
Abstract
Aims:
This study aimed to delineate a novel mechanistic axis linking hyperglycemia-driven glycolytic reprogramming to ferroptotic death in lens epithelial cells (LECs) and to determine its therapeutic significance in diabetic cataract (DC). Specifically, we sought to define the integration of metabolic, epigenetic (histone lactylation), and post-translational (fucosylation) pathways in DC pathogenesis.
Results:
Under hyperglycemic conditions, LECs exhibited robust glycolytic activation and lactate accumulation. This metabolic shift drove selective histone H3K18 lactylation at the promoter of theTSTA3 gene, leading to its increased transcription. The upregulated TSTA3 protein then promoted the core fucosylation of the NF-κB p50 subunit, which facilitated its nuclear translocation. Inside the nucleus, p50 transcriptionally activatedNOX1, resulting in excessive reactive oxygen species (ROS) production and subsequent ferroptotic cell death. Critically, both pharmacological inhibition of glycolysis and genetic silencing ofTSTA3 effectively attenuated oxidative stress, restored redox balance, and ameliorated cataract severity in a diabetic rat model.
Innovation:
This work identifies a previously unrecognized pathogenic cascade—the glycolysis–histone lactylation–fucosylation–ferroptosis axis—that directly links metabolic flux to epigenetic and signaling control in DC. By positioning TSTA3 as a central, druggable node within this axis, our study redefines cataract pathogenesis beyond simple oxidative damage, integrating multiple layers of cellular regulation.
Conclusion:
The glycolysis–histone lactylation–TSTA3–fucosylation–NOX1–ferroptosis axis is a critical driver of LEC death in diabetic cataract. Targeting this newly defined pathway, particularly the TSTA3 node, offers novel opportunities for mechanism-based therapeutic interventions and biomarker development, with potential implications for other complications of metabolic disease.
Introduction
Diabetic cataract (DC) is a major cause of vision loss in patients with diabetes, characterized by progressive lens opacity (Pollreisz and Schmidt-Erfurth, 2010). Death of lens epithelial cells (LECs) is a critical event in DC pathogenesis, driven by hyperglycemia-induced metabolic disturbances such as oxidative stress (Yang et al., 2017) and aberrant protein modifications (Lu et al., 2014). Moreover, ferroptosis, an iron-dependent form of regulated cell death driven by lipid peroxidation, has recently been implicated in various diabetic complications (Li et al., 2021; Stockwell et al., 2017; Wang et al., 2022; Yang et al., 2022b).
In the diabetic milieu, increased glycolysis results in lactate accumulation (Fan and Monnier, 2021; Min et al., 2021; Tan et al., 2018). Lactate serves as a substrate for histone lysine lactylation (Kla), a newly identified epigenetic marker that directly regulates gene transcription (Zhang et al., 2019b). Concurrently, protein fucosylation, a key post-translational modification mediated by fucosyltransferases such as tissue-specific transplantation antigen P35B (TSTA3), plays vital roles in diverse pathological processes, including the regulation of cell death pathways (Yu et al., 2022; Zhang et al., 2019a, 2020).
Nevertheless, the potential interaction between these processes—specifically, whether glycolysis-driven histone lactylation can modulate fucosylation to affect ferroptosis in LECs—remains completely unknown. We hypothesized that under hyperglycemic conditions, lactate accumulation promotes histone lactylation at the TSTA3 promoter, thereby upregulating its expression. The resulting increase in the TSTA3-mediated fucosylation of NF-κB p50 facilitates its nuclear translocation and activation of NADPH oxidase 1 (NOX1), causing excessive reactive oxygen species (ROS) production and subsequent ferroptosis in LECs, thereby accelerating DC progression (Fig. 1). Hence, we conducted this study to define this novel mechanistic axis linking metabolic reprogramming to epigenetic and post-translational control of cell death in DC.
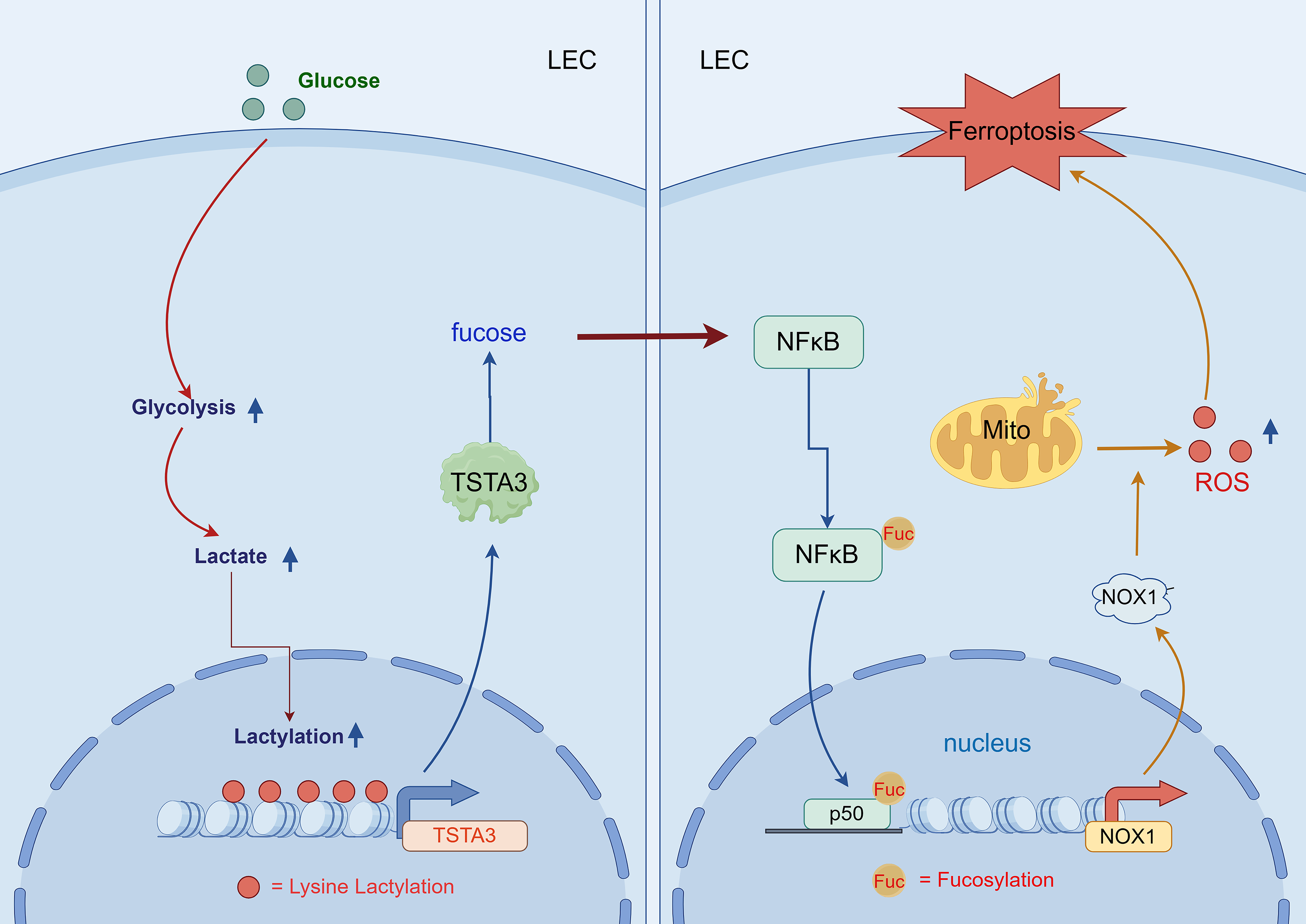
Innovation
This study uncovers a novel metabolic–epigenetic–post-translational axis in DC, where it first identifies histone H3K18 lactylation in LECs under hyperglycemia, linking glycolysis-driven lactate accumulation to TSTA3 upregulation; second, it reveals TSTA3-mediated fucosylation of NF-κB1 p50 as a key trigger for NOX1-dependent ferroptosis—a previously unrecognized role in cataractogenesis; and finally, it validates the TSTA3–NF-κB1 p50–NOX1 cascade as a druggable target, with both 2-DG and TSTA3 knockdown alleviating cataract in vivo. This study redefines DC pathogenesis beyond oxidative stress, providing novel biomarkers (e.g., H3K18la and fucosylated NF-κB1 p50) and therapeutic strategies targeting the metabolism–epigenetics interface.
