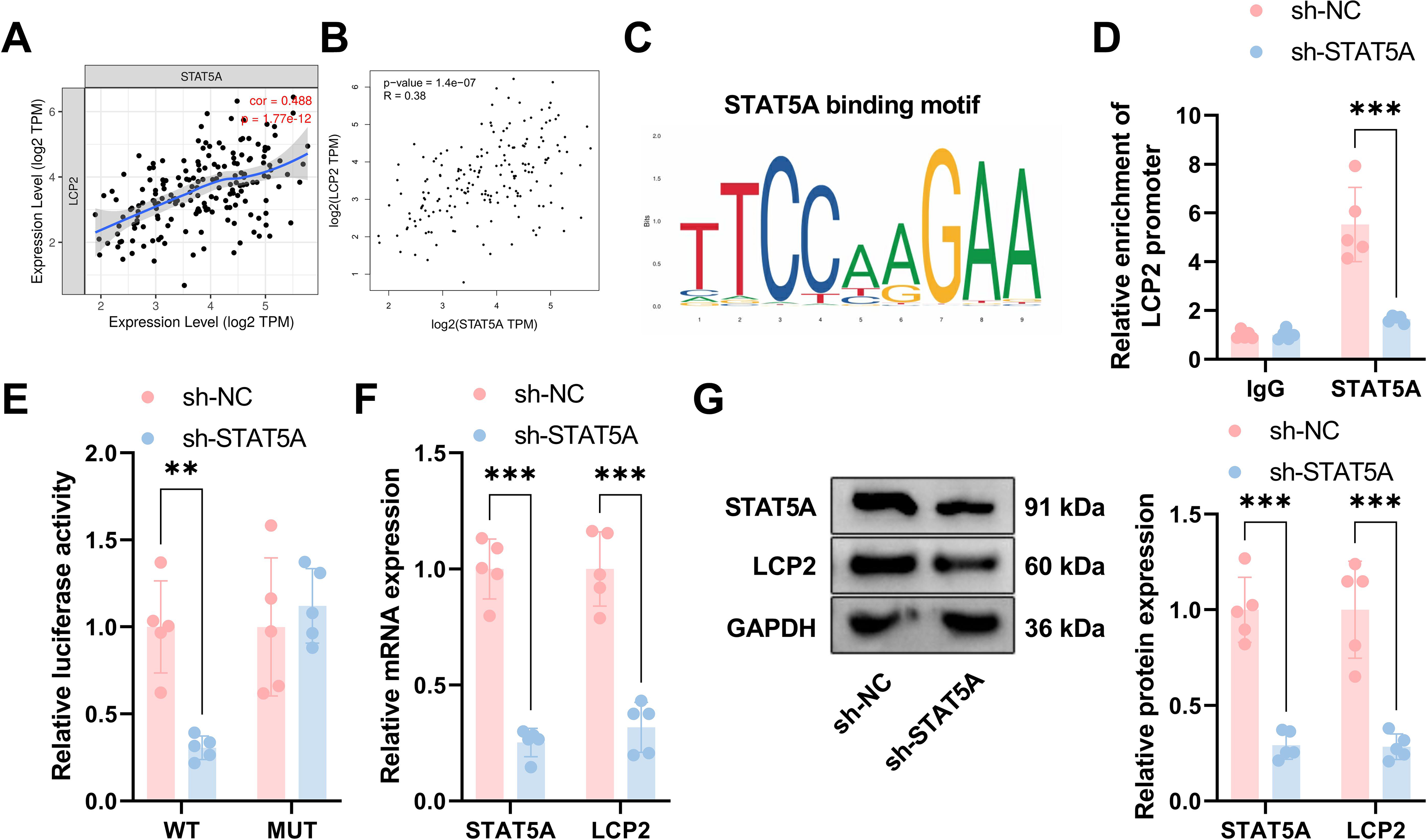
The transcription factor STAT5A directly regulates LCP2 expression
To identify the upstream transcriptional regulator responsible for tumor-intrinsic LCP2 upregulation in EpCAM+ tumor fractions of ESCA, we combined bioinformatic prediction of transcription factor binding sites with mechanistic validation using Chromatin Immunoprecipitation (ChIP) and dual-luciferase reporter assays, which provide gold-standard evidence for direct transcriptional regulation. Analysis from the public database (TCGA) revealed that tumor-intrinsic LCP2 was significantly positively correlated with STAT5A expression (Fig. 7A, correlation coefficient = 0.488, p = 1.77e-12); Figure 7B revealed a positive correlation between tumor-intrinsic LCP2 and STAT5A expression (R = 0.38, p = 1.4e-07). The STAT5A binding motif is TTCCAAGAA (Fig. 7C). ChIP assays demonstrated direct binding of STAT5A to the tumor-intrinsic LCP2 promoter, with significant enrichment in the sh-NC group and markedly reduced enrichment in the sh-STAT5A group (Fig. 7D). We performed dual-luciferase reporter assays to verify the transcriptional regulation of tumor-intrinsic LCP2 by STAT5A. For the wild-type LCP2 promoter construct, STAT5A knockdown significantly reduced relative luciferase activity. For the mutant promoter construct, no significant difference in luciferase activity was observed between the sh-STAT5A and sh-NC groups (Fig. 7E). These results confirm that STAT5A enhances tumor-intrinsic LCP2 promoter activity by binding to this specific sequence. Functional validation demonstrated significantly reduced STAT5A and tumor-intrinsic LCP2 expression at both mRNA (Fig. 7F) and protein (Fig. 7G) levels in sh-STAT5A compared with sh-NC groups. These findings confirm that STAT5A binds the tumor-intrinsic LCP2 promoter and transcriptionally regulates tumor-intrinsic LCP2 expression.
