
Abstract
Aims:
Doxorubicin (DOX) remains a cornerstone of cancer therapy but is limited by dose-dependent cardiotoxicity with inadequate protective strategies. Nerve injury-induced protein 1 (NINJ1), a regulator of inflammation and cell death, has not been explored in this context. We sought to define the role of NINJ1 in DOX-induced cardiotoxicity and evaluate its translational potential.
Results:
Using complementary genetic, pharmacologic, and transcriptomic approaches, we demonstrate that NINJ1 is markedly upregulated in DOX-treated murine hearts and cardiomyocytes. Cardiomyocyte-specific NINJ1 deletion confers robust protection against cardiac dysfunction, oxidative stress, and apoptosis, whereas NINJ1 overexpression exacerbates injury. Mechanistically, NINJ1 suppresses AMP-activated protein kinase (AMPK) activation, promoting ubiquitin-mediated degradation of hypoxia-inducible factor-1α (HIF-1α), thereby impairing antioxidant gene programs. Multilevel evidence, including RNA sequencing, pathway enrichment, and gain- and loss-of-function models, establishes the NINJ1-AMPK-HIF-1α axis as a central regulator of redox homeostasis. Pharmacologic inhibition of NINJ1 with phenyl-β-D-glucopyranoside attenuates cardiac injury in vivo and in vitro without compromising DOX antitumor efficacy, supporting pathway specificity and therapeutic feasibility.
Innovation:
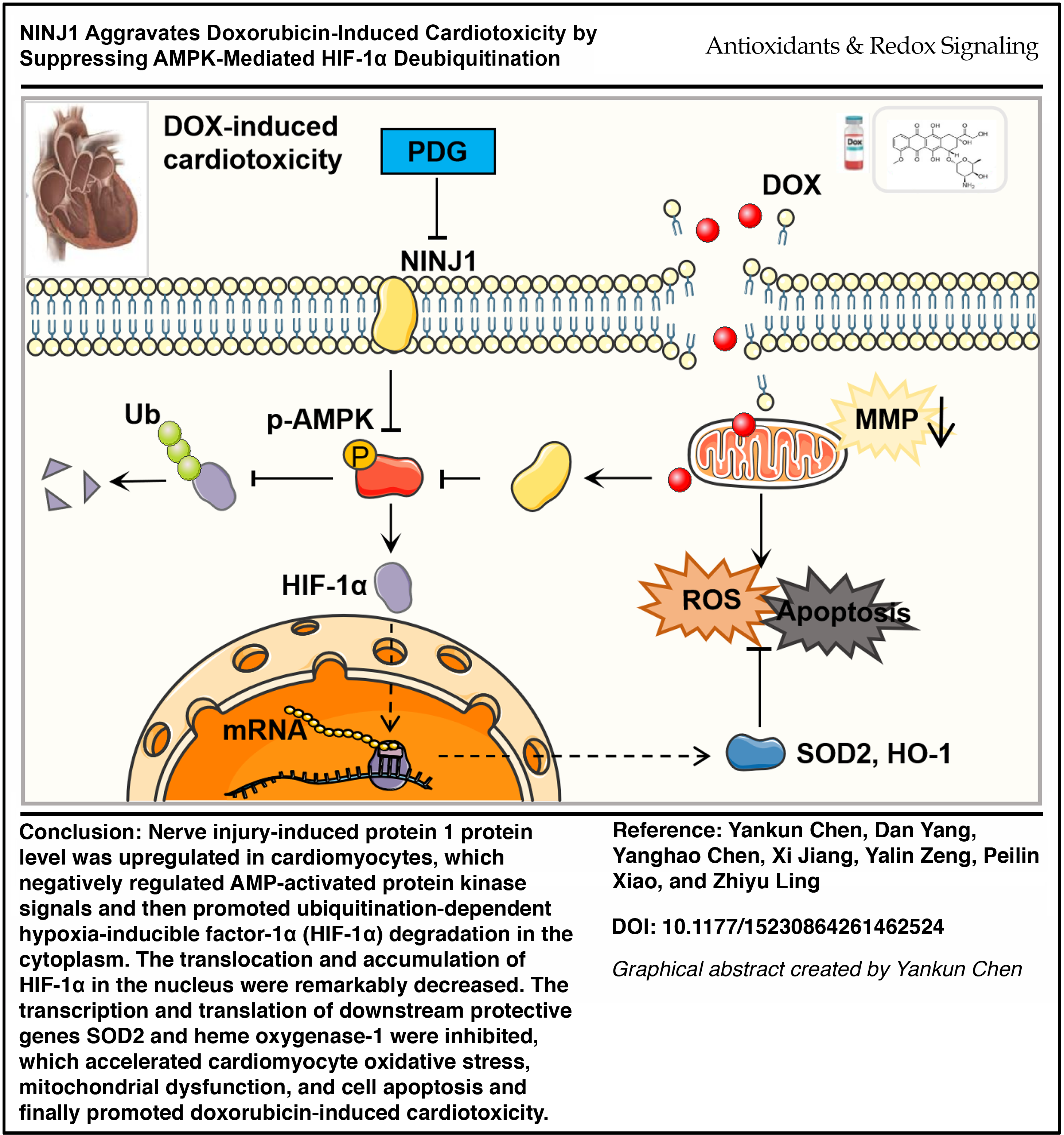
This study identifies NINJ1 as a previously unrecognized driver of anthracycline cardiotoxicity and uncovers a novel signaling axis linking membrane injury signaling to metabolic control of HIF-1α stability.
Conclusions:
NINJ1 promotes DOX-induced cardiotoxicity by destabilizing HIF-1α via AMPK inhibition. Targeting NINJ1 represents a promising cardioprotective strategy.
Clinical Significance:
Therapeutic inhibition of NINJ1 protects the heart while preserving anticancer efficacy, offering a potential strategy to enhance the safety of anthracycline-based chemotherapy and improve outcomes in cancer patients. Antioxid. Redox Signal. 00, 000–000.
Introduction
Doxorubicin (DOX), an anthracycline antibiotic, is among the most frequently prescribed chemotherapeutic agents for the treatment of a broad spectrum of malignancies. Despite its potent antitumor efficacy, the clinical application of DOX is severely limited by cumulative dose-dependent cardiotoxicity (Mohammadi et al., 2020). DOX-associated cardiovascular adverse effects include hypotension, arrhythmias, tachycardia, and most notably, congestive heart failure (Cardinale et al., 2015). It is estimated that 26% of the patients receiving DOX treatment suffer heart failure when the cumulative dose exceeds 550 mg/m2 (Wallace et al., 2020). However, the precise molecular mechanisms underlying DOX-induced cardiotoxicity remain incompletely understood, and effective therapeutic targets to prevent or mitigate cardiac injury during DOX chemotherapy are urgently needed. Recently, it is noted that cell apoptosis and oxidative stress were identified as critical targets to attenuate DOX-induced cardiotoxicity.
Nerve injury-induced protein 1 (NINJ1) is a cell-surface adhesion molecule initially identified for its role in promoting axonal growth following peripheral nerve injury(Araki and Milbrandt, 1996; Lee et al., 2010). NINJ1 is widely expressed across various tissues during embryogenesis and adulthood (Araki and Milbrandt, 1996). Among these tissues, the liver exhibits the highest level of NINJ1 transcripts, whereas its expression in the brain is relatively low and restricted primarily to specific glial cell populations (Ekanayake et al., 2019). Notably, appreciable expression levels of NINJ1 have also been detected in the heart, thymus, adrenal gland, and spleen (Araki et al., 1997). Emerging evidence indicates that NINJ1 is a multifunctional protein involved in diverse biological processes, including cell growth, inflammation, apoptosis, and oxidative stress. Recent studies reveal a critical role for NINJ1 in inflammasome-induced plasma membrane rupture (PMR) during ferroptosis, parthanatos, and H2O2-induced necrosis, leading to the release of macromolecules such as lactate dehydrogenase (LDH) and High mobility group box 1 (HMGB1) (Ahn et al., 2009). NINJ1 is involved in inflammatory response, which can enhance macrophage activation and worsen inflammatory lesions in different tissues (Liu et al., 2020). Antibody-mediated inhibition of NINJ1 has been shown to attenuate tissue injury by preventing PMR and fewer release of proinflammatory substances such as LDH, heat-shock protein, and HMGB1 (Kayagaki et al., 2023). Furthermore, Se et al. found that loss of NINJ1 alleviates oxidative stress-induced liver injury via the AMP-activated protein kinase (AMPK)–nuclear factor erythroid 2-related factor 2 (NRF2) signaling pathway (Park et al., 2024). In the context of apoptosis, NINJ1 has been implicated in Bcl-2-associated X protein (BAX)/Bcl-2 homologous antagonist/killer (BAK)-dependent mitochondrial membrane injury and apoptosome formation, particularly when apoptotic cells are inadequately cleared (Ramos et al., 2024). Moreover, Wu and his colleagues determined that NINJ1 is involved in the formation of abdominal aortic aneurysm through blocking Toll-like receptor 4 (TLR4)–Annexin A2 (ANXA2) interaction and enhancing macrophage infiltration (Wu et al., 2024). Accumulating evidence further suggests a potential role for NINJ1 in cardiovascular diseases. Silencing NINJ1 attenuates oscillating glucose-induced inflammatory stress and reactive oxygen species (ROS) production in human endothelial cells (Toma et al., 2023). Moreover, systemic deletion of NINJ1 confers protection against atherosclerosis in mice fed a western diet for 5–23 weeks (Jeon et al., 2020). These findings highlight the involvement of NINJ1 in cardiovascular pathophysiology and prompt further investigation into its specific roles in the heart.
Currently, dexrazoxane is the only protective intervention approved by the Food and Drug Administration (FDA) for the treatment of DOX-induced cardiotoxicity. Dexrazoxane reduces the production of the oxygen-free radicals within cardiomyocytes by binding iron and blocks the formation of DOX-topoisomerase Ⅱb-DNA complex, preventing the double-strand DNA break-mediated cell death (Upshaw et al., 2024). However, the therapeutic effect is not satisfactory due to insufficient clearance of endogenous ROS and the induction of secondary reactions that activate the downstream apoptosis pathway (Chen et al., 2022; Krüger et al., 2024; Mody et al., 2023). Substantial evidence indicates that excessive ROS generation in cardiomyocytes represents the central pathogenic mechanism underlying DOX-induced cardiotoxicity (Mukhopadhyay et al., 2010). Besides, cardiomyocytes exhibit heightened sensitivity to DOX compared with cardiac fibroblasts and endothelial cells, rendering them particularly vulnerable to DOX-induced injury and ultimately leading to multiple forms of regulated cell death, such as apoptosis, pyroptosis, and ferroptosis (Zhang et al., 2012). Consistent with prior studies, oxidative stress and cardiomyocyte apoptosis have emerged as critical therapeutic targets for protecting the heart against DOX-induced injury and improving clinical outcomes in cancer patients. Although the function of NINJ1 in cardiovascular diseases, particularly in the context of DOX-induced cardiotoxicity, remains largely unexplored, its documented involvement in oxidative stress regulation and cell death across multiple pathological conditions strongly implicates NINJ1 as a potential mediator of DOX-induced cardiac injury. In this study, we aimed to reveal the molecular basis of NINJ1 and identify the NINJ1-AMPK-hypoxia-inducible factor-1α (HIF-1α) axis as a novel regulatory pathway in the DOX-induced chronic cardiomyopathy model.