Results
Aberrant glycolytic activation underlies oxidative stress in DCs
To define the metabolic basis of DC, we first characterized the pathological and metabolic features in a streptozotocin (STZ)-induced rat model. Slit-lamp examination and histological analysis revealed that diabetic rats developed progressive cortical opacities and significant damage to LECs, including cellular disarray and loss, compared with that in normal controls (NCs) (Fig. 2A–C). This structural damage was accompanied by a state of severe oxidative stress, as evidenced by markedly increased levels of ROS and superoxide anions in the epithelial cells of diabetic lenses (Fig. 2D, E).
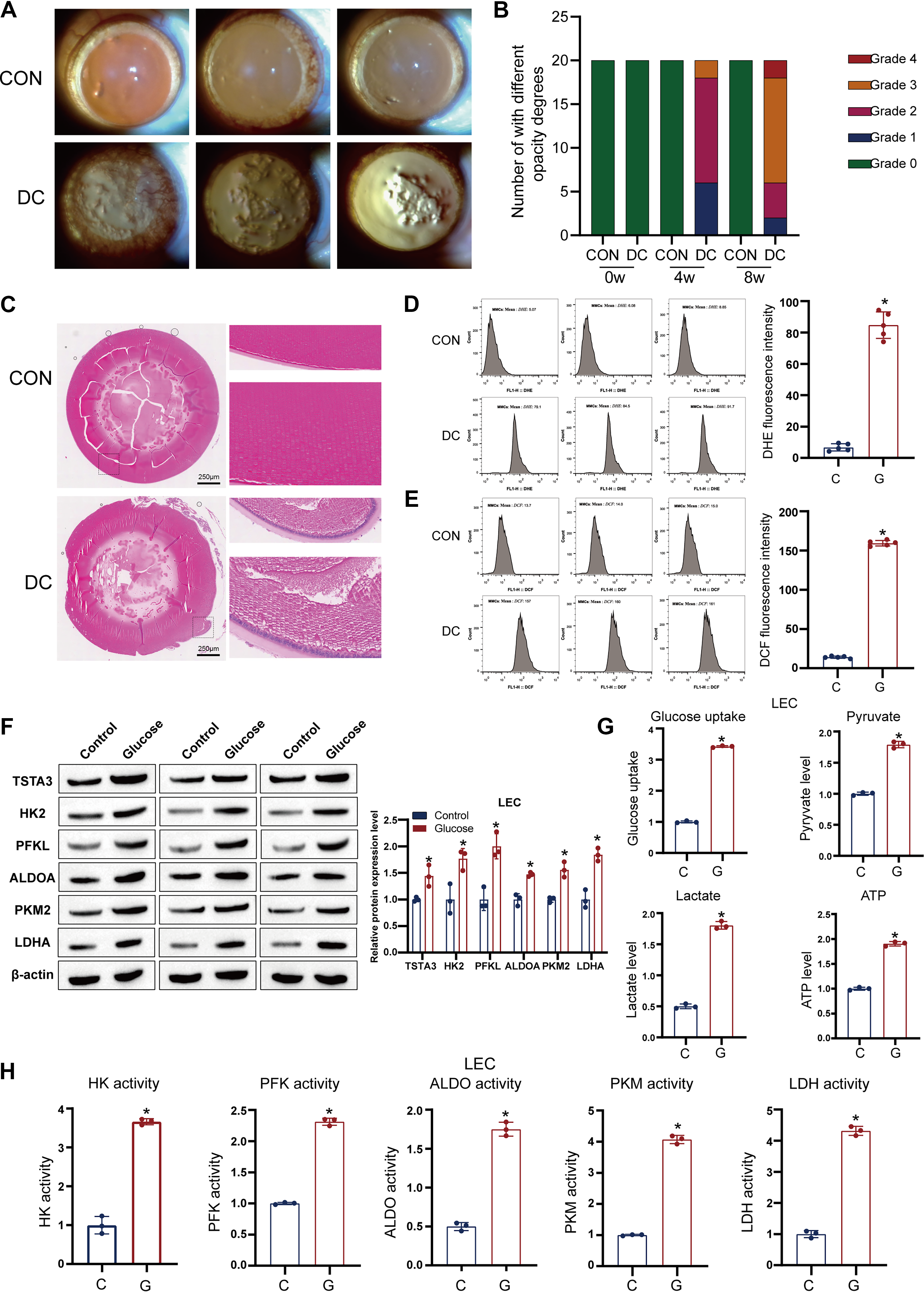
Considering that hyperglycemia is a known driver of glycolysis (Stitt, 2001), we hypothesized that aberrant glycolytic flux contributes to this pathology, and to test this hypothesis, we evaluated the glycolytic status of primary rat LECs cultured under high-glucose (HG) conditions. We found that HG treatment robustly upregulated the protein expression of key glycolytic enzymes, including HK2, PFKL, ALDOA, PKM2, and LDHA (Fig. 2F). Functionally, this upregulation translated into a significant increase in glucose uptake, lactate production, and Adenosine triphosphate (ATP) generation (Fig. 2G, H). These metabolic alterations were also detected in vivo, where diabetic lenses showed increased lactate and ATP levels compared with those in controls (Supplementary Fig. S1A, S1B). Overall, these findings establish that hyperglycemia induces a profound and sustained activation of glycolysis in LECs, which correlates with oxidative stress and cellular damage in DC.