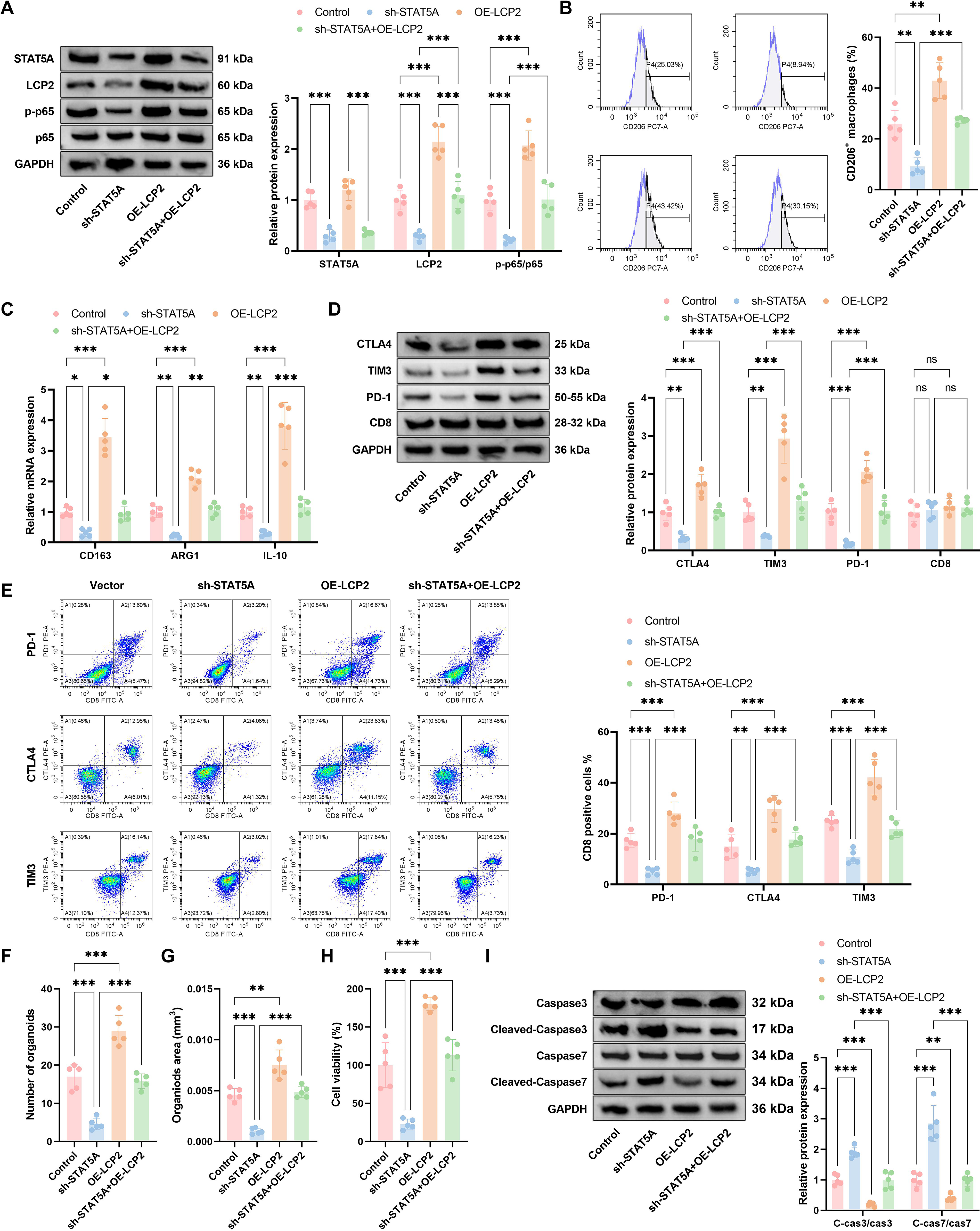
The STAT5A–LCP2–NF-κB axis reshapes the tumor immune landscape and promotes tumor progression
To functionally validate the complete STAT5A–tumor-intrinsic LCP2–NF-κB regulatory axis and confirm that tumor-intrinsic LCP2 acts as the critical intermediate hub for STAT5A-mediated immunosuppression, we performed genetic rescue experiments with STAT5A knockdown and concurrent tumor-intrinsic LCP2 overexpression in both in vitro PDO coculture systems and in vivo xenograft models. Given that sustained activation of STAT5A can induce sorted CD8+ T cell exhaustion (as reported) and promote M2 macrophage polarization (Liu et al., 2025), this study examined the functional significance of the STAT5A–tumor-intrinsic LCP2–NF-κB axis. Western blot analysis (Fig. 8A) revealed decreased STAT5A and tumor-intrinsic LCP2 protein levels, along with reduced p65 phosphorylation, in sh-STAT5A compared with control groups, whereas these markers were elevated in OE-LCP2 cells; and in the sh-STAT5A + OE-LCP2 group, OE-LCP2 partially reversed the downregulation caused by sh-STAT5A. Flow cytometry (Fig. 8B) and qPCR (Fig. 8C) analyses indicated that the proportion of CD206+ macrophages and the expression of M2 markers (CD163, ARG1, IL-10) were significantly reduced in the sh-STAT5A group, significantly increased in the OE-LCP2 group, and partially restored in the sh-STAT5A + OE-LCP2 group. Western blot analysis of fluorescence-activated cell sorting (FACS)-sorted CD8+ T cells from the coculture system (Fig. 8D) and flow cytometric analysis of gated CD45+CD3+CD8+ T cells (Fig. 8E) revealed decreased expression of exhaustion markers (CTLA-4, TIM-3, PD-1) and reduced positive T cell proportions in sh-STAT5A cells (CD8 unchanged), elevated levels in OE-LCP2 groups, and partial restoration in sh-STAT5A + OE-LCP2 cohorts. Organoid assays demonstrated significantly reduced organoid number (Fig. 8F), area (Fig. 8G), and viability (Fig. 8H) in the sh-STAT5A group versus control. Western blot analysis showed increased cleaved-caspase3/caspase3 and cleaved-caspase7/caspase7 ratios in the sh-STAT5A group, whereas the OE-LCP2 group exhibited the opposite trend; all these markers were substantially reversed in the sh-STAT5A + OE-LCP2 group (Fig. 8I).
The STAT5A–
Animal model validation showed that the tumor weight in the sh-STAT5A group was significantly lower than that in the control group (Fig. 9A and B); the OE-LCP2 group was slightly higher than the control group, while the sh-STAT5A + OE-LCP2 group increased compared with the sh-STAT5A group. Figure 9D to F are the quantitative results of TUNEL, Ki67, and cleaved-caspase3 staining in Figure 9C, respectively, showing that the apoptosis rate (TUNEL, Fig. 9D) in the sh-STAT5A group was significantly increased, the proliferation index (Ki67, Fig. 9E) was significantly decreased, and the proportion of cleaved-caspase3-positive cells (Fig. 9F) was significantly decreased. The OE-LCP2 group showed the opposite trend. Compared with the sh-STAT5A group, the sh-STAT5A + OE-LCP2 group had a decreased apoptosis rate and an increased proliferation index, showing a partial recovery effect. Mechanistically, sh-STAT5A cells exhibited reduced p65 phosphorylation (Fig. 9G) and diminished expression of immune checkpoint molecules (PD-1, PD-L1, CTLA-4; Fig. 9J), alongside increased CD8+ T cell infiltration (Fig. 9H and I) and elevated effector cytokine levels (GZMB, IL-2, TNF-α; Fig. 9K). The OE-LCP2 group showed the opposite trend. Compared with the sh-STAT5A group, all indicators in the sh-STAT5A + OE-LCP2 group were significantly reversed. These data validate that the STAT5A–LCP2–NF-κB axis regulates macrophage polarization, T cell function, and ESCA tumor growth in vitro and in vivo (Fig. 1).