Results
NINJ1 expression was elevated in DOX-induced murine hearts and cultured cardiomyocytes
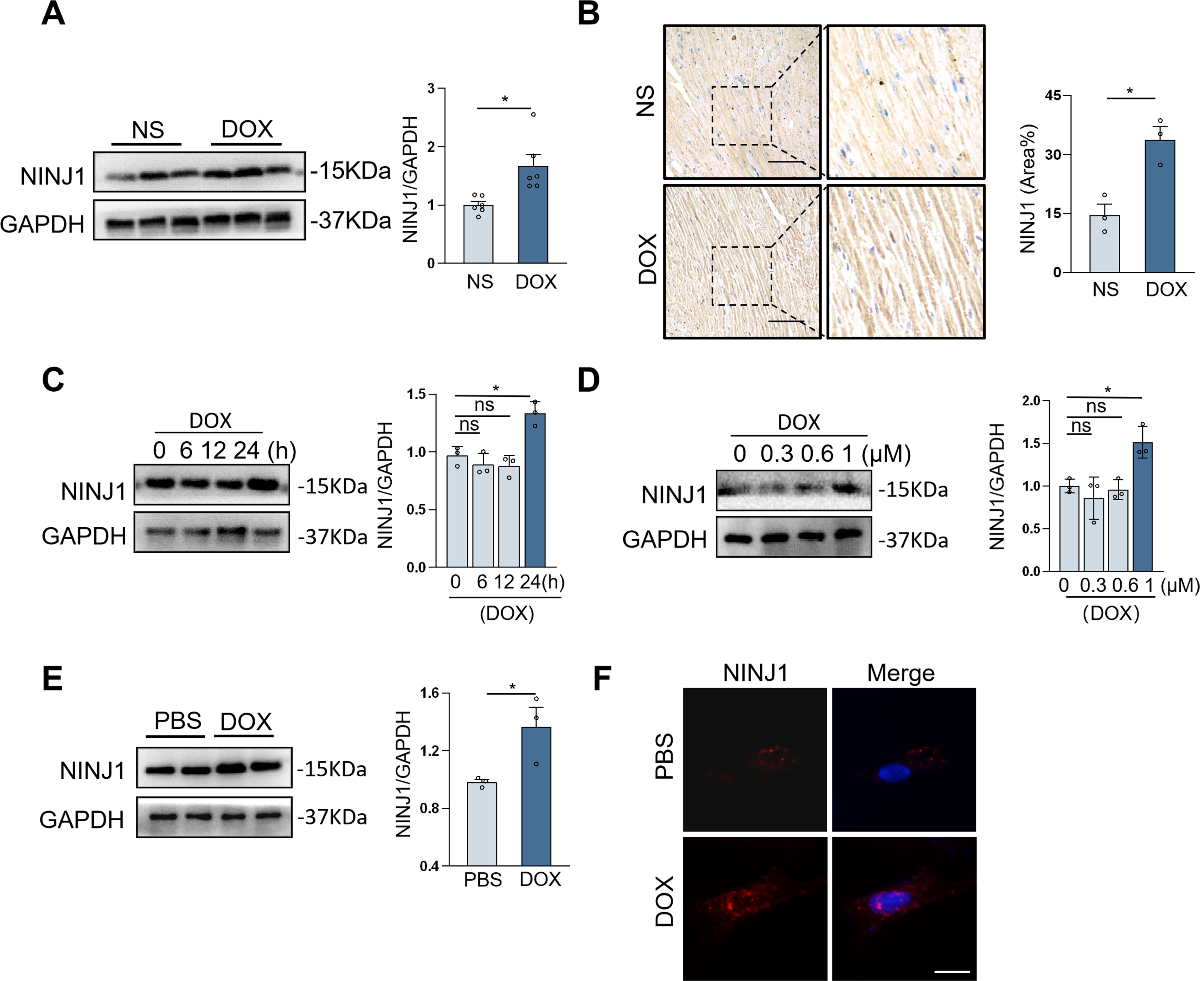
To investigate the relevance of NINJ1 in DOX-associated cardiotoxicity, the chronic cardiotoxicity model was established, and the protein level of NINJ1 was detected. Western blot illustrated that the protein expression of NINJ1 was significantly elevated in DOX-injured murine hearts (Fig. 1A). Meanwhile, immunohistochemical (IHC) staining analysis proved the increased NINJ1 protein level in the mouse hearts following DOX treatment (Fig. 1B). NINJ1 protein expression in H9c2 cells (rat embryonic cardiomyocyte cell line), showing the most pronounced elevation, was observed after treatment with 1 μM for 24 h (Fig. 1C and D). Then, we separated neonatal rat ventricular myocytes (NRVMs) and observed similar upregulation in DOX-treated NRVMs (Fig. 1E). Immunofluorescence staining in H9c2 further revealed that upregulation of NINJ1 during DOX treatment and NINJ1 localized both in the plasma membrane and cytoplasm of cardiomyocytes (Fig. 1F). Taken together, these findings have revealed that NINJ1 expression was elevated during DOX treatment.
Cardiomyocyte-specific NINJ1 deficiency improved DOX-induced heart injury in vivo
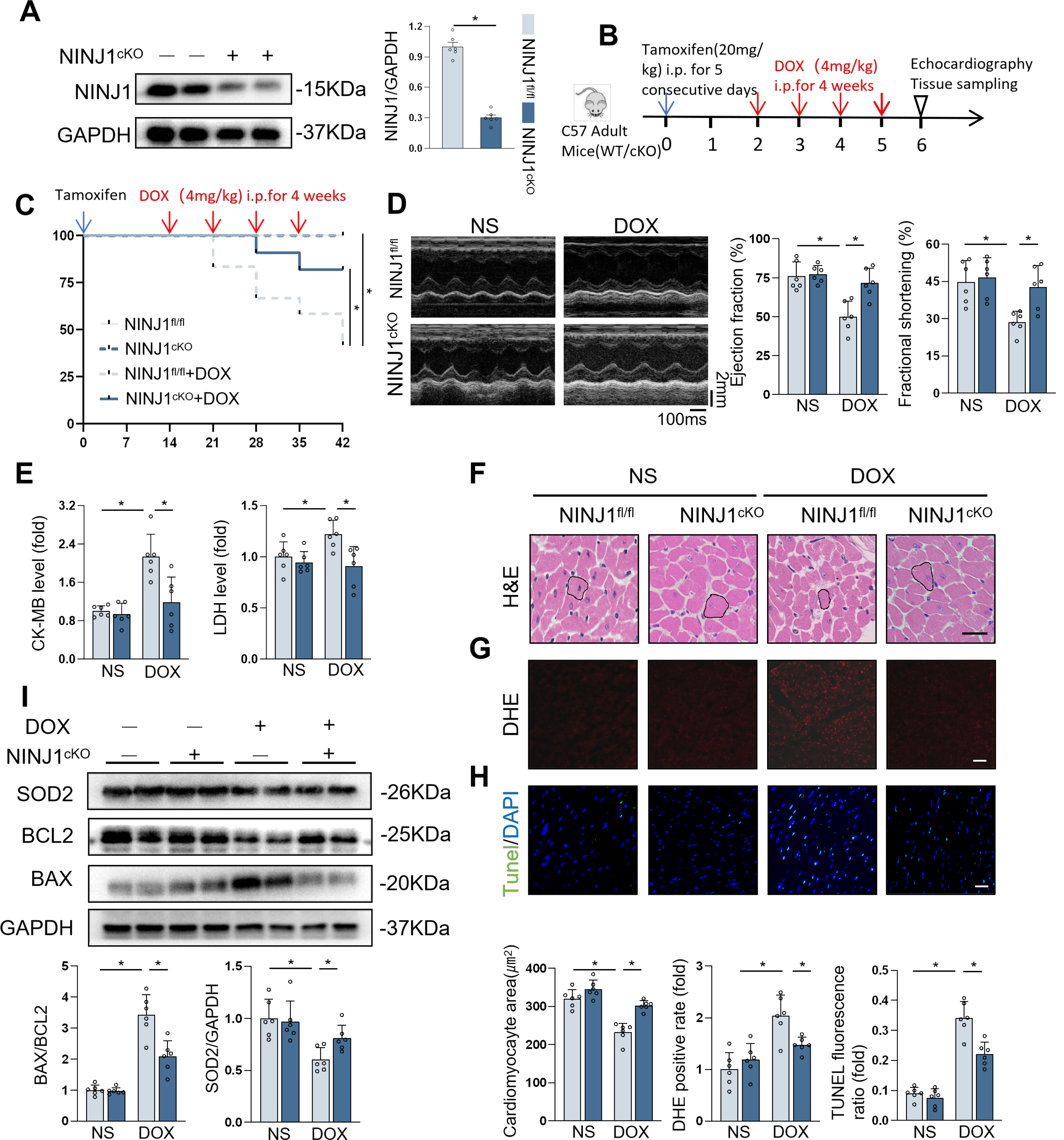
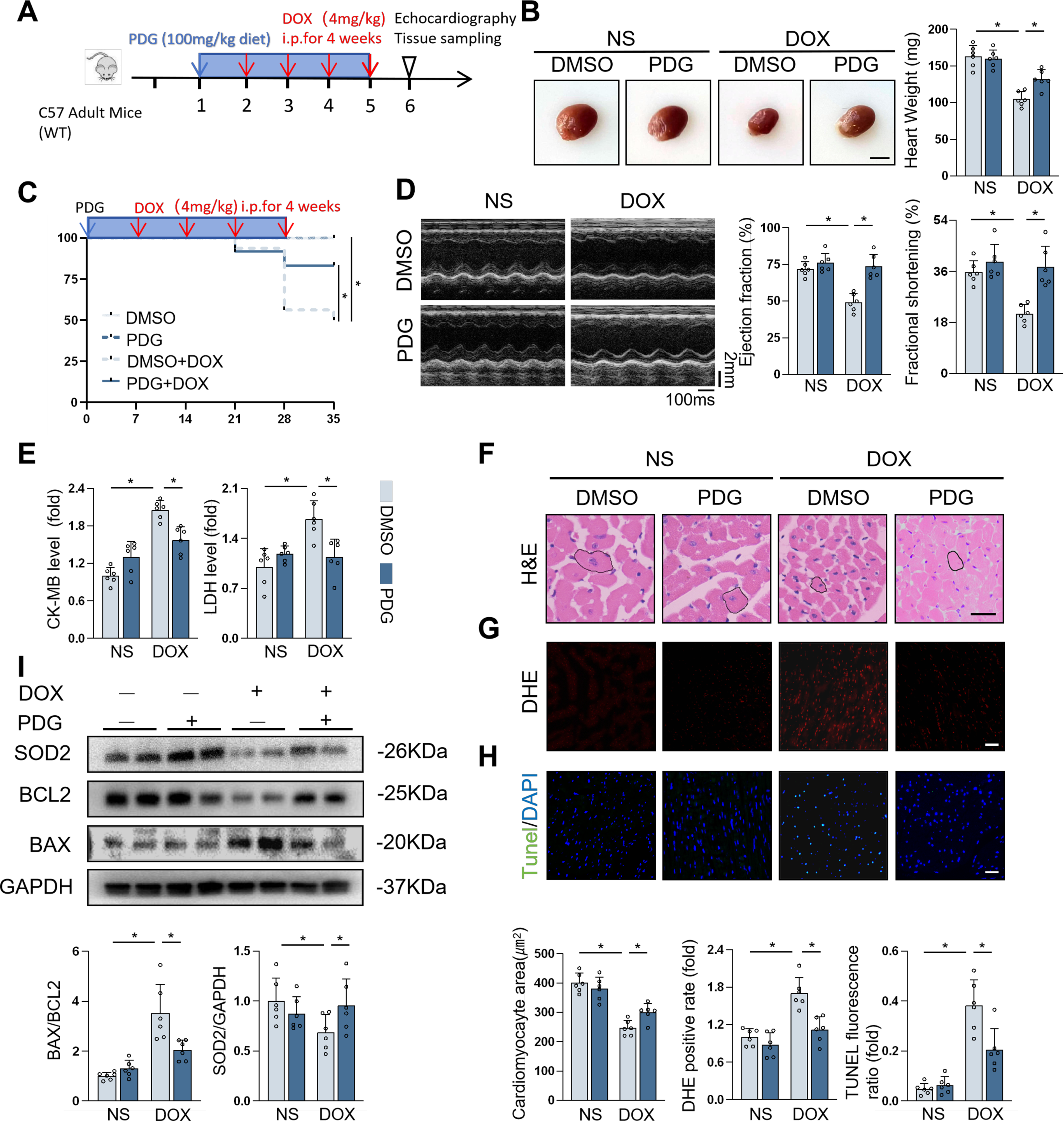
We then established cardiac-specific deletion of NINJ1 to clarify function of NINJ1 (Fig. 2A). In Mice Experiment 1 (Fig. 2B), survival analysis showed improved survival in NINJ1cKO mice than in NINJ1fl/fl mice after DOX treatment (Fig. 2C). Interestingly, NINJ1cKO mice showed an improvement in cardiac function during DOX treatment, as confirmed by increased ejection fraction and fractional shortening, compared with NINJ1fl/fl mice (Fig. 2D). Additionally, serum levels of creatine kinase-myocardial band (CK-MB), LDH, common marker for heart injury, were lower in NINJ1cKO mice treated with DOX than in NINJ1fl/fl mice treated with DOX (Fig. 2E). DOX-treated NINJ1fl/fl mice exhibited remarkable reduction in cardiomyocyte size; however, this effect was significantly prevented in DOX-treated NINJ1cKO mice (Fig. 2F). Cardiomyocyte oxidative stress and apoptosis are hallmark in DOX-induced cardiotoxicity; therefore, they were investigated in our studies (Hu et al., 2020b). Consistent with prior studies, DOX increased intramyocardial ROS production and oxidative damage in the hearts, which were significantly inhibited by cardiomyocyte-specific NINJ1 deficiency (Fig. 2G). We next examined the level of DNA damage and cell apoptosis by TdT-mediated dUTP nick-end labeling (TUNEL) staining, demonstrating that DOX treatment increased the TUNEL fluorescence ratio in both NINJ1fl/fl and NINJ1cKO mice. However, the increased TUNEL fluorescence ratio by DOX in NINJ1fl/fl was greater than in NINJ1cKO (Fig. 2H). As expected, the ameliorated DOX-induced cell apoptosis and oxidative damage were further confirmed by Western blot via the reduction in levels of BAX/BCL2 and Superoxide dismutase 2 (SOD2) (Fig. 2I). These data confirm that NINJ1 deficiency reduced ROS and apoptosis in vivo.