Glycolysis-derived lactate induces H3K18 lactylation to activate TSTA3 transcription
We next explored the mechanism by which glycolytic activation causes changes in pathogenic gene expression. As lactate is a major end-product of glycolysis and a known substrate for histone lactylation (Wang et al., 2021; Yang et al., 2022a), we investigated its epigenetic role. Immunoblotting and immunofluorescence (IF) analyses revealed that HG treatment significantly increased both global histone lactylation and the specific modification at lysine 18 of histone H3 (H3K18la) in LECs (Supplementary Fig. S2A–S2D). This increase was dependent on glycolytic activity, because it was effectively reversed by treatment with the glycolysis inhibitor 2-deoxy-
To identify the direct transcriptional targets of this epigenetic mark, we conducted chromatin immunoprecipitation followed by quantitative polymerase chain reaction (ChIP-qPCR), which revealed a highly specific enrichment of H3K18la at the promoter region of the TSTA3 gene under HG conditions. In contrast, no significant enrichment was found for other lactylation markers, such as H4K8la and H3K9la (Supplementary Fig. S3A). The dependency of this enrichment on both glycolysis and the lactyltransferase EP300 was confirmed by demonstrating that 2-DG or EP300 knockdown (siEP300) abolished H3K18la accumulation at the TSTA3 promoter (Supplementary Fig. S3B, S3C).
Consequently, both HG and direct lactate treatment resulted in a significant upregulation of TSTA3 mRNA and protein levels (Supplementary Fig. S3D–S3F). Conversely, inhibiting glycolysis using 2-DG suppressed the expression of TSTA3, an effect that could be partially rescued by the addition of exogenous sodium lactate (Supplementary Fig. S3G, S3H). Most critically, knockdown of EP300 completely abrogated the ability of exogenous lactate to induce the upregulation of both H3K18la and TSTA3 (Supplementary Fig. S3I). These results demonstrate that glycolysis promotes TSTA3 transcription through an EP300-dependent, H3K18-specific lactylation mechanism.
TSTA3 is required for ferroptosis in LECs through NOX1 upregulation
After establishing TSTA3 as a key downstream effector of glycolytic signaling, we aimed to define its functional role in the fate of LECs. Considering the observed oxidative stress, we focused on ferroptosis. Knockdown of TSTA3 using specific siRNA (siTSTA3) in HG-treated LECs conferred significant protection against cell death, as measured by MTT and calcein–AM/PI assays (Fig. 3A–C). This protective effect was associated with a marked alleviation of ferroptotic markers, namely, reduction of intracellular ROS and malondialdehyde (MDA) levels, and restoration of glutathione (GSH) levels (Fig. 3D, E; Supplementary Fig. S4A–S4C).
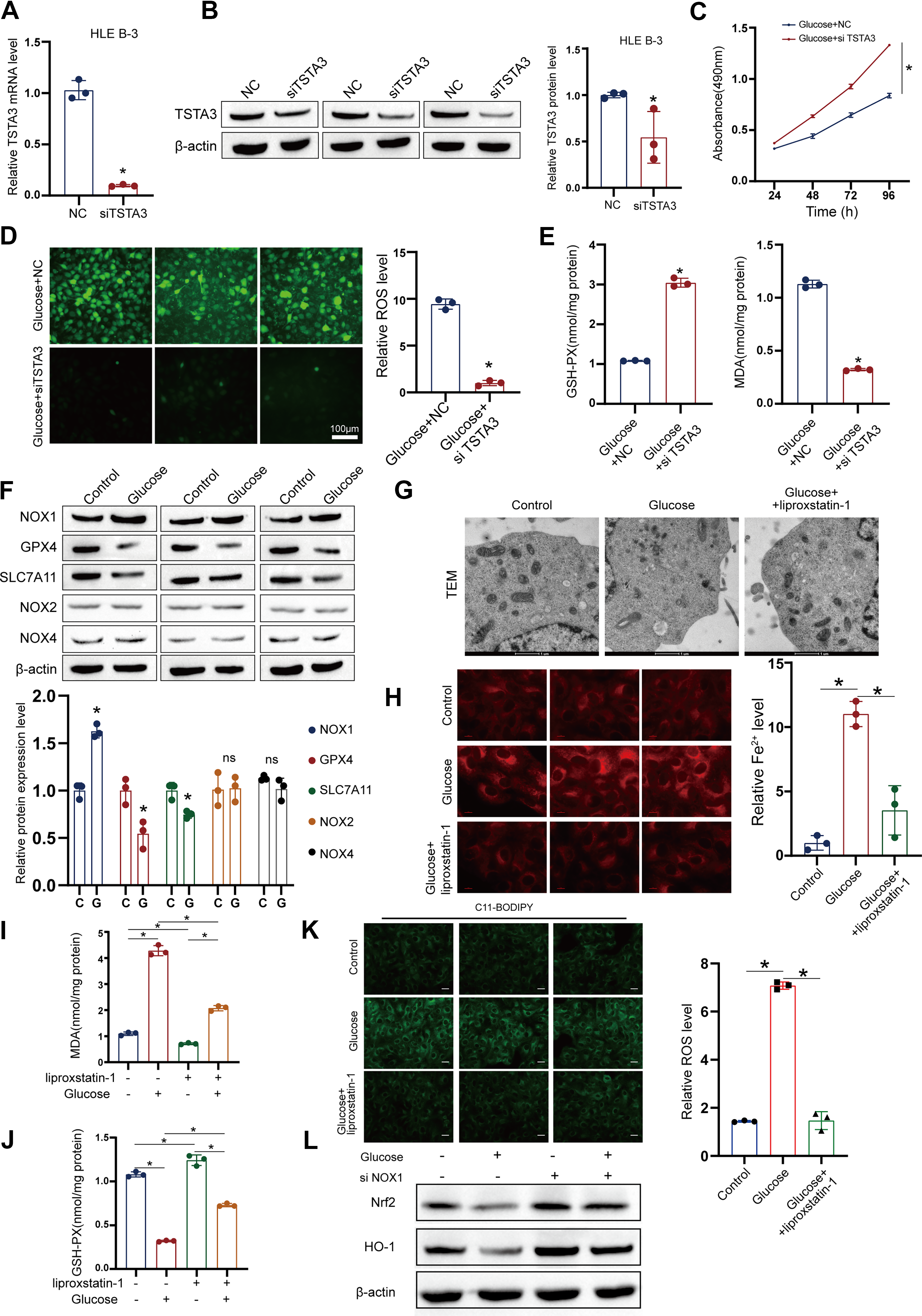
HG treatment induced the canonical hallmarks of ferroptosis in LECs, including upregulation of the pro-oxidant enzyme NOX1, downregulation of the key ferroptosis suppressors GSH peroxidase 4 (GPX4) and SLC7A11, and characteristic mitochondrial damage (shrinkage and increased membrane density) (Fig. 3F, G). However, all these pathological alterations were effectively reversed by the specific ferroptosis inhibitor liproxstatin-1 (Fig. 3H–K), thereby confirming ferroptosis as the primary mode of cell death. We next examined the status of the Nrf2 antioxidant pathway in the context of NOX1 upregulation, for which we investigated the expression of Nrf2 and its downstream target genes by Western blotting. NOX1-driven sustained stress results in Nrf2 inhibition (Fig. 3L). Mechanistically, we found that TSTA3 knockdown directly reduced NOX1 protein expression (Fig. 4A).