Validation of core findings in the additional ESCA cell line Eca109
To validate the robustness and generalizability of our core findings across distinct genetic backgrounds of ESCA, we replicated all key experiments from our PDO models in the well-characterized human ESCA cell line Eca109. We first validated the successful establishment of tumor-intrinsic LCP2-knockdown Eca109 cells via Western blot analysis (Fig. 10A). Consistent with our findings in PDOs, tumor-intrinsic LCP2 knockdown significantly inhibited the phosphorylation of p65 (Fig. 10A). We next validated the regulatory role of tumor-intrinsic LCP2 in the immunosuppressive tumor microenvironment in Eca109 cells. In the Transwell coculture system, LCP2 knockdown in Eca109 cells significantly reduced the proportion of CD206+ M2-polarized macrophages, downregulated the mRNA levels of M2 markers (CD163, ARG1, IL-10), and decreased the secretion of immunosuppressive cytokines IL-10 and TGF-β (Fig. 10B–D). Furthermore, sh-LCP2 significantly reduced the expression of exhaustion markers (PD-1, CTLA-4, TIM-3) on cocultured CD8+ T cells, which was consistent with our findings in PDOs (Fig. 10E and F). We subsequently confirmed the upstream regulatory relationship between STAT5A and tumor-intrinsic LCP2 in Eca109 cells. STAT5A knockdown significantly downregulated LCP2 expression (Fig. 10G). These data validate the robustness of our core findings in the independent ESCA cell line Eca109.
Discussion
In this study, we identify the STAT5A–LCP2–NF-κB axis as a previously unrecognized tumor-intrinsic regulatory cascade that drives PD-1 resistance in ESCA. Specifically, we demonstrate that STAT5A transcriptionally activates LCP2 expression by directly binding its promoter and that tumor-intrinsic LCP2 in EpCAM+ tumor fractions subsequently activates NF-κB signaling to promote M2 macrophage polarization and CD8+ T cell exhaustion. Genetic or pharmacological disruption of this axis suppresses immunosuppressive TME remodeling, restores CD8+ T cell effector function, and synergizes with PD-1 blockade to inhibit ESCA progression in preclinical models.
Biologically, these findings redefine the directionality of immune regulation in ESCA by establishing that malignant epithelial cells actively initiate immunosuppression through a tumor-intrinsic signaling hub, rather than being passive recipients of immune editing. While prior studies have focused on immune cell–intrinsic mechanisms of PD-1 resistance, our work demonstrates that tumor cell–intrinsic LCP2, co-opted from its canonical role as a hematopoietic adaptor, coordinates both M2 macrophage polarization and CD8+ T cell exhaustion via NF-κB pathway activation. This paradigm shift repositions the tumor epithelial cell as the primary architect of immune suppression in ESCA.
Clinically, these findings address a critical unmet need: only ∼20% of ESCA patients achieve durable responses to anti-PD-1 therapy, with resistance driven largely by an immunosuppressive TME (Jin and Zhao, 2023; Ren et al., 2023; Yang et al., 2025; Zhang and Wu, 2025). Our data provide preclinical proof-of-concept that LCP2 targeting can reprogram the TME—reducing M2 macrophage enrichment and T cell exhaustion—to sensitize tumors to PD-1 blockade. Furthermore, the strong association between LCP2 expression and advanced tumor stage, lymph node metastasis, and poor survival supports its potential as both a prognostic biomarker to stratify patients and a tractable therapeutic target to guide combination immunotherapy strategies in ESCA.
The upregulation of tumor-intrinsic LCP2 in ESCA clinical specimens, and its association with aggressive clinicopathological features and poor survival, support its preliminary potential as a candidate prognostic biomarker for ESCA, though definitive validation requires a larger, multicenter cohort. Notably, our IF staining confirmed tumor-intrinsic LCP2 expression in ESCA epithelial cells, ruling out that its upregulation merely reflects immune cell infiltration. The statistical robustness of our survival analysis is not affected by the small sample size of stage IV patients in the TCGA-ESCA cohort, consistent with established analytical standards for ESCA prognostic biomarkers. Although traditionally recognized as an immune adaptor protein critical for TCR-mediated lymphocyte activation, our findings suggest a broader oncogenic role (Abraham et al., 2012). However, the role of LCP2 in solid tumors remains poorly characterized, with scarce evidence implicating its dysregulation in promoting aggressive phenotypes in malignancies such as breast and lung cancers (Hu et al., 2024). These findings establish that the aberrant upregulation of this co-opted immune adaptor LCP2 in ESCA is tightly associated with aggressive clinicopathological features, including advanced tumor stage, high histological grade, and lymph node metastasis. This supports the model wherein LCP2 drives disease progression by functioning as an oncogenic immune modulatory hub to augment the immune evasion capabilities of ESCA cells.
The immunosuppressive state of the TME is a key factor in immune escape and treatment resistance in ESCA, with macrophage polarization and T cell exhaustion being central events in TME immune remodeling (Song et al., 2022). This investigation revealed a positive association between LCP2 expression and M2 macrophage markers. Furthermore, LCP2 knockdown markedly decreased the prevalence of CD206+ macrophages and suppressed immunosuppressive cytokine secretion, suggesting LCP2 facilitates immunosuppressive microenvironment formation by driving M2 polarization. As tumor-promoting macrophages, the M2 phenotype suppresses effector T cell activity through the secretion of IL-10 and TGF-β, while simultaneously facilitating angiogenesis and metastatic progression (Boutilier and Elsawa, 2021). Following LCP2 knockdown, the expression of M2 macrophage functional markers (CD163, ARG1) was significantly downregulated in ESCA-PDOs, an effect that was more substantial under coculture conditions. This suggests LCP2 may either directly modulate macrophage phenotype or indirectly affect polarization through tumor-derived soluble factors. This finding provides a new perspective on understanding the mechanisms underlying M2 macrophage accumulation in ESCA. Furthermore, this study verified a positive association between LCP2 and immune checkpoint molecule expression, demonstrating that LCP2 depletion substantially lowered their levels in T cells. CD8+ T cell exhaustion represents a primary mechanism of immunotherapy resistance in ESCA, marked by persistent overexpression of inhibitory receptors, including PD-1 and CTLA-4, resulting in compromised cytotoxic activity (Mahuron et al., 2025; Zhang et al., 2025). In this study, following LCP2 knockdown, the secretion of cytokines from CD8+ T cells increased, confirming that LCP2 weakens antitumor immunity by inducing T cell exhaustion. In summary, the co-opted tumor-intrinsic immune adaptor LCP2 remodels the immunosuppressive TME through the coordinated dual regulation of M2 macrophage polarization and CD8+ T cell exhaustion, firmly positioning it as a central oncogenic immune modulatory hub governing immune escape and PD-1 resistance in ESCA.