Cardiomyocyte-specific NINJ1 overexpression aggravated DOX-induced cardiac injury in vivo
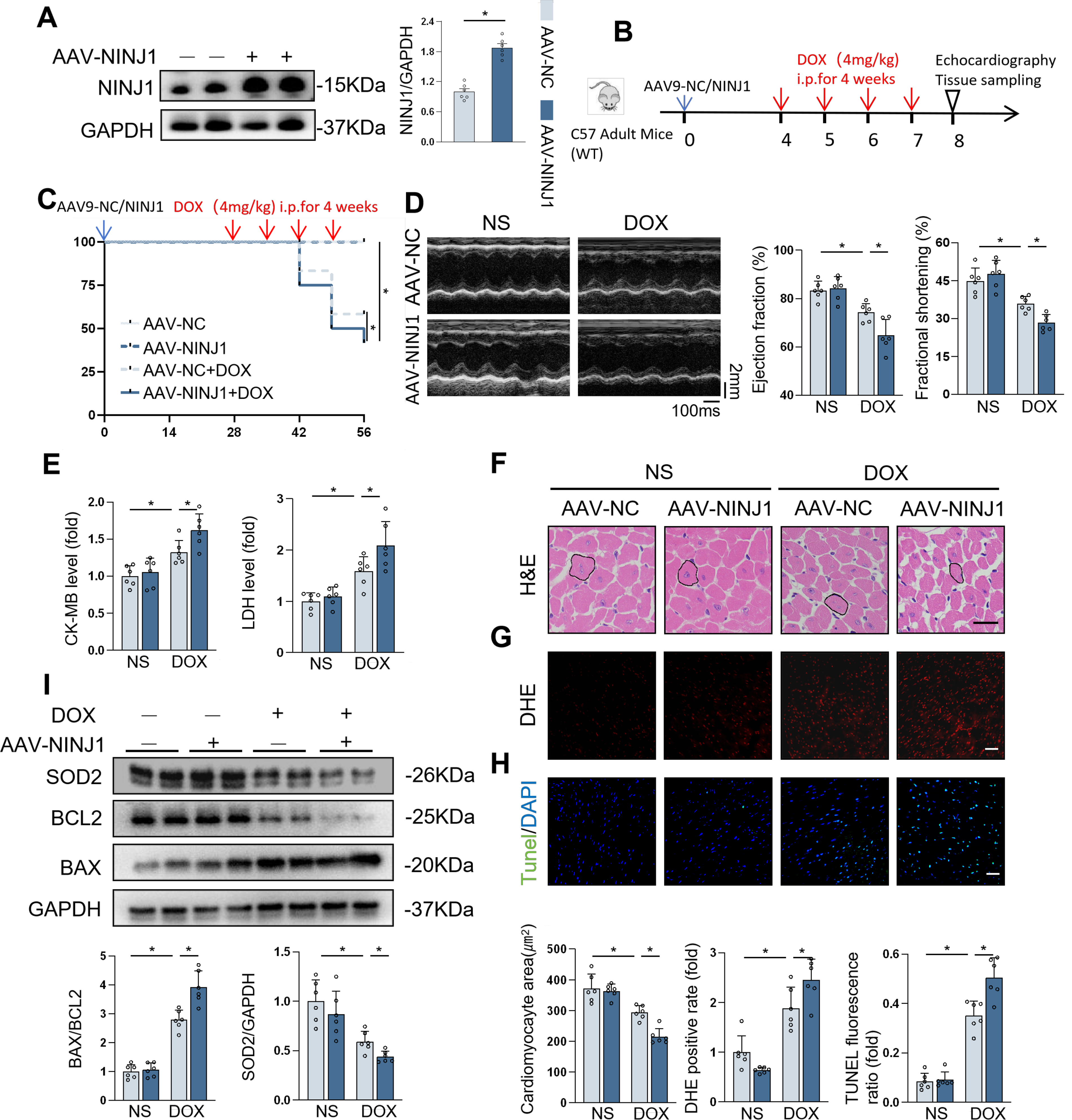
To further decipher the role of NINJ1, we generated adeno-associated virus 9 (AAV9) vectors containing a cardiomyocyte-specific cTnT promoter with NINJ1 gene sequence to overexpress NINJ1 in the mice cardiomyocytes (Fig. 3A). In Mice Experiment 2 (Fig. 3B), the survival rate was lower in mice with NINJ1 overexpression after DOX stimulation (Fig. 3C). Intriguingly, NINJ1 overexpression mice had deteriorated cardiac function (Fig. 3D), higher serum levels of myocardial injury markers (Fig. 3E), and smaller cardiomyocyte size (Fig. 3F) compared with the control mice after DOX treatment. DOX-induced oxidative stress and cell apoptosis in the myocardium were also aggravated by NINJ1 overexpression, as determined by a higher level of Dihydroethidium (DHE) positive rate (Fig. 3G), TUNEL + nuclei (Fig. 3H), and Western blot analysis (Fig. 3I). Thus, overexpression of NINJ1 deteriorated DOX-induced cardiotoxicity in mice.
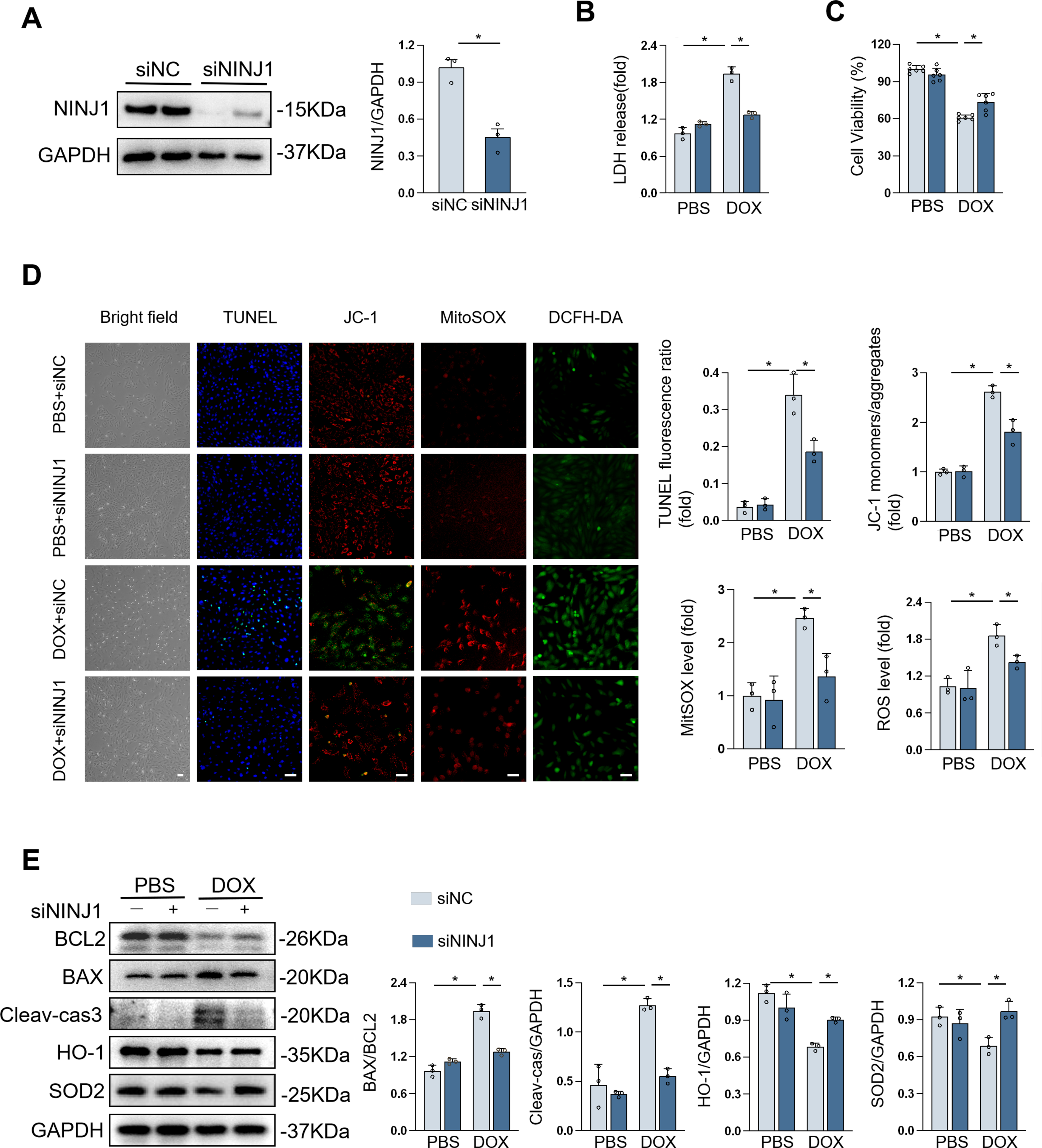
NINJ1 knockdown prevented DOX-induced cardiomyocyte injury in vitro
H9c2 cells were used to verify their response to DOX in vitro, and the NINJ1 protein level was dramatically decreased by small interfering RNA targeting NINJ1 (siNINJ1; Fig. 4A). Silencing NINJ1 restored DOX-triggered cell apoptosis, as assessed by the decreased LDH release and increased cell viability (Fig. 4B and C). Similarly, DOX-caused cell apoptosis was notably blocked by NINJ1 silencing, as evidenced by TUNEL staining (Fig. 4D). It is well known that DOX leads to oxidative stress, which is deemed the major cause of cardiotoxicity. tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and MitoSOX staining showed that NINJ1 knockdown prevented the reduction of mitochondrial membrane potential (MMP) and increased mitochondrial ROS in H9c2 during DOX stimulation (Fig. 4D). Dichlorodihydrofluorescein diacetate (DCFH-DA) staining showed that DOX-triggered ROS generation was significantly upregulated post-DOX treatment compared with control, while NINJ1–siRNA significantly reduced ROS accumulation (Fig. 4D). Therefore, NINJ1 knockdown markedly prevented DOX-induced mitochondrial dysfunction and oxidative stress. Western blot demonstrated that NINJ1 depletion increased the levels of BCL2/BAX, SOD2, and heme oxygenase-1 (HO-1), while decreasing the levels of Cleaved-caspase3 after DOX exposure (Fig. 4E). Collectively, NINJ1 knockdown reduced DOX-related cell apoptosis and ROS generation in vitro.