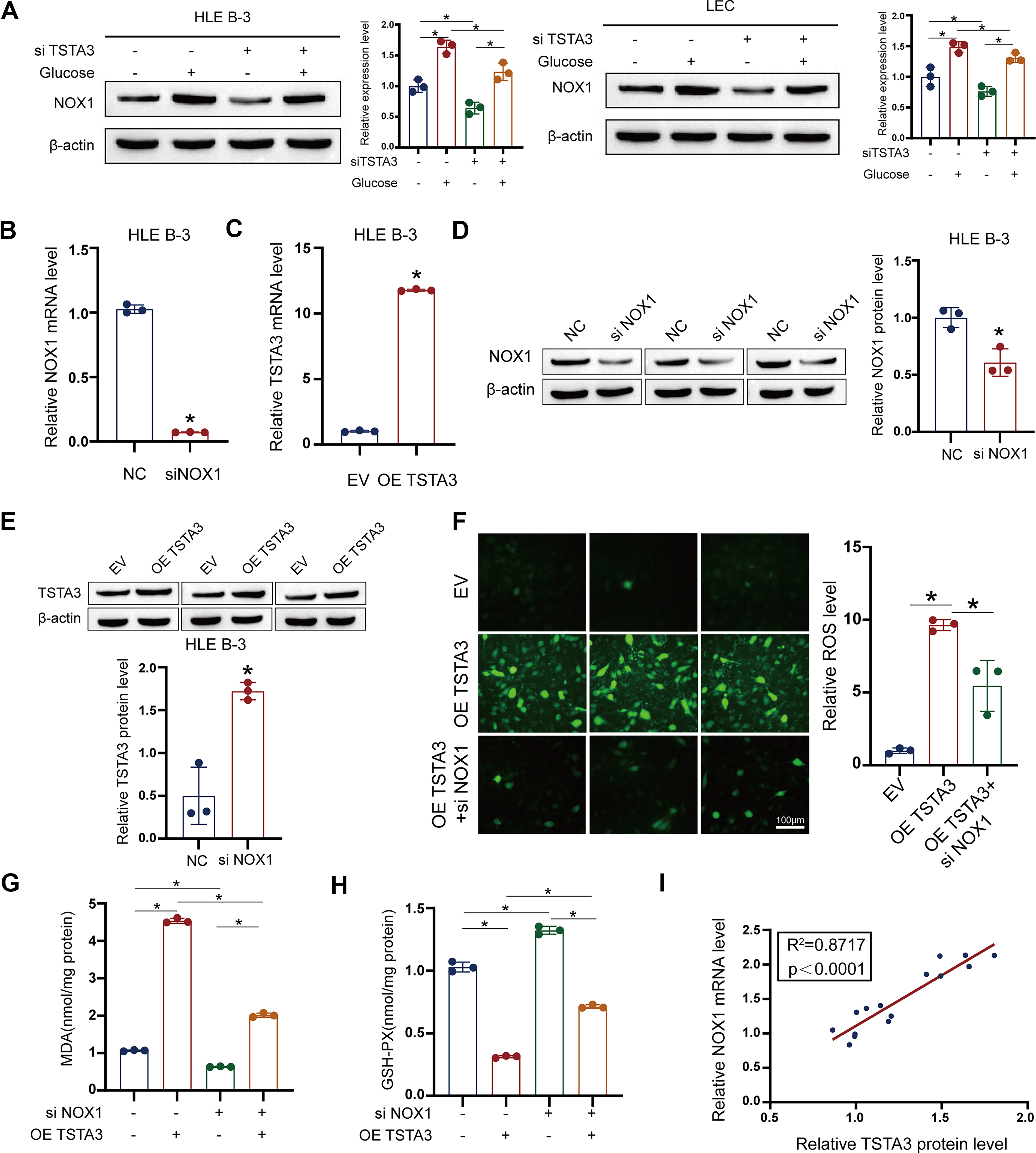
To establish a causal relationship between TSTA3 and ferroptosis, we conducted critical rescue experiments. Importantly, siRNA-mediated knockdown of NOX1 completely reversed the pro-ferroptotic effects of TSTA3 overexpression. In TSTA3-overexpressing cells, NOX1 knockdown normalized ROS and MDA levels; restored GSH, GPX4, and SLC7A11 expression; and prevented mitochondrial damage (Fig. 4B–H). And there is a strong correlation between TSTA3 and NOX1 expression (Fig. 4I). Moreover, TSTA3 overexpression sensitized LECs to the ferroptosis inducer RSL3, exacerbating cell death and lipid peroxidation (Fig. 5A–E). Collectively, these results firmly establish TSTA3 as an essential driver of ferroptosis in LECs, acting primarily through NOX1 upregulation.