The NF-κB pathway is a core signaling axis regulating the TME; its aberrant activation promotes the expression of immunosuppressive molecules and the secretion of cytokines, thereby contributing to tumor immune escape (Shen et al., 2019). This study is the first to demonstrate that tumor-intrinsic LCP2 in EpCAM+ tumor fractions is associated with the regulation of the immune microenvironment in ESCA, with functional evidence showing that its proimmunosuppressive effects are dependent on NF-κB pathway activation. Mechanistically, as a classical immune adaptor protein, LCP2 exerts its biological functions mainly through protein–protein interactions via its multiple functional domains. In hematopoietic cells, LCP2 has been well documented to form a complex with ζ-chain-associated protein kinase 70 (ZAP70) and GADS, which in turn triggers the phosphorylation and activation of the IKK complex, the core upstream kinase of the NF-κB cascade, ultimately promoting p65 phosphorylation, nuclear translocation, and subsequent NF-κB transcriptional activity (Meri et al., 2025). In ESCA tumor cells, our data confirmed that LCP2 overexpression was positively correlated with enhanced p65 phosphorylation, nuclear translocation, and NF-κB transcriptional activity. Meanwhile, both pharmacological inhibition (via BAY 11-7082) and orthogonal genetic silencing of RELA/p65 consistently abolished LCP2-driven M2 macrophage polarization and CD8+ T cell exhaustion, confirming that the proimmunosuppressive effects of tumor-intrinsic LCP2 are dependent on NF-κB pathway activation. We further validated that LCP2 activates the NF-κB pathway to upregulate the expression of canonical downstream target genes (including immunosuppressive chemokines IL-8, CCL2, and CXCL10), which in turn shape the immunosuppressive tumor microenvironment. This aligns with established findings that LCP2 modulates immune function through NF-κB pathway activation, but our study extends this understanding to the noncanonical function of tumor-intrinsic LCP2 in regulating the solid TME. Notably, the ZAP70-GADS axis may act as the key intermediate mediator of LCP2-induced NF-κB activation in ESCA cells, which warrants further systematic validation in future studies. This aligns with established findings that LCP2 modulates immune function through NF-κB pathway activation (Li et al., 2024), but our study extends this understanding to the regulation of the solid tumor microenvironment. Moreover, current evidence indicates that NF-κB activation upregulates the expression of various molecules, including PD-L1 and IL-10 (Deng et al., 2021). Our discovery that LCP2-mediated upregulation of CTLA-4, TIM-3, and PD-1 depends on NF-κB pathway activation provides new evidence for the role of this pathway in regulating immune checkpoints. Notably, LCP2-induced NF-κB activation leaves CD8 expression unaltered while predominantly upregulating inhibitory molecules, implying that LCP2 contributes to immunosuppression via selective modulation of NF-κB downstream targets. This specificity provides a theoretical basis for targeted interventions—suppressing the LCP2-NF-κB axis may enhance antitumor immunity without damaging CD8+ T cells themselves.
Beyond its classical roles in inflammatory signaling, NF-κB is now recognized as a pivotal redox-sensitive transcriptional node, whose activation state is bidirectionally regulated by reactive oxygen species (ROS). Oxidative modification of IKKβ, NF-κB essential modulator (NEMO), and the p65 subunit—including ROS-dependent phosphorylation of RelA at Ser276 and glutathionylation of IκBα—can either amplify or attenuate NF-κB transcriptional output in a context-dependent manner (Mao et al., 2025). Notably, elevated ROS levels in the tumor microenvironment have been linked to sustained NF-κB activation, which in turn promotes M2 macrophage polarization and immunosuppressive cytokine secretion (Yu et al., 2024). In the context of the STAT5A–LCP2–NF-κB axis described here, we hypothesize that tumor-intrinsic ROS may act as upstream amplifiers of this immunosuppressive cascade: oxidative stress could potentiate STAT5A phosphorylation—consistent with evidence that JAK/STAT signaling is redox-sensitive (Liang et al., 2025)—thereby increasing LCP2 transcription and further driving NF-κB-dependent remodeling of the TME. While this ROS–STAT5A–LCP2–NF-κB coupling remains to be experimentally validated in ESCA, it aligns with the known capacity of the oxidized tumor milieu to sustain immunotherapy resistance. Future studies should incorporate ROS quantification (e.g., CellROX assay, mitochondrial superoxide detection), antioxidant rescue experiments (e.g., N-acetylcysteine treatment), and assessment of oxidative damage markers (8-OHdG, 4-HNE) to determine whether redox regulation acts as an upstream amplifier of LCP2-mediated NF-κB activation and macrophage polarization in ESCA. Such an investigation would firmly position this axis within the redox biology of esophageal cancer immunotherapy resistance. It is important to note that, at present, direct experimental evidence linking ROS levels to STAT5A or LCP2 activity in ESCA cells is lacking in this study. The tumor microenvironment of ESCA is known to be highly oxidized, with elevated ROS levels reported to sustain NF-κB activation and promote M2 macrophage polarization in gastrointestinal cancers (Liang et al., 2025). Whether ROS function as upstream amplifiers of the STAT5A–LCP2–NF-κB cascade in ESCA specifically remains an open and important question for future investigation.
Although we only observed a moderate positive correlation between STAT5A and LCP2 expression in the TCGA-ESCA cohort, ChIP and dual-luciferase reporter assays provided solid direct evidence that the transcription factor STAT5A specifically binds to the LCP2 promoter and transcriptionally activates its expression, thus defining a STAT5A–LCP2–NF-κB signaling axis. This finding provides a direct upstream mechanistic explanation for the abnormal overexpression of LCP2 in ESCA tumor cells. As a pivotal component of the JAK/STAT pathway, STAT5A is commonly dysregulated in malignancies and contributes to processes including cell proliferation and immune evasion through the regulation of downstream target genes (Maninang et al., 2023). Recent studies have shown that STAT5A activation can promote M2 macrophage polarization and T cell exhaustion (Liu et al., 2025). Our study reveals that STAT5A’s regulation of the immune microenvironment is dependent on LCP2: knockdown of STAT5A mimics the phenotypes observed in LCP2 knockdown (e.g., reduced CD206+ macrophages and lower expression of T cell exhaustion markers), while the overexpression of LCP2 partially reverses the effects of STAT5A knockdown. This suggests that STAT5A may exert its immunoregulatory function through LCP2 acting as an “intermediate hub.” Notably, we observed that LCP2 overexpression only partially rescued the immunosuppressive phenotypes induced by STAT5A knockdown, which suggests that STAT5A exerts LCP2-independent biological effects in regulating the TME. As a pleiotropic transcription factor, STAT5A has been reported to directly transcriptionally regulate multiple genes involved in tumor immunosuppression, including PD-L1, TGF-β, and IL-6, in addition to LCP2 (Suske et al., 2024). Furthermore, STAT5A activation in tumor cells can also regulate the expression of chemokines that mediate immune cell recruitment, independently of the LCP2-NF-κB axis (Wu et al., 2025). These LCP2-independent regulatory effects of STAT5A may collectively explain the partial rescue phenotype observed in our study, which also indicates that targeting the STAT5A–LCP2 axis can block the core immunosuppressive cascade, while combined STAT5A inhibition may achieve a more comprehensive immunosuppression reversal effect.