NINJ1 overexpression promoted DOX-induced cardiomyocyte injury in vitro
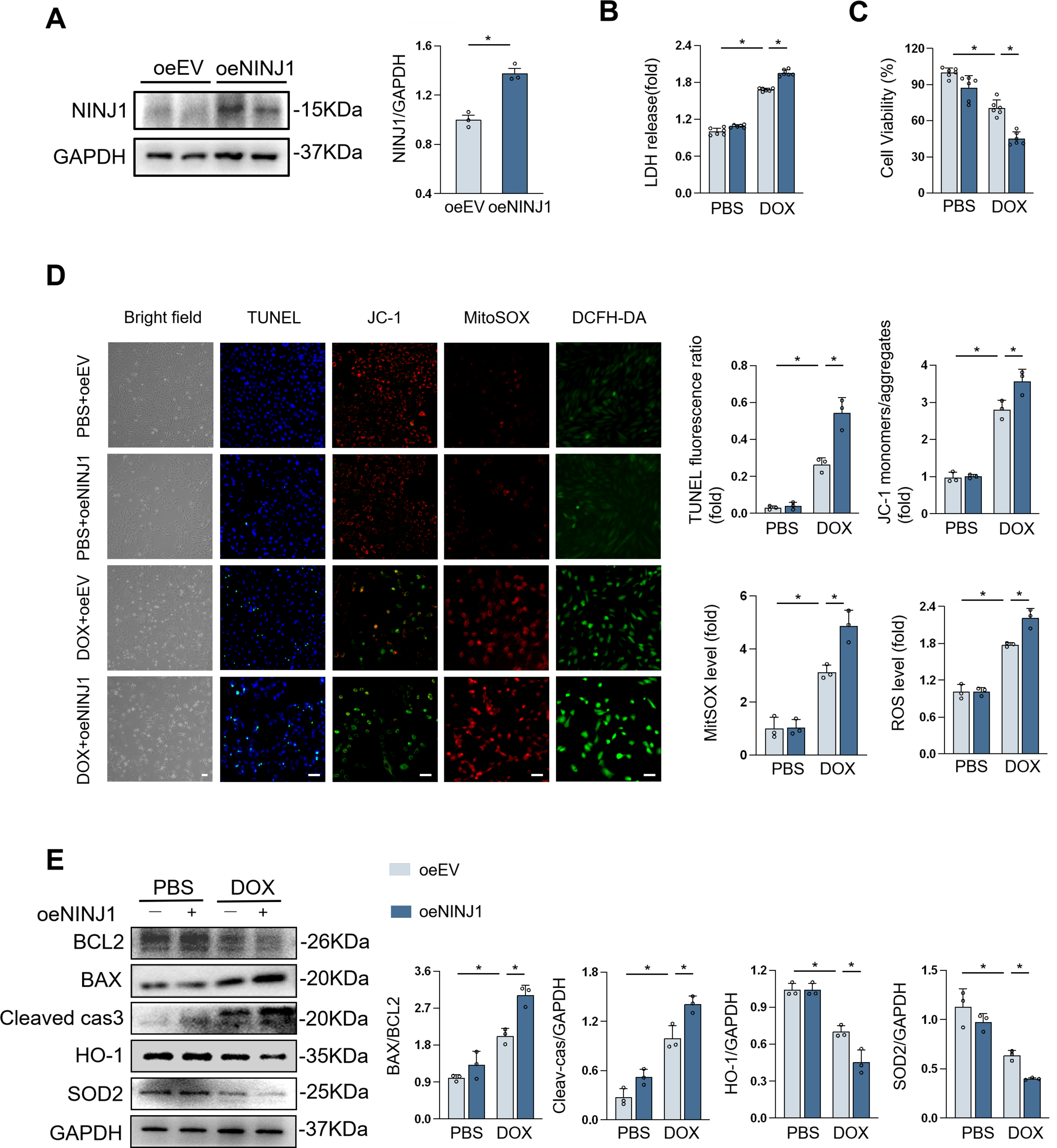
Likewise, we also investigated the function of NINJ1 overexpression, achieved through plasmid transfection, in H9c2 cells under DOX stimulation (Fig. 5A). Predictably, overexpression of NINJ1 enhanced cell susceptibility to DOX, as determined by LDH released assay, cell counting kit-8 (CCK8) kits, and cell apoptosis staining (Fig. 5B–D). Compared with the empty vector-treated group, worse mitochondrial dysfunction can be observed in the overexpression group after DOX stimulation (Fig. 5D). Correspondingly, NINJ1 overexpression increased Cleaved-caspase3 levels and decreased BCL2/BAX, SOD2, and HO-1 levels in H9c2 with DOX treatment (Fig. 5E). These results indicated that NINJ1-overexpressing cardiomyocytes were more sensitive to DOX-induced damage.
NINJ1 deficiency ameliorated DOX-induced cardio injury through activating HIF-1α in vivo and in vitro
To elucidate the underlying molecular mechanisms by which DOX caused cardiotoxicity in adult mice, we performed RNA sequencing (RNA-seq) of the hearts from NINJ1fl/fl mice and NINJ1cKO mice after DOX stimulation. Bioinformatics analysis of RNA-seq showed 575 of the differentially expressed genes (DEGs) were upregulated, and 486 were downregulated after knockdown of NINJ1 (Supplementary Fig. S2A). Specifically, further analysis of RNA-seq data shown in a heatmap revealed that 113 key genes were expressed differently; 69 of these genes were upregulated, whereas 44 of these were downregulated (Fig. 6A and Supplementary Fig. S2B). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis based on RNA-seq revealed that upregulated DEGs were enriched in the cardioprotective HIF-1 signaling pathway, which was in line with cellular response to hypoxia by gene set enrichment analysis (GSEA) (Fig. 6B and C). In addition, upregulated genes in the heart of NINJ1cKO mice were enriched in detoxification of ROS, NRF2 signaling, positive regulation of nitric oxide (NO) synthase activity, oxidative phosphorylation, mitochondrial respiratory chain complex I assembly, and negative regulation of apoptotic signaling pathway (Supplementary Fig. S2C–E), confirming that NINJ1 deficiency may render cardiomyocytes more resistant to oxidative stress.
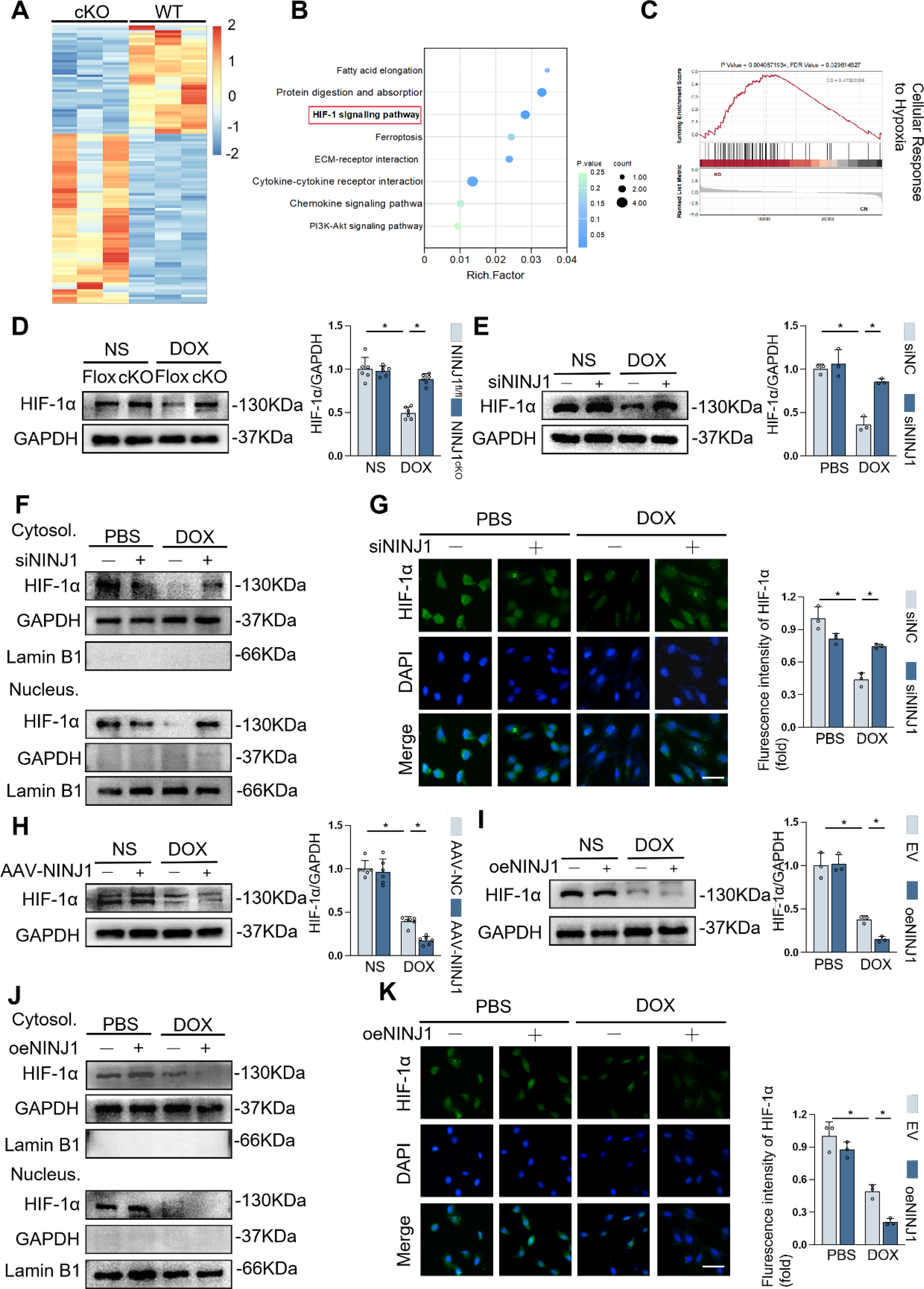
HIF-1α serves as a master transcription factor governing systemic oxygen homeostasis, which is responsible for resistence to oxidantive stress and promoting cell survival during DOX stimulation (Zhang et al., 2026). Therefore, we determined whether targeting NINJ1 impaired DOX-induced cardiotoxicity through preserving HIF-1α. As shown in Figure 6D and E, HIF-1α protein level was increased by NINJ1 deficiency in DOX-stimulated heart samples and H9c2 cells, in line with IHC staining results (Supplementary Fig. S2F). HIF-1α can translocate into the nucleus and function as a transcription factor. Thus, it is necessary to detect protein levels of HIF-1α in different patterns by isolating cytoplasmic and nuclear proteins in H9c2 cells. Here, we found that siNINJ1 treatment restored HIF-1α protein level in both cytoplasm and nucleus with DOX treatment, which was consistent with the immunofluorescent findings (Fig. 6F and G). In contrast, the total, cytoplasmic, and nuclear protein expression levels of HIF-1α were further decreased by NINJ1 overexpression in the presence of DOX (Fig. 6H–K and Supplementary Fig. S2F). HIF-1α was accumulated in the nucleus to activate antioxidative transcription program, such as superoxide dismutase (SOD), HO-1. Silencing of HIF-1α with siRNA reduced DOX-stimulated SOD2 or HO-1 expression at protein levels in NINJ1-knockdown H9c2 (Supplementary Fig. S2G). These results indicated NINJ1 promoted oxidative stress through suppressing HIF-1α and its nuclear accumulation.