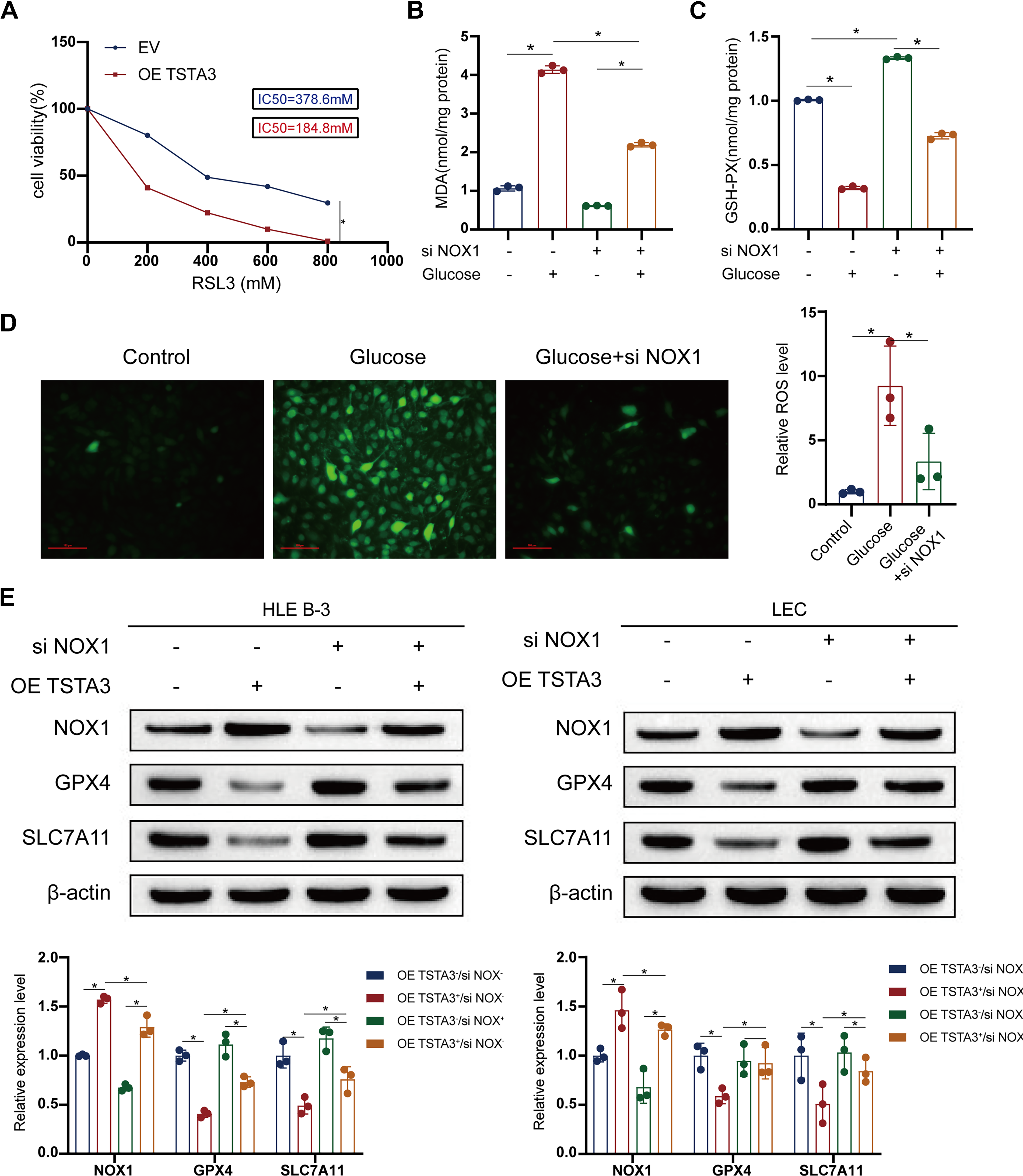
TSTA3 mediates NF-κB p50 fucosylation to enable its nuclear translocation and NOX1 transactivation
To clarify the molecular mechanism by which TSTA3 regulates NOX1, we investigated its potential role as a fucosyltransferase in modifying transcription factors (Zhang et al., 2020). Bioinformatic analysis of the NOX1 promoter identified binding sites for several transcription factors, with NF-κB p50 emerging as the top candidate based on prediction scores (Fig. 6A, B). After HG treatment, the expression levels of NF-κB p50 and NOX1 increased (Fig. 6C, D). This prediction was validated experimentally, where the results of ChIP-qPCR confirmed that NF-κB p50 binds directly to the NOX1 promoter (Fig. 6E), and a luciferase reporter assay demonstrated that NF-κB p50 overexpression significantly increased NOX1 promoter activity (Fig. 6F, G). Fluorescence in situ hybridization detected the interaction between NF-κB p50 protein and the NOX1 gene in LECs treated with HG or a fucosylation inhibitor (2 F-Fuc) and measurement of NF-κB1 transcriptional activity using an NF-κB1-dependent luciferase reporter construct (4xkB-Luc) in LECs (Fig. 6H, I).
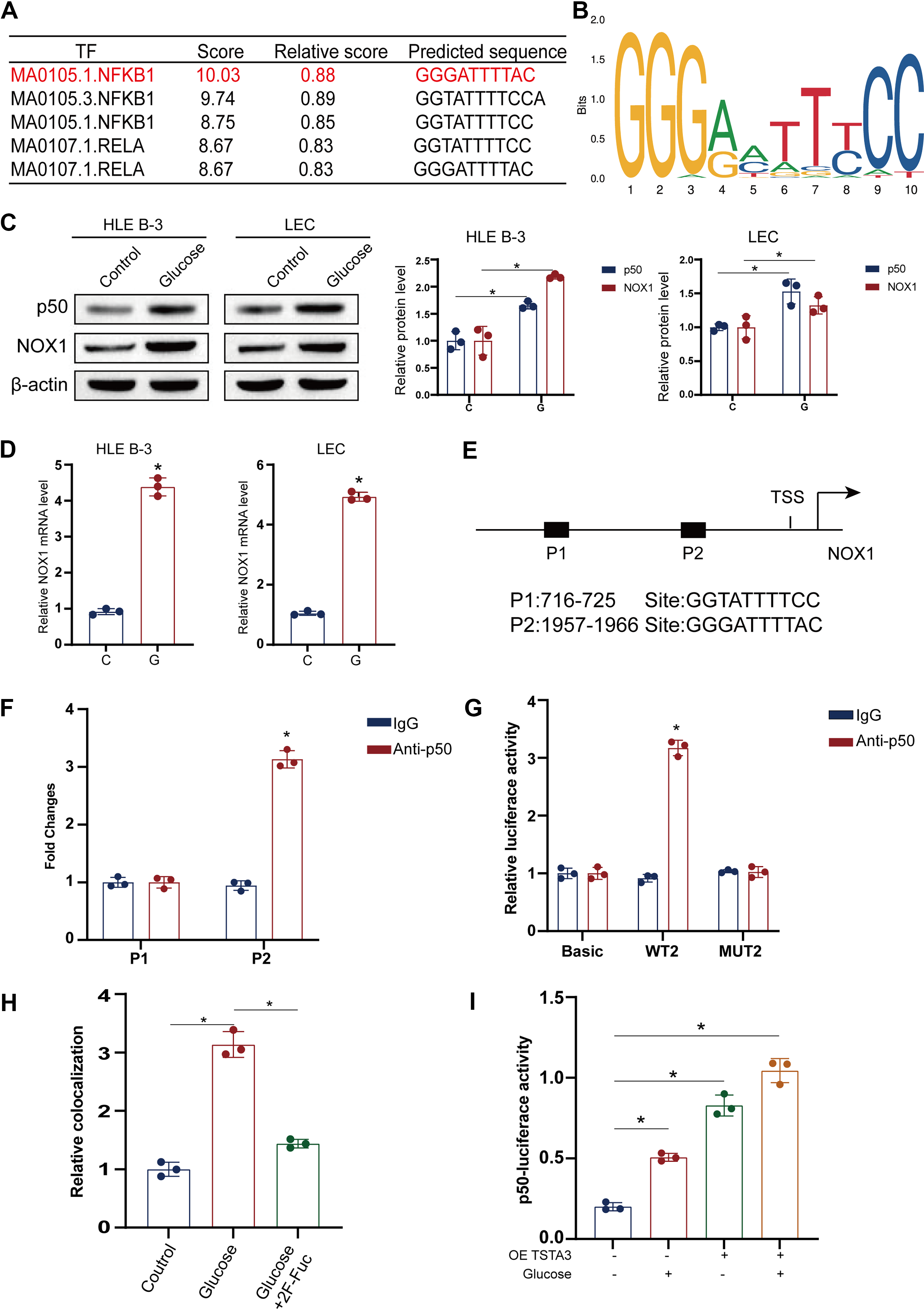