The clinical response rate to PD-1 inhibitors in esophageal cancer remains below 20%. This clinical bottleneck underscores the urgent need to elucidate the mechanisms of immunotherapy resistance (Cheng et al., 2022). In this study, tumor-intrinsic LCP2 knockdown significantly enhanced the sensitivity of ESCA xenografts to anti-PD-1 therapy. This sensitization effect was manifested by reduced tumor volume and increased intratumoral sorted CD8+ T cell infiltration. These findings confirm that LCP2 is associated with PD-1 resistance in ESCA, and its proimmunosuppressive effects are required for the reduced immunotherapy response. Mechanistically, LCP2 depletion downregulates immune checkpoint molecules (e.g., PD-1, CTLA-4) while enhancing CD8+ T cell infiltration and cytokine production, potentially establishing a more permissive microenvironment for PD-1 blockade to alleviate T cell exhaustion. Clinical studies have shown that insufficient CD8+ T cell infiltration or functional exhaustion in the tumor microenvironment are major causes of resistance to PD-1 inhibitors (Shi et al., 2022). The current study demonstrates that LCP2 knockdown improves both of these deficiencies, suggesting that targeting LCP2 may reverse resistance through a “dual effect” (reducing immune checkpoints and enhancing T cell function). In addition, organoid and animal experiments in this study confirm that the antitumor effect of LCP2 knockdown can be partially reversed by anti-CD8 antibody treatment, indicating that its antitumor activity depends on the functional restoration of CD8+ T cells. This further supports that LCP2 operates primarily through immune modulation rather than direct tumor cell proliferation, justifying combined therapeutic strategies. Notably, LCP2 inhibition can be strategically combined with PD-1 blockade or NF-κB inhibitors to maximize clinical efficacy. When paired with PD-1 inhibitors, LCP2 targeting reverses the immunosuppressive TME (reduced M2 polarization and T cell exhaustion) to “prime” tumors for immunotherapy, while PD-1 blockade relieves T cell inhibitory signals—creating a synergistic effect validated in our xenograft model (Fig. 6). Combining LCP2-targeted agents with NF-κB inhibitors (e.g., BAY 11-7082) offers another promising approach: NF-κB inhibition directly blocks downstream immunosuppressive signaling, while LCP2 targeting abrogates upstream pathway activation, forming a “double blockade” of the STAT5A–LCP2–NF-κB axis. Clinically, these combinations could benefit ESCA patients with high LCP2 expression (a potential predictive biomarker) who fail single-agent PD-1 therapy.
This study has certain limitations. First, the clinical validation of LCP2 expression and its correlation with clinicopathological features was performed in a single-center cohort with a relatively small sample size of 27 paired ESCA specimens. Although our functional experiments confirmed that the proimmunosuppressive effects of LCP2 are strictly dependent on NF-κB pathway activation, we did not perform experiments to verify the direct molecular interactions between LCP2 and NF-κB signaling components (e.g., coimmunoprecipitation to detect protein–protein interactions) and thus cannot confirm a direct mechanistic activation of NF-κB by LCP2. The direct molecular mechanism underlying the association between LCP2 and NF-κB pathway activation requires further systematic validation in future studies. The small sample size of our clinical cohort is insufficient to definitively establish the prognostic significance and clinical applicability of LCP2 as a predictive or prognostic biomarker for ESCA patients. The clinical relevance of LCP2 needs to be systematically verified in a large-scale, multicenter, prospective clinical cohort in future studies. Second, although organoid and animal models can simulate the tumor microenvironment, they still differ from the complex human immune network, and further validation using patient-derived xenograft models is warranted. Third, the precise molecular details of how LCP2 regulates the NF-κB pathway have not been fully elucidated. Notably, NF-κB is a well-recognized redox-sensitive transcription factor, and oxidative stress has been reported to modulate NF-κB activation to regulate macrophage polarization and immunosuppressive microenvironment formation in ESCA. However, the current study focuses on the regulatory effect of the STAT5A–LCP2–NF-κB axis on macrophage polarization and PD-1 resistance, and the potential crosstalk between this axis and oxidative stress/redox signaling has not been experimentally verified. Fourth, our findings are based on organoid and xenograft models, which do not fully recapitulate clinical tumor heterogeneity. Furthermore, the translational potential of LCP2-targeted therapies requires confirmation in prospective clinical trials to assess safety and efficacy in ESCA patients.
Future directions prioritize the following key areas: (1) the development of LCP2 as a predictive biomarker, with prospective cohorts validating whether high LCP2 expression correlates with PD-1 inhibitor response to stratify patients for combination therapy; (2) further preclinical trials of LCP2-targeted agents (e.g., small-molecule inhibitors, siRNA) in combination with PD-1 or NF-κB inhibitors to optimize dosing strategies and safety profiles; (3) systematic investigation of the crosstalk between the STAT5A–LCP2–NF-κB axis and oxidative stress/redox signaling, including ROS detection, antioxidant rescue experiments, and validation of oxidative damage markers, to elucidate whether redox regulation acts as an upstream mechanism of LCP2-mediated NF-κB activation and macrophage polarization in ESCA; (4) the launch of clinical trials to evaluate the efficacy of LCP2-targeted combinatorial regimens in ESCA patients with PD-1 resistance, translating preclinical findings into actionable therapeutic options.
In summary, our study identifies the STAT5A–LCP2–NF-κB axis as a key mediator of PD-1 resistance in ESCA, which is associated with immunosuppressive TME remodeling via M2 macrophage polarization and CD8+ T cell exhaustion that depend on axis activation. These findings suggest LCP2 as a promising candidate prognostic biomarker and a therapeutic target for overcoming anti-PD-1 resistance in ESCA, providing a preclinical rationale for LCP2-targeted combination immunotherapy.