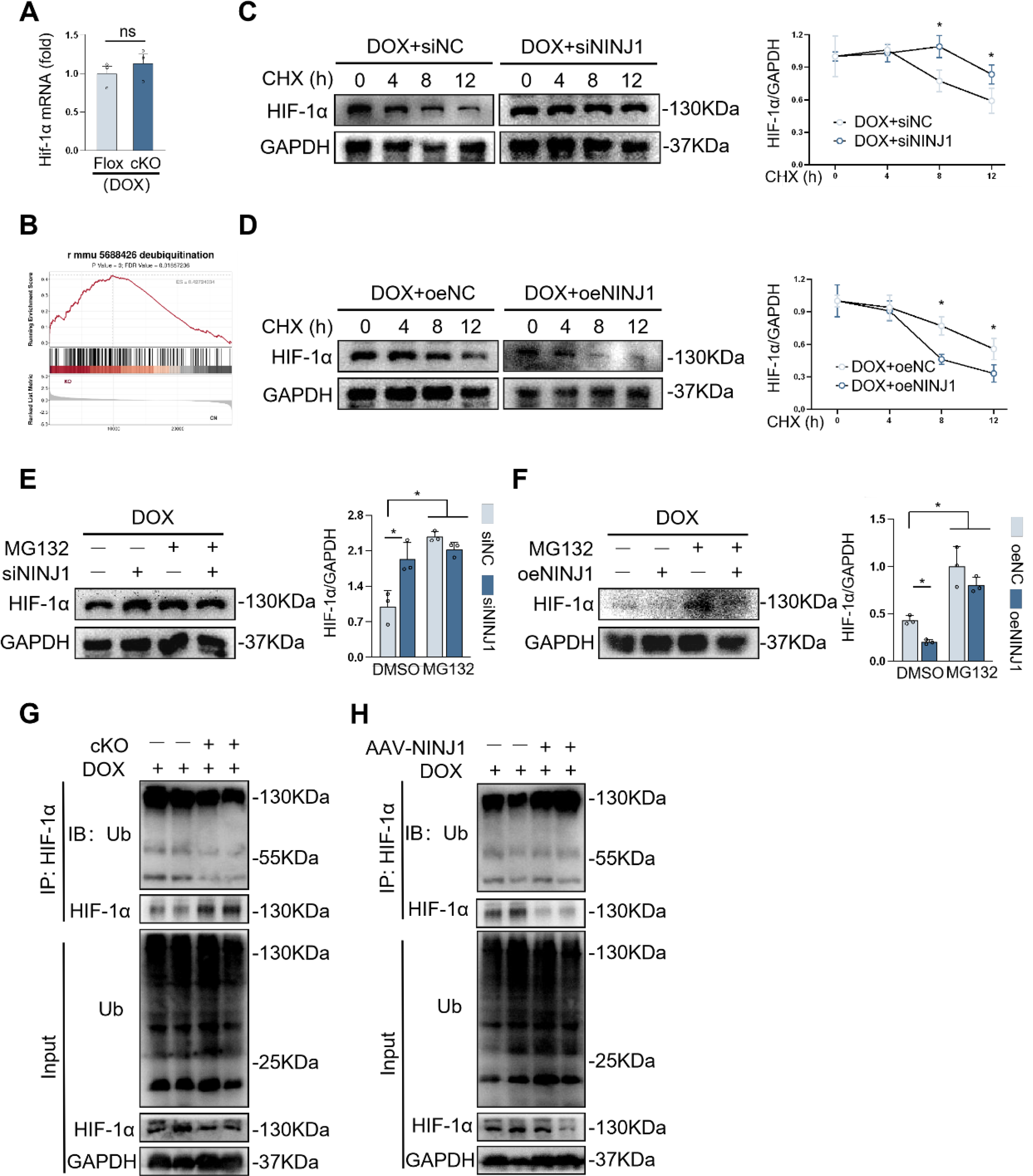
NINJ1 promoted ubiquitination-mediated HIF-1α degradation during DOX-induced cardiotoxicity
Next, we studied the underlying mechanism by which NINJ1 participated in modulating HIF-1α protein level. In accordance with RNA-seq results, HIF-1α mRNA level was surprisingly unchanged in the heart of NINJ1cKO mice after DOX treatment (Fig. 7A), indicating that NINJ1 did not regulate HIF-1α at the transcriptional level. In addition, GSEA revealed a significant enrichment of deubiquitination-related pathways in the NINJ1-deficient group (Fig. 7B). Therefore, we speculated that NINJ1 inhibited HIF-1α protein levels by promoting its ubiquitin-mediated proteasome pathway. To test this hypothesis, cycloheximide (CHX) chase assays were performed to assess the half-life and stability of HIF-1α. As expected, DOX treatment notably shortened the half-life of HIF-1α, whereas NINJ1 knockdown significantly delayed HIF-1α degradation, thereby prolonging its half-life and enhancing protein stability (Fig. 7C). In contrast, NINJ1 overexpression further accelerated HIF-1α turnover and reduced its stability (Fig. 7D). To further confirm the involvement of the proteasome pathway, H9c2 cells were pretreated with the proteasome inhibitor MG132. Under MG132 treatment, HIF-1α protein levels were comparable between control and NINJ1-knockdown cells (Fig. 7E), whereas the reduction of HIF-1α protein induced by NINJ1 overexpression was effectively rescued (Fig. 7F). These results suggested NINJ1 promoted proteasome-dependent degradation of HIF-1α. Moreover, cardiomyocyte-specific NINJ1 deletion markedly reduced polyubiquitination of HIF-1α in DOX-treated hearts, while NINJ1 overexpression significantly increased HIF-1α ubiquitination (Fig. 7G and H). Collectively, our results indicated that NINJ1 inhibited HIF-1α by promoting ubiquitin-dependent proteasomal degradation.
NINJ1 suppressed HIF-1α by regulating AMPK signaling pathway during DOX-induced cardiotoxicity
Previous research indicated that the AMPK signaling pathway is involved in the various biological processes of NINJ1, including oxidative stress, inflammation, and activation of autophagy (Park et al., 2024). To understand whether NINJ1 regulated AMPK pathway, we tested AMPK and phosphorylation of AMPK (p-AMPK) protein expression in vivo and in vitro. Compared with the DOX treatment group, NINJ1 deficiency significantly elevated the levels of p-AMPK, and NINJ1 overexpression further minimized p-AMPK levels during DOX stimulation, in line with IHC results (Fig. 8A and Supplementary Fig. S3A-B). Consistent alterations of protein abundance were observed in vitro when exposed to DOX (Fig. 8B and Supplementary Fig. S3C). Altogether, these results highlighted NINJ1 deficiency in vivo and in vitro activated AMPK pathway against DOX-induced cardiotoxicity.
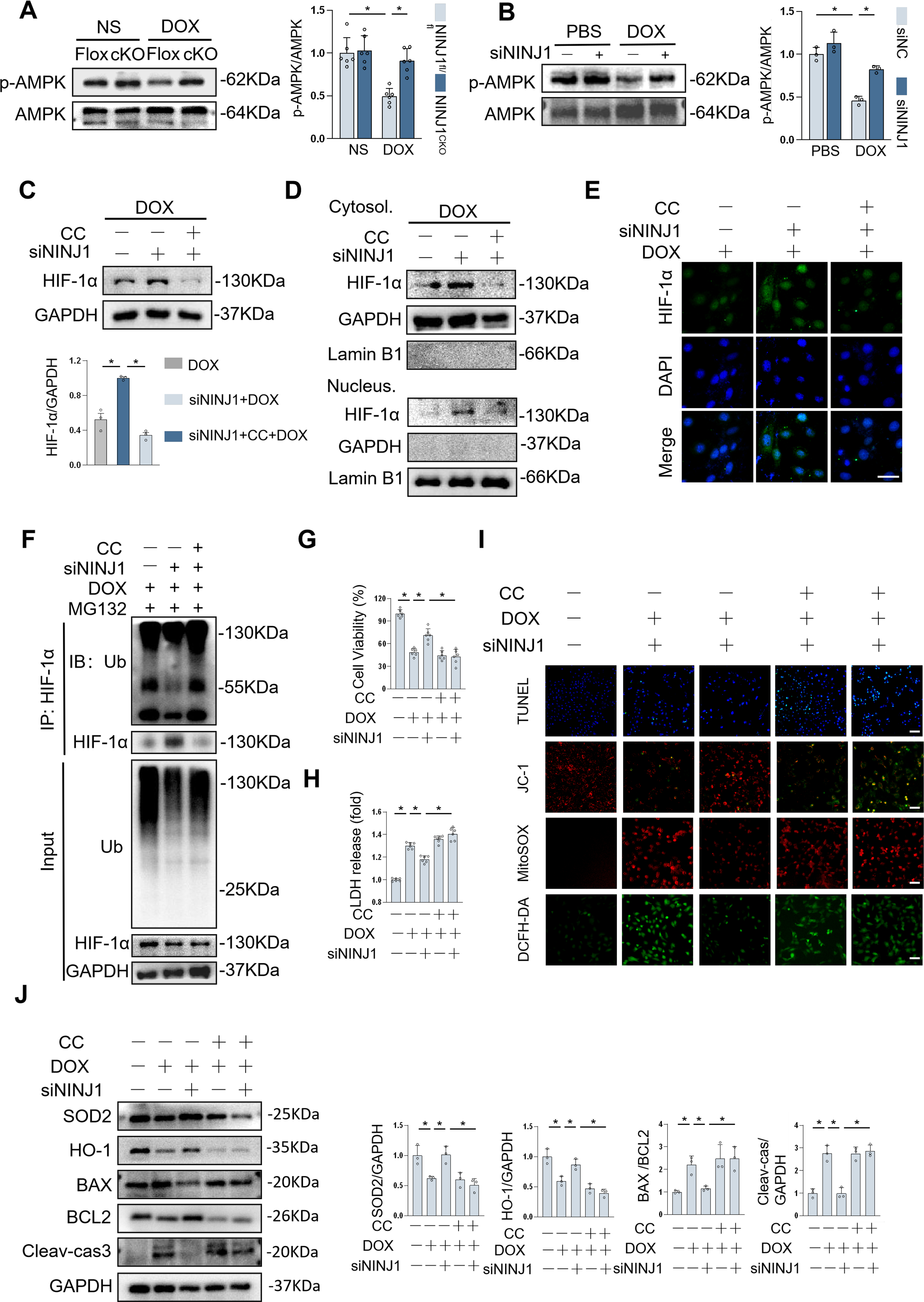
AMPK has been found to be involved in the regulation of HIF-1α expression and transactivation (Chen et al., 2014; Lee et al., 2021; Yun et al., 2023). To determine whether this effect is AMPK-dependent, we administrated H9c2 cells with either AMPK inhibitor compound C (CC) or AMPK agonist acadesine (AICAR). The data indicated that NINJ1 deficiency enhanced total cellular and nuclear accumulation of HIF-1α protein during DOX stimulation, which was significantly abolished by AMPK inhibition (Fig. 8C–E). On the contrary, AICAR administration in H9c2 cells notably attenuated the decrease of total and intranuclear HIF-1α protein expression induced by NINJ1 overexpression under DOX stimulation (Supplementary Fig. S3D–F).