Based on the known function of TSTA3 (Fan and Monnier, 2021; Zhang et al., 2020), we hypothesized that it fucosylates NF-κB p50 to regulate its activity. First, we predicted the N-glycosylation sites in NF-κB p50 using the NetNGlyc1.0 server (Fig. 7A). The results of lectin affinity chromatography performed using lens culinaris agglutinin (LCA), which recognizes core fucosylation, confirmed that TSTA3 overexpression specifically increased the fucosylation level of NF-κB p50 without changing its total protein abundance (Fig. 7B, C). Next, through site-directed mutagenesis of the predicted N-glycosylation sites, we identified asparagine residues N247, N323, and N370 as the critical sites for TSTA3-mediated fucosylation (Fig. 7D). This interaction was specific, because it could be completed by free fucose and abolished by deglycosylation (PNGase F) (Fig. 7E–G).
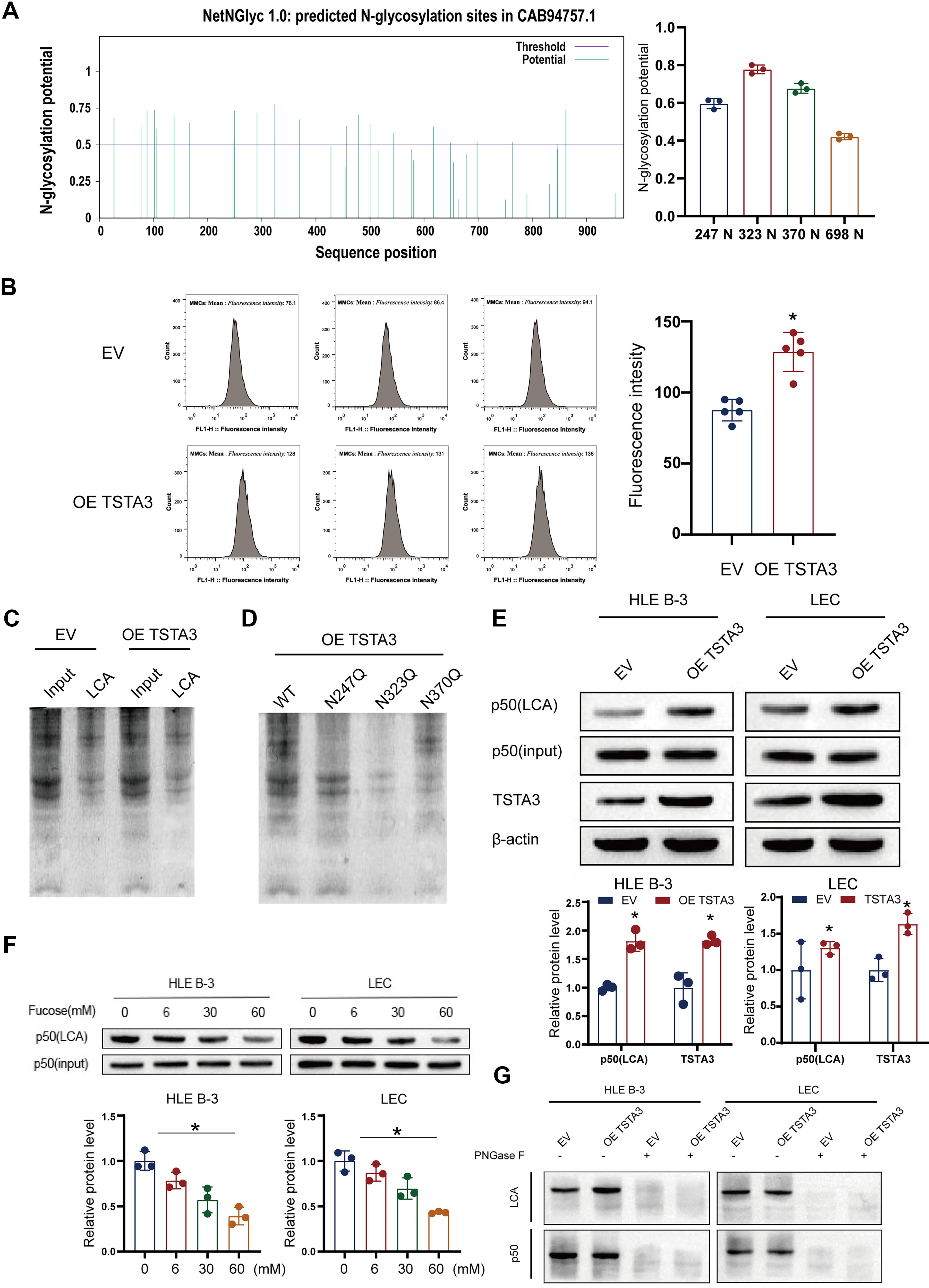
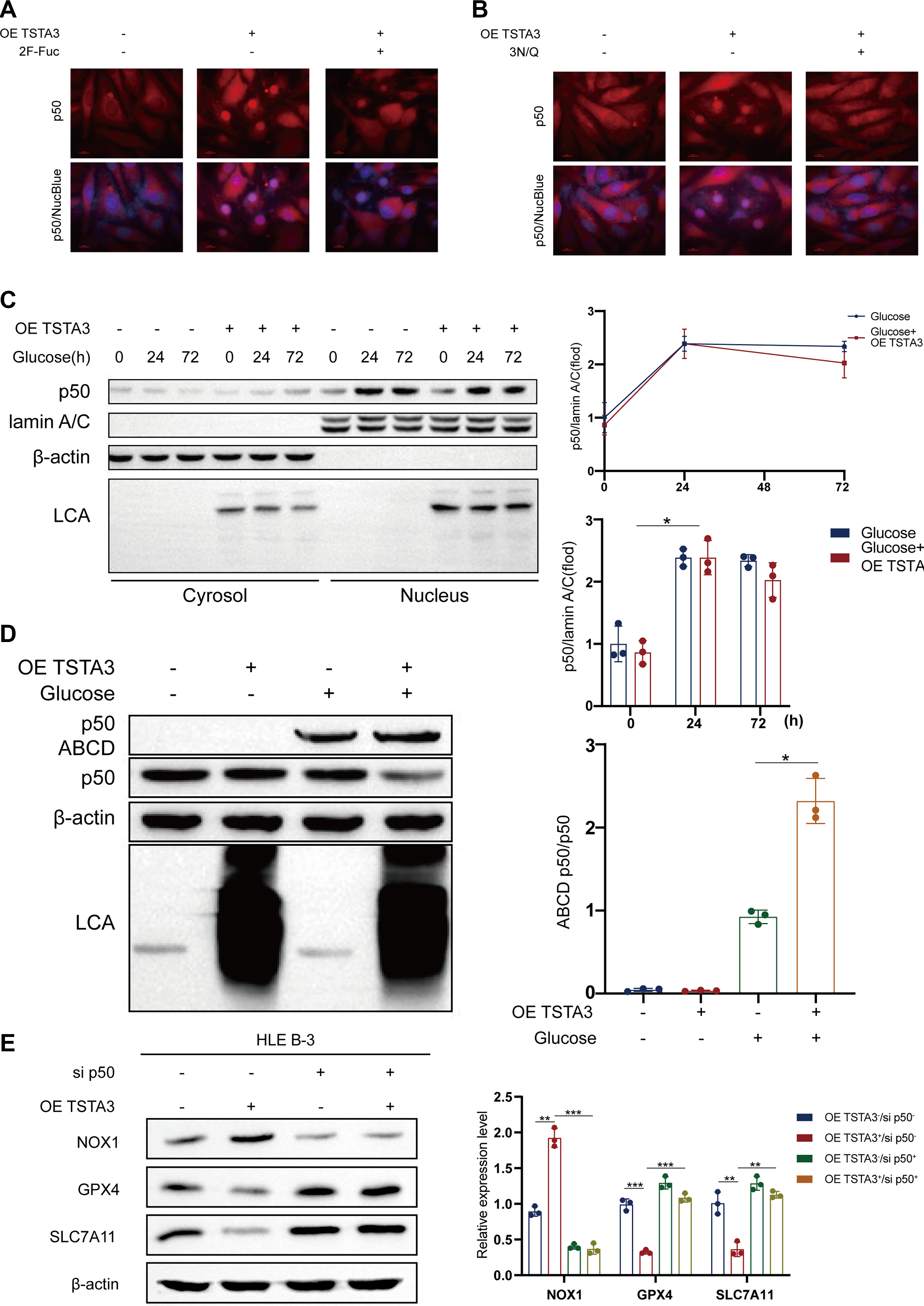
Functionally, this fucosylation event was vital for the activity of NF-κB p50. Both TSTA3 overexpression and HG treatment promoted the nuclear translocation of NF-κB p50, an effect that was blocked by treatment with a fucosylation inhibitor (Fig. 8A–C). Furthermore, TSTA3 improved the DNA-binding affinity of NF-κB p50 to the NOX1 promoter and its overall transcriptional activity, as demonstrated by an electrophoretic mobility shift assay and an NF-κB p50-responsive luciferase reporter, respectively (Fig. 8D; Fig. 