Materials and Methods
Database analysis
LCP2 mRNA expression in various cancer types was assessed using the TCGA Pan-Cancer Atlas via the TIMER2.0 web platform (http://cistrome.org/TIMER). The UALCAN database (http://ualcan.path.uab.edu) was employed to assess LCP2 expression in ESCA versus normal tissues and its correlation with tumor stage, lymph node metastasis, and pathological grade. LCP2 mRNA data from the TCGA-ESCA cohort were retrieved using GEPIA2 (http://gepia2.cancer-pku.cn). Patients were stratified into high- and low-expression groups based on median expression. Kaplan–Meier survival curves were constructed, and differences between groups were assessed with the log-rank test.
Clinical sample collection
Surgical specimens from 27 treatment-naive ESCA patients who underwent radical surgery in our hospital between January 2022 and December 2024 were collected, including paired primary tumor tissues and matched adjacent noncancerous esophageal tissues (>5 cm from the tumor margin). All patients were confirmed with ESCA by postoperative pathological examination. None of the patients received neoadjuvant radiotherapy, chemotherapy, or immunotherapy before surgery. Three representative paired samples were randomly selected for Western blot validation, while all 27 paired specimens were used for qRT-PCR analysis of LCP2 expression. This study was approved by the Medical Ethics Committee of Shaanxi Provincial People’s Hospital (No. 2025R064), and written informed consent was obtained from all enrolled patients. The collection and analysis of human clinical samples were conducted in compliance with the Strengthening the Reporting of Observational Studies in Epidemiology guidelines, ensuring the standardization and reproducibility of clinical data.
Cell and organoid culture
Cell lines: The human ESCA line Eca109 was acquired from the Cell Bank of the Chinese Academy of Sciences. Cells were maintained in Roswell Park Memorial Institute 1640 (RPMI-1640) medium (Gibco) containing 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 humidified atmosphere.
ESCA-PDO: Fresh ESCA tumor specimens were minced and enzymatically digested in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) medium containing 2 mg/mL collagenase IV (Sigma) for 1 h at 37°C. After filtration to generate a single-cell suspension, cells were embedded in Matrigel (Corning) and seeded into 24-well plates. Following polymerization, complete ESCA-PDO culture medium based on advanced DMEM/F12 (Gibco) was added, supplemented with the following components at their final concentrations: 1 × B27 Supplement minus vitamin A (Gibco, cat. no. 12587010), 1 × N2 Supplement (Gibco, cat. no. 17502048), 50 ng/mL epidermal growth factor (PeproTech), 50 ng/mL fibroblast growth factor 10 (PeproTech), 100 ng/mL Noggin (PeproTech), 500 ng/mL R-spondin 1 (PeproTech), 10 μM Y-27632 (PeproTech), 1.25 mM N-acetylcysteine (Sigma-Aldrich), 10 nM gastrin I (Sigma-Aldrich), 500 nM A83-01 (Tocris), and 100 U/mL penicillin + 100 μg/mL streptomycin (Gibco). The medium was refreshed every 2 to 3 days, and organoids were subcultured weekly. The medium was refreshed every 2 to 3 days, and organoids were subcultured weekly. BAY 11-7082 (Selleck), an NF-κB inhibitor, was prepared in dimethyl sulfoxide and applied at 10 μM. Cells or organoids were exposed to the compound 48 h after transfection and harvested 24 h later for further analysis.
Plasmid construction and cell transfection
Tumor-intrinsic LCP2 Knockout (LCP2-KO): A single-guide RNA (sgRNA) targeting LCP2 (NM_005560.4; sequence: 5′-GCTGATGTTGGTGCTGATGA-3′) was designed and inserted into the pSpCas9(BB)-2A-Puro vector (Addgene) for knockout in EpCAM+ ESCA tumor fractions. Following sequence confirmation, the plasmid was introduced into ESCA-PDO or Eca109 cells via Lipofectamine 3000 (Invitrogen). Forty-eight hours after transfection, cells were selected with 2 μg/mL puromycin. Knockout efficiency was verified via Western blot, using a nontargeting sgRNA vector (Scramble) as the control.
LCP2 Overexpression (OE-LCP2): The full-length LCP2 coding sequence was PCR-amplified and cloned into the pcDNA3.1 vector (Invitrogen) to generate the overexpression construct. The empty vector served as the control (vector). Transfection was performed as previously described, and overexpression efficiency was confirmed by qRT-PCR and Western blot.
STAT5A Knockdown (sh-STAT5A): short hairpin RNA (shRNA) directed against STAT5A (sequence: 5′-GATCCGGTGAAGATGTAGACCTTAATTCAAGAGATTAAGGTCTACATCTTCACCCTTTTTG-3′) was synthesized and cloned into the pLKO.1 vector (Addgene). Lentiviral particles were produced and used for cellular infection, followed by selection of stable knockdown lines with 2 μg/mL puromycin. A nontargeting shRNA construct (sh-NC) was used as control. Knockdown efficiency was evaluated via qRT-PCR and Western blot analysis.
RELA/p65 Knockdown: shRNA targeting human RELA (encoding p65, sequence: 5′-CCGGAGAGGACATTGAGGTGTATTTCTCGAGAAATACACCTCAATGTCCTCTTTTTTG-3′) was synthesized and cloned into the pLKO.1 vector (Addgene). Lentiviral particles were produced and used to infect ESCA-PDO cells, followed by selection with 2 μg/mL puromycin. A nontargeting shRNA (sh-NC) was used as the negative control, and knockdown efficiency was verified via qRT-PCR and Western blot.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from tissues or cells using TRIzol reagent (Invitrogen), followed by cDNA synthesis with the PrimeScript RT Kit (TaKaRa). qRT-PCR was performed using SYBR Green PCR Master Mix (TaKaRa) on a CFX96 Real-Time PCR System (Bio-Rad), normalizing to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The following primer sequences were employed:
LCP2: F 5′-AGAAAGCCACGAAGAGGACA-3′, R 5′-GAGCTTCCTCGTCATTGGAG-3′
CD163: F 5′-TGGAGGAACAGACAAGG-3′, R 5′-GATCCATCCAAATGCGT-3′
ARG1: F 5′-ATTGAGAAAGGCTGGTCTGC-3′, R 5′-CATTAGGGATGTCAGCAAAGG-3′
IL-10: F 5′-AAGGACCAGCTGGACAACAT-3′, R 5′-AGACACCTTTGTCTTGGAGCTTA-3′
STAT5A: F 5′-AAATGGCGGGCTGGATCCAGG-3′, R 5′-AGCGTGGGATTCAAACATTC-3′
GAPDH: F 5′-GCACCGTCAAGGCTGAGAAC-3′, R 5′-TGGTGAAGACGCCAGTGGA-3′
Relative gene expression was calculated using the 2−ΔΔCT method.