To find out whether the AMPK pathway was essential to the stability of HIF-1α, the level of HIF-1α ubiquitination was detected in H9c2 with CC or AICAR treatment. As expected, the block of AMPK reversed NINJ1 deficiency-regulated downregulation of HIF-1α ubiquitination level (Fig. 8F). Likewise, we also observed that the ubiquitination and degradation of HIF-1α were alleviated in NINJ1 overexpressed H9c2 cells by AMPK activation following DOX treatment (Supplementary Fig. S3G). The above endogenous assay results indicated that AMPK may serve as a primary downstream mediator responsible for HIF-1α stability in cardiomyocytes under DOX stimulation.
To further confirm the precise role of AMPK in NINJ1 regulating cardiomyocyte oxidative stress and apoptosis during DOX treatment, we incubated H9c2 cells with CC or AICAR, respectively. As anticipated, AMPK inhibition abolished protection by NINJ1 deficiency and failed to improve apoptosis and oxidative stress under DOX challenge (Fig. 8I–J). In contrast, AMPK activation reversed NINJ1 overexpression-induced adverse effects in cells under DOX treatment (Supplementary Fig. S3H–K). However, protein binding assays verified that NINJ1 failed to directly bind to AMPK, and NINJ1-AMPK interaction still remained unresolved. This study demonstrated that AMPK activation was responsible for NINJ1 deficiency-mediated protective role in DOX-induced cardiotoxicity.
Pharmacological inhibition of NINJ1 attenuated DOX-induced cardiac injury in vivo
Finally, in order to evaluate whether the administration of NINJ1 inhibitor as a therapeutic target for DOX-induced cardiac injury, a traditional medicine, phenyl-β-D-glucopyranoside (PDG) for anti-inflammatory, which has been shown to attenuate NINJ1 expression, was employed in mice during DOX treatment. In Mice Experiment 3 (Fig. 9A), mice treated with PDG notably suppressed the reductions in heart weight and survival rate (Fig. 9B and C). PDG-treated mice presented improved cardiac function and fewer cardiac injury indicators (Fig. 9D and E). DOX-induced cardiomyocyte atrophy was also relieved by therapy of PDG (Fig. 9F). Additionally, DHE fluorescence and cardiomyocyte apoptosis were alleviated in PDG-treated mice (Fig. 9G and H). Similar Western blot findings were observed in the PDG-treated group under DOX challenge (Fig. 9I). Thus, these data suggested that therapeutic administration of PDG was efficacious in alleviating DOX-induced cardiac injury in vivo.
PDG alleviated DOX-induced cardiomyocyte injury in vitro
To explore the antioxidative role of PDG in vitro, H9c2 cells were incubated with PDG during DOX stimulation. Increased cell viability, lower LDH release, and decreased TUNEL fluorescence ratio revealed that PDG attenuated cardiomyocyte apoptosis caused by DOX treatment (Supplementary Fig. S4A-C). JC-1, MitoSOX, and ROS staining identified improved mitochondrial function and oxidative stress after PDG administration (Supplementary Fig. S4C). Consistently, the protective effects of PDG against apoptosis and oxidative stress were demonstrated by Western blot analysis (Supplementary Fig. S4D). We next investigated whether PDG could achieve antiapoptosis and oxidative stress effects in NINJ1-overexpressed H9c2 cells. Unexpectedly, overexpression by plasmid reversed the protective effects of PDG against DOX-induced cardiomyocyte injury (Supplementary Fig. S4E). These data indicated that PDG protected cardiomyocytes during DOX treatment in a NINJ1-dependent way.
PDG did not affect DOX-induced cytotoxicity in HeLa cells
Finally, to evaluate whether NINJ1 deficiency affects the efficacy of DOX in suppressing cancer cells’ proliferation and survival, we used human cervical cancer cells HeLa. DOX triggered a decrease in cell viability and apoptosis, while PDG did not change the tumor-killing action of DOX, as evidenced by CCK8 assay, TUNEL staining, and immunoblots (Supplementary Fig. S5A–C). Compared with the dimethyl sulfoxide (DMSO) control group, PDG-treated HeLa cells presented lower cell proliferation in response to DOX stimulation, as assessed by the results of Ki67 and EdU staining (Supplementary Fig. S5D and E). We further investigated whether PDG affected cell migration in DOX-treated HeLa cells, and the wound scratch assay showed that DOX-induced inhibition of cell migration in HeLa remained unchanged after PDG administration (Supplementary Fig. S5F). Taken together, these results suggested that PDG was sufficient to attenuate cardiotoxicity without reducing its anticancer efficacy.
Discussion
In the present study, we identify the NINJ1-AMPK-HIF-1α axis as a novel regulatory pathway governing oxidative stress and apoptosis in DOX-induced cardiotoxicity. Genetic silencing or pharmacological inhibition of NINJ1 significantly mitigated these detrimental effects. Importantly, PDG confers prominent cardioprotection without compromising the anticancer potency of DOX. These findings demonstrate that targeting NINJ1 may complement existing cardioprotective strategies in oncology, which offers new opportunities for improving clinical outcomes in cancer patients receiving DOX-based chemotherapy.
The clinical utility of DOX is severely constrained by cumulative, dose-dependent cardiotoxicity, for which effective cardioprotective strategies remain insufficient. Among the multiple mechanisms implicated, apoptosis and oxidative stress are widely recognized as dominant contributors to DOX-induced cardiac injury (Kuzu et al., 2018). Several studies have demonstrated that DOX induces excessive production of ROS and reactive nitrogen species, leading to mitochondrial dysfunction and suppression of endogenous antioxidant defenses, including glutathione peroxidase, SOD, and NRF2 (Doroshow et al., 1980; Songbo et al., 2019). In parallel, DOX disrupts mitochondrial membrane integrity, resulting in cytochrome c release into the cytosol and subsequent activation of caspase 3-dependent apoptotic cascades, ultimately causing nuclear DNA fragmentation(Kitazumi and Tsukahara, 2011). In addition, DOX-topoisomerase 2β (Top2β)-DNA complex formation has been identified as a major driver of cardiomyocyte DNA damage (Lyu et al., 2007), triggering p53 phosphorylation and cell death (Lin et al., 2018).
NINJ1 is a 16-kDa transmembrane cell-surface protein containing two membrane-spanning domains with extracellular N- and C-termini (Degen et al., 2023). It is ubiquitously expressed and has been implicated in diverse pathological conditions, including cardiovascular diseases, diabetes, inflammation, and cancer (Araki et al., 1997). Here, we demonstrate that NINJ1 is expressed in cardiomyocytes and is significantly upregulated during DOX-induced cardiomyopathy. Previous studies have linked NINJ1 to apoptotic regulation in multiple biological contexts. For instance, NINJ1 promoted apoptosis in vascular endothelial cells during early ocular development (Lee et al., 2009), whereas its knockdown enhanced trophoblast proliferation, migration, and invasion, while inhibiting apoptosis through signal transducer and activator of transcription 3 activation (Zhang et al., 2022). In pancreatic acinar cells, NINJ1-mediated PMR has been associated with Ca2+ overload, leading to mitochondrial dysfunction, oxidative stress, and subsequent cellular injury (Lee et al., 2023; Marolt et al., 2022). Consistent with these findings, our data demonstrate that NINJ1 inhibition in cardiomyocytes markedly attenuates DOX-induced apoptosis both in vivo and in vitro, supporting a conserved pro-apoptotic role for NINJ1 under stress conditions.
Beyond apoptosis, NINJ1 has been implicated in plasma membrane permeabilization and inflammatory cell death. NINJ1 deficiency has been reported to reduce gasdermin D-dependent LDH release in response to either Lipopolysaccharide (LPS) or nigericin stimulation (Evavold et al., 2018; Kayagaki et al., 2011; Shi et al., 2015). In line with this, we observed decreased LDH release in H9c2 cells following NINJ1 silencing under DOX treatment. Besides, it is well documented that the intracellular oxidative stress can be a strong trigger for inflammatory stress and even cell apoptosis (Mittal et al., 2014). Existing evidence confirms that NINJ1 deficiency improves oxidative injury by modulating the AMPK/HIF-1 axis in acetaminophen-induced liver injury, further supporting its indirect regulation of redox balance (Park et al., 2024). Our findings extend these observations by demonstrating that NINJ1 knockdown significantly reduces ROS accumulation and cardiomyocyte death in a chronic DOX-induced cardiotoxicity model, underscoring the critical role of NINJ1 in oxidative stress-driven cardiac injury.