6I). Importantly, the siRNA-mediated silencing of NF-κB p50 completely abolished the ability of TSTA3 overexpression to upregulate NOX1 (Fig. 8E). These findings demonstrate that TSTA3 acts as a critical regulator of the NF-κB p50–NOX1 axis by mediating NF-κB p50 fucosylation, which is indispensable for its nuclear function and the subsequent transactivation of NOX1.
In vivo targeting of glycolysis or TSTA3 ameliorates DCs
Finally, we investigated the therapeutic potential of targeting this newly identified axis in vivo, for which we treated diabetic rats with either the glycolysis inhibitor 2-DG or subjected them to adeno-associated virus (AAV)-mediated shRNA knockdown of TSTA3 via intravitreal injection. Both interventions significantly attenuated the progression of lens opacification, preserved the morphology of LECs, and reduced the levels of ROS compared with those in vehicle-treated diabetic controls (Fig. 9A–E). Furthermore, molecular analysis of the lens tissue revealed that both treatments effectively suppressed the entire pathogenic cascade, resulting in reduced expression of glycolytic markers, decreased levels of H3K18la, and downregulation of TSTA3, NF-κB p50, and ferroptosis indicators (Fig. 9F). These protective effects were dose-dependent and caused no observable systemic toxicity (Supplementary Fig. S5A–S5D).