Western blot
Total protein was extracted from tissues or cultured cells and quantified using a bicinchoninic acid assay. Proteins (30 μg per lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred electrophoretically to polyvinylidene fluoride membranes (Millipore). Following a 1-h block with 5% nonfat milk at room temperature, membranes were incubated with primary antibodies overnight at 4°C. Following washes, horse radish peroxidase-conjugated secondary antibodies were applied for 1 h at room temperature. Blots were developed using an enhanced chemiluminescence detection kit (Millipore), and band intensities were quantified with ImageJ software. β-Actin was used as the loading control for normalization. The following antibodies were employed: LCP2 (Abcam, ab124933), CD163 (CST, #93498), CTLA-4 (CST, #11179), TIM-3 (CST, #45208), PD-1 (CST, #86163), CD8 (CST, #98941), p65 (CST, #8242), p-p65 (CST, #3033), cleaved-caspase3 (CST, #9664), caspase3 (CST, #9662), cleaved-caspase7 (CST, #8438), Caspase7 (CST, #12827), STAT5A (CST, #9363), β-actin (CST, #4970), and goat antirabbit IgG secondary antibody (CST, #7074).
Enzyme-linked immunosorbent assay
Cell culture supernatants and tumor tissue homogenates were collected, and the levels of TGF-β (R&D, DY240), IL-10 (R&D, DY217B), GZMB (R&D, DY1860), IL-2 (R&D, DY202), TNF-α (R&D, DY210), and IFN-γ (R&D, DY285) were quantified following the manufacturer’s protocols. Absorbance was measured at 450 nm using a microplate reader (Bio-Rad), and standard curves were employed to calculate analyte concentrations.
Flow cytometry
Cell suspensions from tumors or single-cell preparations were washed with phosphate-buffered saline (PBS) and stained with fluorescent-conjugated antibodies for 30 min at 4°C in the dark. Following two PBS washes, samples were acquired on a BD FACSCanto II flow cytometer. Antibodies used included: CD206-PE (BD, 562122), PD-1-APC (BD, 564424), CTLA-4-FITC (BD, 555852), TIM-3-PE-Cy7 (BD, 564684), CD45-APC-Cy7 (BD, 560583), and CD8-PE (BD, 555366). The proportion of positive cells was determined using FlowJo software.
Complete gating strategy: All flow cytometry assays followed a standardized gating workflow: (1) FSC-A/SSC-A gating to exclude debris and select the total cell population; (2) FSC-H/FSC-A gating to exclude doublets and select single cells; (3) Live/dead cell discrimination via Zombie NIR dye (BioLegend) staining to select viable cells; (4) For macrophage analysis: CD45+ cell gating followed by CD206+ cell quantification; (5) For T cell analysis: CD45+CD3+ cell gating followed by CD8+ cell subset gating and subsequent quantification of PD-1+, CTLA-4+, and TIM-3+ cells within the CD8+ T cell population.
Animal experiments
Syngeneic xenograft model: Six- to eight-week-old male immunocompetent C57BL/6J mice (SPF grade, cat. no. 219, Beijing Vital River Laboratory Animal Technology Co., Ltd.) were used for this study. This strain has a complete adaptive immune system with functional mature CD3+CD8+ cytotoxic T lymphocytes, which is a standardized model for evaluating anti-PD-1 immunotherapy efficacy and T cell-dependent antitumor immune mechanisms in esophageal cancer (Yang et al., 2024b). Mice were randomly allocated to experimental groups (n = 5 per group) and inoculated subcutaneously in the right flank with ESCA-PDOs to establish the immunocompetent tumor-bearing model. Tumor volume (length × width2/2) was measured every 3 days from day 7 after inoculation. Mice were euthanized on day 28 after inoculation, and tumors were excised, weighed, and prepared for subsequent flow cytometry, histopathological, and molecular biology analyses. The entire experimental timeline is as follows: day 0: cell inoculation; day 7: start of antibody administration and tumor measurement; day 28: euthanasia and sample collection.
CD8+ T cell depletion experiment: Beginning 7 days after cell inoculation, mice were intraperitoneally injected with antimouse CD8α neutralizing antibody (clone 2.43, cat. no. BE0061, BioXCell, West Lebanon, NH, USA; 200 μg/mouse, twice weekly) to achieve specific depletion of functional CD8+ T cells in vivo. Mice in the control group received an equivalent dose of rat IgG2b isotype control (cat. no. BE0090, BioXCell). The depletion efficiency of CD8+ T cells in peripheral blood was verified via flow cytometry 72 h after the first antibody administration, with a depletion rate >95% considered as successful modeling (Najar et al., 2026).
Anti-PD-1 immunotherapy experiment: From day 7 after cell inoculation, mice were intraperitoneally injected with antimouse PD-1 monoclonal antibody (clone RMP1-14, cat. no. BE0146, BioXCell; 200 μg/mouse, twice weekly), which is a species-matched, functionally validated reagent for C57BL/6J mouse models. Mice in the control group received an equivalent dose of rat IgG2a isotype control (cat. no. BE0089, BioXCell). Tumor volume was measured every 3 days until the end of the experiment on day 28.
All procedures involving animals were approved by Shaanxi Provincial People’s Hospital Medical Ethics Committee. All animal experiments were designed and reported in accordance with the Animal Research: Reporting of In Vivo Experiments guidelines to ensure the rigor and transparency of in vivo data.
Histopathological examination
TUNEL Staining: Paraffin-embedded tumor sections were deparaffinized and rehydrated. Apoptotic cells were identified via TUNEL assay (Roche TUNEL Kit) according to manufacturer protocols. Signal development employed DAB, with subsequent hematoxylin nuclear counterstaining. The percentage of TUNEL-positive cells was quantified in five random fields per sample with Image-Pro Plus software.
Immunohistochemistry: Following deparaffinization, rehydration, and antigen retrieval, tissue sections were treated with 3% H2O2 for 10 min to quench endogenous peroxidase and then blocked with 5% BSA for 1 h. Primary antibodies against Ki67 (Abcam, ab15580) and cleaved-caspase3 (CST, #9664) were applied and incubated overnight at 4°C. After washing, a secondary antibody was applied for 30 min, followed by DAB development and hematoxylin counterstaining. Positive cells were counted in five random fields per section using Image-Pro Plus for quantitative analysis.
p65 Nuclear Translocation IF Staining: ESCA-PDO cells were seeded on glass coverslips, fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 for 10 min, and blocked with 5% BSA for 1 h at room temperature. Cells were incubated with anti-p65 primary antibody (CST, #8242) overnight at 4°C, followed by incubation with Alexa Fluor 594-conjugated secondary antibody (Invitrogen) for 1 h at room temperature in the dark. Nuclei were counterstained with 4,6-diamino-2-phenyl indole (DAPI). Images were captured using a laser scanning confocal microscope (Zeiss LSM 880). The nuclear translocation rate of p65 was quantified as the percentage of cells with p65 fluorescence predominantly localized in the nucleus, with at least five random fields per sample analyzed.