HIF-1 is a heterodimeric transcription factor composed of an oxygen-sensitive α subunit and a constitutively active β subunit (Wang et al., 1995). Under hypoxic or stress conditions, stabilized HIF-1α translocates into the nucleus, dimerizes with HIF-1β, and binds to the hypoxia response element [5′-(A/G) CGTG-3′] within target gene promoters to regulate transcription (Semenza, 2020). HIF-1 plays pivotal roles in immune regulation, oxidative stress resistance, anti-apoptosis processes, metabolic reprogramming, angiogenesis, inflammation, tumor progression (Choudhry and Harris, 2018; Halligan et al., 2016; Warbrick and Rabkin, 2019), and cardioprotection, particularly during ischemia-reperfusion injury (Gu et al., 2014). Notably, DOX-induced cardiotoxicity has been associated with oxidative stress and ROS generation that culminate in cardiomyocyte injury and apoptosis (Yarmohammadi and Karimi, 2026). Activation of HIF-1α promotes transcription of antioxidant genes and protects cardiomyocytes from oxidative injury, suggesting that impaired HIF-1α signaling may contribute to DOX-induced cardiac damage (Syukri et al., 2022). AMPK, a central cellular energy sensor, is activated in response to energetic stress and orchestrates metabolic adaptation and cell survival (Steinberg and Hardie, 2023). Accumulating evidence indicates that AMPK serves as an important upstream regulator of HIF-1α stability, nuclear accumulation, and transcriptional activity under metabolic stress conditions. Chen et al. found that increased AMPKα expression and activity are accompanied by upregulation of HIF-1α in human ovarian cells (Chen et al., 2014). Similarly, under hypoxia or low-glucose conditions, AMPK activation facilitates nuclear accumulation and functional activation of HIF-1α, revealing a previously unrecognized regulatory link between AMPK and HIF-1 signaling (Chen et al., 2015). Consistently, Malik reported that dysregulation of AMPK signaling compromises vascular repair by destabilizing HIF-1α (Abdel Malik et al., 2017). Moreover, serine- and glycine-deficient conditions have been shown to enhance ROS–AMPK–HIF-1α signaling, thereby promoting glioblastoma cell survival (Yun et al., 2023). Beyond transcriptional regulation, AMPK has emerged as a critical modulator of protein stability through ubiquitin–proteasome-dependent mechanisms. Activation of AMPK by metformin was shown to stabilize von Hippel–Lindau protein by indirectly modulating its ubiquitination status, leading to suppressed cell proliferation and enhanced chemosensitivity in pancreatic ductal adenocarcinoma (Li et al., 2026). In contrast, AMPK activation by the antipsychotic drug, penfluridol, promoted the ubiquitin proteasomal-mediated degradation of PD-L1, thereby augmenting antitumor immunity in colorectal cancer (Wang et al., 2025). In contrast, we demonstrate that NINJ1 silencing enhances AMPK activation and selectively increases HIF-1α protein levels without affecting its mRNA expression. This effect is abolished by pharmacological inhibition of AMPK, indicating that NINJ1 negatively regulates HIF-1α stability through AMPK inactivation. These findings suggest that ubiquitin-dependent proteasomal degradation represents a primary mechanism by which NINJ1 suppresses HIF-1α signaling, thereby exacerbating oxidative stress and cardiomyocyte injury during DOX treatment.
Pharmacological targeting of NINJ1 further underscores its translational potential. The NINJ1 inhibitor PDG, a glycoside compound composed of glucose and a phenyl group, exhibits well-documented anti-inflammatory and anticancer properties. PDG inhibits the expression of cyclooxygenase-2 and inducible NO synthase, enzymes responsible for the production of NO (Choi et al., 2014). It also suppresses nuclear translocation of Nuclear factor kappa-B (NF-κB), one of the most important transcription factors involved in the inflammatory process (Hwang and Lee, 2015). Furthermore, PDG has been implicated in attenuating the expression of NINJ1 and abolishing the activity of matrix metalloproteinase (MMP) under endotoxin stimulation. Sheng et al. found that administration of PDG markedly alleviated thoracic aortic dissection severity in mice, as evidenced by reduced fibrosis, preservation of elastic fibers, and decreased infiltration of T cells and macrophages (Sheng et al., 2023). Consistent with these findings, we observed that PDG treatment significantly alleviated DOX-induced oxidative stress, cardiomyocyte apoptosis, and cardiac dysfunction, supporting its potential utility as a cardioprotective agent during anthracycline chemotherapy.
Importantly, we further evaluated the combined effects of DOX and PDG on tumor cells to assess potential interference with antitumor efficacy. It is interesting to find that NINJ1 inhibition protected cardiomyocytes from DOX-induced injury while preserving, and in some cases enhancing, the antitumor efficacy of DOX in HeLa cervical cancer cells, including suppression of tumor cell migration and growth. Previous studies have revealed a context-dependent role of NINJ1 in cancer biology, functioning as either a tumor promoter or suppressor depending on tumor type, genetic background, and microenvironmental conditions (Chen et al., 2025). NINJ1 deficiency has been shown to attenuate tumor progression in non-small cell lung cancer (Hyun et al., 2022) and colorectal cancer (Song et al., 2025), whereas elevated NINJ1 expression has been reported as a diagnostic biomarker in hepatocellular carcinoma (Kim et al., 2001) and B-cell acute lymphoblastic leukemia (Chen et al., 2001). Conversely, several studies revealed that NINJ1 suppresses tumor development and growth in colon cancer (Woo et al., 2016), and lower expression of NINJ1 may be associated with worse prognosis in serous ovarian cancer (Berkel and Cacan, 2023). These divergent effects highlight the complexity of NINJ1 signaling in oncology and suggest that NINJ1 inhibition may be particularly advantageous in cancers that depend on NINJ1 for survival or progression, while simultaneously reducing DOX-associated cardiotoxicity.
Several limitations of the present study should be acknowledged. First, NINJ1 is expressed not only in cardiomyocytes but also in fibroblasts, endothelial cells, and immune cells; thus, the contribution of NINJ1 signaling in these nonmyocyte populations to DOX-induced cardiotoxicity warrants further investigation. Second, the precise molecular mechanisms underlying the direct or indirect interaction between NINJ1 and AMPK remain to be fully elucidated. Third, although our data suggest that NINJ1 inhibition does not compromise the antitumor efficacy of DOX, in vivo tumor-bearing models are required to definitively assess therapeutic safety and efficacy. Finally, our in vitro experiments were performed using H9c2 rat cardiomyoblasts, which differ from adult human cardiomyocytes; future studies employing human-induced pluripotent stem cell-derived cardiomyocytes will further strengthen translational relevance.
Materials and Methods
Reagents and antibodies
The following primary antibodies were obtained from Proteintech (Wuhan, China): GAPDH monoclonal antibody (60004-1-lg), BAX polyclonal antibody (50599-2-lg), BCL2 polyclonal antibody (26593-1-AP), Cleaved caspase3 polyclonal antibody (25128-1-AP), SOD2 polyclonal antibody (24127-1-AP), HO-1 recombinant antibody (81281-1-RR), HIF-1 alpha polyclonal antibody (20960-1-AP), AMPK alpha polyclonal antibody (10929-2-AP), and ubiquitin polyclonal antibody (10201-2-AP), whereas anti-BCL-2 antibody (ab32124) was purchased from abcam (Cambridge UK), Phospho-AMPKα (Thr172) (#50081) and Ki-67 (D3B5) Rabbit mAb were obtained from Cell Signaling Technology (Boston, USA), and NINJ1 antibody was obtained from Santa Cruz Biotechnology (Danvers, USA). DOX, PDG, DMSO, tamoxifen, PEG3000, dorsomorphin (CC), AICAR, 5-BrdU, MG132, and CHX were purchased from MCE (New Jersey, USA). CCK8, LDH release assay kit, MMP assay kit, EdU-594 cell proliferation assay kit, and ROS assay kit with CM-H2DCFDA were obtained from Beyotime Biotechnology (Shanghai, China). CF488 TUNEL cell apoptosis detection kit, assay kits for detecting organized ROS, hematoxylin and eosin (HE) staining solution, and paraformaldehyde fixative solution were obtained from Servicebio (Wuhan, China). Cardiomyocyte-specific AAV9 to induce NINJ1 overexpression was constructed by Cyagen Biosciences (Suzhou, China), whereas the blank AAV9 vector without any tags was regarded as the negative control. siNINJ1 and a negative control were synthesized by RiboBio (Guangzhou, China).
Animals and treatments
All experiments involving animals adhered to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and received approval from the Laboratory Animal Center, the Second Affiliated Hospital of Chongqing Medical University (approval no. IACUC–SAHCQMU-2024-00056). C57BL/6 mice (8 weeks old) were obtained from Enbokang Bioscience (Chengdu, China), housed in an Specific pathogen-free (SPF) barrier system under a regular 12 h light/dark cycle with free access to the standard laboratory chow diet.
In Mice Experiment 1, NINJ1fl/fl mice, Myh6-Cre/Esr1-Tg mice, and wild type C57BL/6J were purchased from model organisms (Shanghai, China). Cardiomyocyte-specific NINJ1 knockout mice were generated by NINJ1fl/fl mice crossed with Myh6-Cre/Esr1-Tg mice and then intraperitoneally injected with tamoxifen (20 mg/kg) daily for 5 days (Supplementary Fig. S1A and B). The 12-week-old mice were exposed to DOX treatment for four times to simulate clinical cardiotoxicity. Echocardiography and tissue samples were performed 1 week after the last injection (Fig. 2B).
In Mice Experiment 2, mice were subjected to a single tail vein injection (2 × 1011 viral genome/mouse) of AAV9-NINJ1 or AAV9-NC. One month after the AAV9 was injected through the tail vein, the mice were intraperitoneally administered either normal saline (NS) or DOX (4 mg/kg) every week for four times and evaluated their cardiac function (Fig. 3B).
In Mice Experiment 3, in order to verify the effect of NINJ1 inhibitor PDG, mice (12 weeks old) received PDG by intraperitoneal injection at a dose of 100 mg/kg diet from 1 week before the first DOX treatment to sacrifice (Sheng et al., 2023). Then, male mice were injected with DOX (4 mg/kg, i.p.) for 4 consecutive weeks (Hu et al., 2020a). One week after the final DOX treatment, parameters of cardiac function were recorded (Fig. 9A). Then, all mice were euthanized by inhalation of 3% isoflurane and perfused with 0.9% saline. Part of the heart tissues from sacrificed mice was immediately frozen in liquid nitrogen and stored at −80°C, while other tissues were fixed with 4% paraformaldehyde and stored at 4°C for further research.
ELISA detection
Serum samples collected from sacrificed mice were stored at 4°C before analysis. Then, the samples were centrifuged at 3000 rpm for 15 min at 4°C to collect the supernatant for testing immediately. Serum CK-MB and LDH levels in mice were measured using a mouse standard enzyme-linked immunosorbent assay (ELISA) kit purchased from Servicebio (GM1120&GM1122, Wuhan, China), according to the manufacturer’s guidelines.
Echocardiography
Echocardiographic analysis was evaluated to assess the cardiac function of the DOX-induced cardiomyopathy model mice and control mice in anesthetized mice by transthoracic echocardiography (VINNO 6 LAB ultrasonic diagnostic apparatus, Suzhou, China). Mice were maintained in an anesthetized state using 1.5% isoflurane via inhalation (Zhang et al., 2025). The parameters of cardiac systolic function, including left ventricular ejection fraction and fractional shortening, were measured and averaged from at least five consecutive cardiac cycles by ventricular M-mode ultrasound.
Histological analysis
Myocardial samples were fixed in 4% paraformaldehyde and subsequently embedded in paraffin. The embedded tissues were sectioned into consecutive 5-μm-thick slices and stained with HE for the detection of cardiomyocyte size. More than 30 fields were included for calculating the cross-sectional area of cardiomyocytes in each group, with at least five cardiomyocytes per field analyzed (Zhang et al., 2024). The chosen fields per slide were analyzed and averaged via Image-Pro Plus 6.0 software.
For IHC staining, paraffin-embedded sections were deparaffinized in xylene, rehydrated through a graded ethanol series, and subjected to heat-induced antigen retrieval using citrate buffer. The sections were then incubated overnight at 4°C with primary antibodies against NINJ1, HIF-1α, p-AMPKα (Thr172), or nonspecific immunoglobulin as a negative control, followed by hematoxylin counterstaining of nuclei. Images were obtained using a light microscope (Leica German) and analyzed by ImageJ.
Frozen sections were subjected to DHE staining to detect superoxide anion. Slices were incubated with 10 μM DHE working solution at 37°C for 30 min in the dark, followed by washing with PBS to remove residual dye. Fluorescent photographs were observed from random fields per section by Nikon Digital Sight 10 (Tokyo, Japan).
Apoptosis was detected using CF488 TUNEL cell apoptosis detection kit at 37°C in the dark for 1 h, followed by 4′,6-diamidino-2-phenylindole (DAPI) incubation. Images were acquired using a Nikon microscope. Six randomly chosen fields per slide were quantitatively analyzed via ImageJ software.
Immunofluorescence staining
Cells were seeded on cell coverslips in 12-well plates at a confluency of nearly 70%. After indicated treatment, cells were washed with PBS and fixed in 4% formaldehyde for 15 min. Next, cells were permeabilized by 0.5% Triton X-100 for 15 min and then blocked with 5% bovine serum albumin at 37°C for 30 min. Cells were incubated with primary antibody overnight at 4°C. After washing with phosphate-buffered saline (PBS) for 5 min for three times, cells were combined with fluorescence wavelength 594 or 488 secondary antibody at 37°C for 1 h. Finally, the antifade reagent with DAPI was used to cover the coverslip and stored in the dark at 4°C. Cell images were observed through Nikon Digital Sight 10, and results were analyzed via ImageJ software.
Cell culture and treatment
H9c2 and HeLa cell lines were provided by Pricella Biotechnology (Wuhan, China). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, US) supplemented with 10% fetal bovine serum (Ncmbio, Suzhou, China) and 1% penicillin/streptomycin (Gibco, US) in a 37°C humidified incubator with 5% CO2. H9c2 cells were treated with 1 μM DOX for different time periods (0, 6, 12, and 24 h) and different DOX concentrations (0, 0.3, 0.6, and 1 μM) to determine the optimal concentration and stimulation time point of DOX treatment. H9c2 cells were stimulated with 1 μM DOX, and HeLa cells were stimulated with 2 μM for 24 h in subsequent experiments. In transfection studies, H9c2 was seeded at a density of 1.0 × 106 cells/well in 6-well plates and incubated at 37°C for 24 h. In order to evaluate the role of NINJ1 silencing in H9c2, small interfering RNA against NINJ1 designed by OriGene (Guiyang, China) was used to knock down the expression of NINJ1, and the control group was transfected with negative control siRNA (siNC). siRNA duplexes were transiently transfected into H9c2 cells using Lipofectamine 2000 reagents (Invitrogen, US). Six hours after siRNA incubation, cells were kept in DMEM supplemented with 10% serum for an additional 24 h and then treated with PBS or 1 μM DOX for 24 h before cell collection. To explore the role of NINJ1 overexpression in H9c2, plasmids for the overexpression of NINJ1 generated by GenScript (New Jersey, US) and a negative control plasmid vector were transfected into H9c2 cells. Plasmid was transfected into H9c2 cells by Lipofectamine 3000 and P3000 (Invitrogen, US). Six hours after plasmid incubation, cells were kept in DMEM supplemented with 10% serum for an additional 24 h and then treated with PBS or 1 μM DOX for 24 h before cell collection. To test the half-life of HIF-1α, H9c2 cardiomyocytes were treated with 10 μM CHX at the indicated time points (0, 4, 8, and 12 h) with 1 μM DOX incubation. To inhibit ubiquitination-dependent degradation of HIF-1α, H9c2 cells were pretreated with 10 μM MG132 for 1 h prior to DOX exposure. In rescue experiments, after being transfected with siRNA or plasmid, H9c2 was pretreated with 5 μM CC or 2 mM AICAR for 1 h to inhibit or activate the AMPK pathway before DOX treatment and then treated with PBS or 1 μM DOX for 24 h before cell collection. To clarify whether the NINJ1 inhibitor PDG affects tumor-killing ability by DOX in vitro, we incubated H9c2 and HeLa with different concentrations (0, 50, 100, 200, and 300 mM) of PDG during DOX stimulation and then selected 100 mM as the optimal concentration for further studies.
The isolation of primary NRVMs
NRVMs were isolated from 2-day-old Sprague-Dawley (SD) rat hearts. After disinfecting the newborn rats with 75% alcohol, the hearts were excised and immersed in cold PBS. The washed ventricular myocardium was sliced into ∼1 mm3 pieces and placed into a 37°C digesting solution containing 0.125% trypsin for 8 min. The supernatant was collected, and digestion was stopped by adding the stop solution (10% DMEM). The above processes were repeated five to six times until the ventricular myocardium was completely digested. Subsequently, the centrifuged cell pellets were resuspended in 10% DMEM in a plate for 1 h in a 37°C incubator with 5% CO2 to let fibroblasts attach. Most of the non-cardiomyocytes were removed by differential adherence, and culture medium for NRVMs was supplemented with 100 μM 5-BrdU to block the proliferation of remaining non-cardiomyocytes. After cell attachment, NRVMs were exposed to 1 μM DOX.
Nuclear and cytoplasmic fractionation
H9c2 cells with the indicated treatment were washed with PBS and scraped off, followed by centrifugation (3000 rpm, 3 min). Cytoplasmic protein extraction reagent A supplemented with phenylmethylsulfonyl fluoride (PMSF) was added to the cell pellet and incubated on ice for 15 min. Then, cytoplasmic protein extraction reagent B was added to the cell pellet and incubated on ice for 4 min. The mixture was centrifuged (12,000 rpm, 5 min) to separate the components and collect the supernatant as the cytoplasmic extract. The nuclear pellet was washed with PBS and then centrifuged (12,000 rpm, 30 s), and the supernatant was discarded as thoroughly as possible. Add nuclear protein extraction reagent containing fresh PMSF to the nuclear pellet, incubate on ice for 10 min, and perform ultrasonication (40 W, 30 s) to obtain the nuclear extract. Nuclear proteins and cytoplasmic proteins were analyzed separately by Western blot.
Cell viability and EdU cell proliferation assay
A total of 1.6 × 105 H9c2 cells were prepared and seeded onto 96-well plates with 100 µL volume per well. After indicated treatments, 10 µL WST-8 was added into each well and incubated for 1 h at 37°C. Then, the absorbance of each well was determined at 450 nm by a microplate reader varioskan lux (Thermoscientific, US). HeLa cells in 12-well plates with a density of 20,000 cells per well were incubated with 10 μM EdU working solution at 37°C for 2 h after the indicated treatment. Cells were washed with PBS three times and fixed in 4% formaldehyde for 15 min. Then, each well was added 0.25% Triton X-100 and incubated at room temperature for 15 min. After permeabilization, cells were reacted with the Click reaction solution at room temperature in the dark for 30 min. Finally, DAPI was added to the cell coverslips to stain the nucleus. Images were acquired on Nikon Digital Sight 10 and analyzed by ImageJ software.
LDH release assay
Cells were cultured at a seeding density of 2000 cells per well in 96-well plates for various treatments. After a determined time, the original medium was removed, and 120 µL cell lysis solution was added and incubated for 30 min. Next, the prepared LDH detection solution was added to each well at a volume of 80 µL and incubated for 30 min in the dark. Finally, the absorbance was determined at 490 nm.
DCFH-DA staining
CM-H2DCFDA was added into the 12-well plate to a final concentration of 5 μM after treatment, incubated for 30 min in the dark, and then excess probes were rinsed with PBS buffer three times. Green fluorescence signal intensity was observed in 495/530 nm on ZEISS microscopy (Oberkochen, Germany).
JC-1 staining
The MMP changes were detected using the MMP assay kit with JC-1 (Beyotime) as described in the product manual. Briefly, the transfected H9c2 cells were exposed to 1 μM DOX for 24 h. Then, wash the cells three times with ice-cooled PBS and incubate with JC-1 working solution for 30 min at 37°C in the dark. H9c2 without JC-1 staining was used as a negative control. Subsequently, cells were washed with PBS three times prior to observation by ZEISS microscopy (Oberkochen, Germany). JC-1 aggregates in mitochondria emit red fluorescence in healthy cells, while the JC-1 monomer released from mitochondria yields green fluorescence in unhealthy cells. Thus, the relative MMP potential was calculated as red fluorescence intensity/green fluorescence intensity.
Wound scratch assay
HeLa cells were seeded at 2.5 × 105 cells/well in 12-well plates at a confluency of nearly 100% and then scraped with a 200 μL disposable pipette tip. Then, the detached cells were removed by PBS washing, and each well plate was filled with 1000 μL DMEM medium containing 2% serum. The wound sizes were recorded at different time points (0, 12, 24, and 48 h) by an inverted microscope. The wound closure (%) was quantified by ImageJ and calculated as [(wound size at 0 h – wound size at different time points)/wound size at 0 h] × 100%.
Co-immunoprecipitation and ubiquitination assay
Mouse heart tissues and H9c2 cells were lysed in RIPA buffer supplemented with protease inhibitor and incubated on ice for 30 min. After centrifugation (12,000 rpm, 20 min), supernatants were collected and incubated with HIF-1α antibody on a rotator at 4°C overnight, followed by incubation with protein A/G magnetic beads for 4 h the next day. Next, the immunocomplexes were washed four times with RIPA buffer or PBST and then boiled in SDS loading buffer (Affinibody) for 3 min. Finally, Western blot was performed to detect the ubiquitination level of HIF-1α protein.
Western blot
Proteins were extracted from mice ventricles or cells with radioimmunoprecipitation assay (RIPA) lysis buffer containing 1% proteinase inhibitors (Boster), 1% PMSF, and 1% phosphatase inhibitor, and concentrations were determined by the BCA kit (Thermo Scientific). Nuclear and cytoplasmic proteins were isolated using a Nuclear and Cytoplasmic Proteins Extraction Kit (Beyotime). Equal amounts (20 μg) of protein samples were subjected to 10% SDS-PAGE separation and then transferred onto the polyvinylidene difluoride membrane (Millipore, Boston). Next, the membrane was blocked with Protein Free Rapid Blocking Buffer (Epizyme, Shanghai) for 10 min. After incubation with the indicated primary antibodies (1:5000 dilution) at 4°C overnight and the HRP-conjugated secondary antibodies (1:10000 dilution) at room temperature for 1 h, the protein bands were detected by enhanced chemiluminescent (NCM Biotech, Suzhou).
RNA-seq and data analysis
The total RNA extracted from the harvested hearts in the different treatment groups was utilized for transcriptome sequencing. Three biological replicates were performed in individual groups. Total RNA was isolated from murine heart tissue using TRIzol reagent. The concentration and purity of the extracted RNA were determined using a NanoDrop 2000, and the RNA integrity was evaluated by denaturing agarose gel electrophoresis. mRNA was isolated from total RNA and then randomly sheared into small fragments of ∼300 bp under appropriate conditions and reverse-transcribed into cDNA. RNA-seq was performed on an Illumina NovaseqTM 6000. Subsequently, DESeq2 was applied to analyze DEGs among multiple samples, with a screening threshold of p < 0.05 and |log2FC| > 1. The clusterProfiler package was used to perform Gene Ontology and KEGG enrichment analyses of DEG to interpret biological significance.
Statistical analysis
All data were reported as means ± standard error of the mean and analyzed with GraphPad 10.0 software. One‐way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was used to compare multiple groups. Statistical differences between two means were evaluated by the two-tailed unpaired Student’s t-test. p < 0.05 was considered statistically significant.
Authors’ Contributions
Z.L., Yankun C., and D.Y. contributed to the literature search and study design. Yankun C. and D.Y. participated in the drafting of the article. X.J. and Yankun C. carried out the experiments. Z.L., Yanghao C., and D.Y. revised the article. Y.Z., D.Y., and P.X. contributed to data collection and analysis.
Footnotes
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by the National Natural Science Foundation of China (grant number 82470267, 82170520, and 82500311), Program for Youth Innovation in Future Medicine, Chongqing Medical University (grant number W0078), and Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University, Chongqing, China.
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.