Tumor organoid-immune cell coculture system
Immune cell isolation and preparation: Human peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood of healthy donors via density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare). CD14+ monocytes were sorted from PBMCs using CD14 MicroBeads (Miltenyi Biotec) for macrophage differentiation, while CD8+ T cells were sorted using CD8 MicroBeads (Miltenyi Biotec) for functional assays. All donor sample collection was approved by the Medical Ethics Committee of Shaanxi Provincial People’s Hospital (No. 2025R064), with written informed consent obtained from all donors.
Macrophage differentiation and polarization: Sorted CD14+ monocytes were cultured in RPMI-1640 medium supplemented with 10% FBS and 50 ng/mL recombinant human M-CSF (PeproTech) for 7 days to induce differentiation into M0 macrophages. For M2 polarization, M0 macrophages were further treated with 20 ng/mL recombinant human IL-4 and 20 ng/mL IL-13 (PeproTech) for 48 h, with M2 phenotype validated via flow cytometry detection of CD206 expression.
Coculture system setup: Tumor cells were seeded in the upper chamber of 0.4 μm Transwell inserts (Corning), with immune cells seeded in the lower chamber at the same 1:3 ratio, for 72 h of coculture to detect tumor cell-derived paracrine effects on immune cell phenotype.
Cell sorting for molecular assays: After coculture, cells were harvested and sorted into two fractions using FACS: EpCAM+ tumor fractions (stained with EpCAM-PE, BD, 555799) and CD45+ immune cells (stained with CD45-APC-Cy7, BD, 560583). Sorted cell purity was verified to be >95% for both fractions via postsort flow cytometry analysis. All Western blot assays for immune-related markers were performed on sorted CD8+ T cells and EpCAM+ tumor fractions separately, not mixed coculture populations.
Organoid functional assays
Organoid Number and Area Measurement: Following 7 days in culture, organoids were imaged with an inverted microscope (Olympus). The count of organoids per field was recorded, and their cross-sectional area was quantified using ImageJ software. Analysis included at least three replicate wells per group, with five fields evaluated per well.
Cell Viability Assay: Viability was assessed using a Cell Counting Kit-8 (CCK-8) kit (Dojindo). Briefly, 10 μL of CCK-8 reagent was added to each well and incubated for 2 h at 37°C, after which absorbance was measured at 450 nm with a microplate reader. Viability was expressed as: (OD experimental/OD control) × 100%.
ChIP assay
Chromatin from ∼1 × 107 Eca109 cells was cross-linked using formaldehyde and fragmented via sonication. Sheared chromatin was immunoprecipitated overnight at 4°C using an anti-STAT5A antibody (CST, #9363) or a control IgG. Protein A/G magnetic beads (Thermo) were used to capture immune complexes, which were then washed and eluted to isolate bound DNA. Enrichment of the LCP2 promoter in immunoprecipitated DNA was quantified via qPCR with the primers:
Forward 5′-TGTGGTGGTGGTGTTGTTGT-3′, Reverse 5′-CCTGCTGCTGCTGTTGTTCT-3′.
Dual-luciferase reporter assay
Wild-type (WT) and mutant (MUT) LCP2 promoter reporter constructs were created in the pGL3-Basic vector (Promega). The STAT5A binding site (TTCCAAGAA) was mutated to TTGGTCCAA in the MUT construct. Eca109 cells were cotransfected with sh-STAT5A or a nontargeting control (sh-NC), a reporter plasmid, and the pRL-TK Renilla luciferase vector. Luciferase activity was measured 48 h posttransfection using the Dual-Luciferase Reporter Assay System (Promega), with promoter activity expressed as the firefly-to-Renilla luminescence ratio.
Statistical analysis
Statistical analyses were conducted using SPSS 26.0 (IBM, USA) and GraphPad Prism 9.0 (GraphPad Software, USA). Continuous variables are presented as mean ± standard deviation (SD), and all error bars in the figures represent SD. For all experiments, five independent biological replicates were performed, with three technical replicates for each biological replicate unless otherwise specified. Normality and variance homogeneity tests: The Shapiro–Wilk test was used to assess the normal distribution of all continuous datasets, and Levene’s test was performed to evaluate the homogeneity of variance between groups. Two-group comparisons: For datasets that met both normal distribution and homogeneity of variance assumptions, intergroup differences were assessed by two-tailed independent samples t-test; for datasets that did not meet the parametric test assumptions, the nonparametric two-tailed Mann–Whitney U test was applied. Multigroup comparisons: For datasets that met parametric test assumptions, one-way analysis of variance was performed, followed by LSD post hoc test for pairwise comparisons with homogeneity of variance and Dunnett’s T3 post hoc test for pairwise comparisons with unequal variance; for datasets that did not meet parametric test assumptions, the nonparametric Kruskal–Wallis H test was used, with Dunn’s post hoc test for pairwise comparisons. Bonferroni correction was applied for multiple pairwise comparisons to control the family-wise error rate. Survival and correlation analysis: Kaplan–Meier survival curves were compared by the log-rank test, and univariable and multivariable Cox regression analyses were performed to identify independent prognostic factors. Pearson’s correlation coefficient was used for correlation analysis of gene expression levels with normally distributed data, while Spearman’s rank correlation coefficient was applied for non-normally distributed data. Sensitivity analysis: Sensitivity analyses were performed by alternating parametric and nonparametric tests for all core endpoint datasets to verify the robustness of our statistical results and core conclusions. A two-tailed p value <0.05 was considered statistically significant for all analyses. All statistical analysis workflows were designed in compliance with international preclinical cancer research statistical reporting guidelines.
A priori power analysis was conducted using G*Power 3.1 software to determine the appropriate sample size. Based on our primary endpoints (protein expression levels and functional assays), with a significance level α = 0.05 and statistical power = 0.8, the minimum required sample size was calculated. The sample size used in this study meets the basic statistical requirements and ensures the reliability of the experimental results.
Electronic laboratory notebook was not used.
Footnotes
Acknowledgments
The authors would like to thank the technical support from the Department of Otorhinolaryngology Head and Neck Surgery, Shaanxi Provincial People’s Hospital.
Author Disclosure Statement
The authors declare no conflicts of interest. No ghostwriters were used in the preparation of this article. All authors have made substantial contributions to the conception, design, data acquisition, analysis, and interpretation of the study and have approved the final version for submission.
Funding Information
This study was supported by the
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.