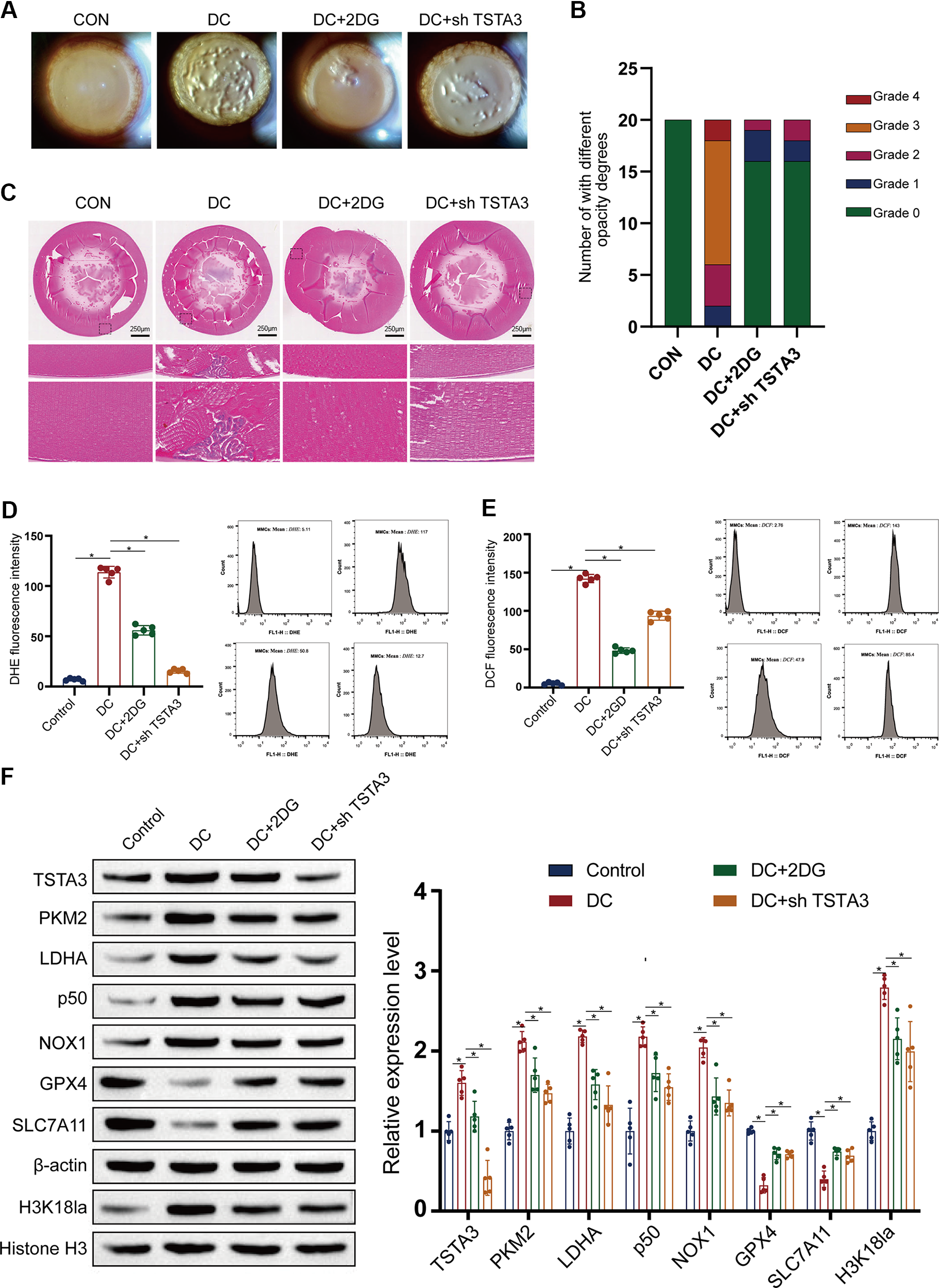
Importantly, to confirm the translational significance of our findings, we recapitulated the key experiments in human LECs (HLE-B3). In these cells, HG-induced ferroptosis, characterized by increased ROS/MDA and depleted GSH/GPX4 levels, was effectively rescued by either 2-DG treatment or TSTA3 knockdown with siRNA (Supplementary Fig. S5E, S5F). This cross-species validation emphasizes the vital role of the glycolysis–TSTA3–ferroptosis axis in human DC pathogenesis and its promise as a target for future therapeutic approaches.
Discussion
Key findings
This study identifies a previously unrecognized mechanistic axis in DC pathogenesis, that is, hyperglycemia-driven glycolysis promotes histone H3K18 lactylation at the TSTA3 promoter, resulting in TSTA3 upregulation, which, in turn, mediates NF-κB p50 fucosylation, nuclear translocation, and subsequent NOX1-dependent ferroptosis in LECs. This study integrates metabolic reprogramming, epigenetic regulation, post-translational modification, and a specific form of regulated cell death into a unified framework, thereby significantly advancing our understanding beyond the classical view of DC as merely a consequence of oxidative damage or osmotic stress.
Mechanistic insights
Our findings firmly establish TSTA3 as a vital, druggable node that actively links glucose metabolism to LEC fate. Although the roles of glycolysis in diabetes (Cai et al., 2020; Min et al., 2021) and ferroptosis in various diabetic complications (Li et al., 2021; Wang et al., 2022; Yang et al., 2022b) are established, the critical bridge connecting these processes—specifically through histone lactylation and protein fucosylation—had not been defined. Our study demonstrates that TSTA3 is not merely a passive marker but an active driver of ferroptosis. The specificity of H3K18 lactylation in regulating TSTA3 transcription was directly evidenced by ChIP-qPCR, where the results revealed its selective enrichment at the TSTA3 promoter under HG conditions (Supplementary Fig. S3A), an effect that is dependent on both glycolysis and the lactyltransferase EP300 (Supplementary Fig. S3B, S3C).
Functionally, the requirement of TSTA3 in this pathogenic cascade is unequivocal. Knockdown of TSTA3 vigorously protected LECs from HG-induced cell death (Fig. 3A–C) and alleviated key ferroptotic markers, including reduced ROS/MDA and increased GSH levels (Fig. 3D, E). Conversely, TSTA3 overexpression sensitized LECs to the ferroptosis inducer RSL3 (Fig. 5A–E). Importantly, we identified the downstream mechanism, that is, increased TSTA3 expression promotes core fucosylation of the NF-κB p50 subunit. The results of lectin affinity assays further confirmed that TSTA3 overexpression specifically increases NF-κB p50 fucosylation without altering its total protein level (Fig. 7B, C). Moreover, site-directed mutagenesis indicated N247, N323, and N370 as the critical fucosylation sites responsible for this modification (Fig. 7D).
This TSTA3-mediated fucosylation is functionally essential for the pro-ferroptotic activity of NF-κB p50. It facilitates the nuclear translocation of NF-κB p50 (Fig. 8A–C) and improves its DNA-binding affinity and transcriptional activity toward the NOX1 promoter, as validated by luciferase reporter assays (Figs. 6I, 8D). Most importantly, the siRNA-mediated silencing of NF-κB p50 completely abolished the TSTA3-driven NOX1 upregulation (Fig. 8E), demonstrating that NF-κB p50 is the indispensable effector linking TSTA3 to NOX1 activation. The entire axis culminates in NOX1-dependent ferroptosis, because genetic ablation of NOX1 completely reversed the pro-ferroptotic effects of TSTA3 overexpression, normalizing redox balance and restoring GPX4/SLC7A11 levels (Fig. 4B–H). This series of rigorous loss-of-function, gain-of-function, and rescue experiments provides compelling evidence for a linear and causal pathway, namely, glycolysis → H3K18la → TSTA3 ↑ → NF-κB p50 fucosylation → NF-κB p50 nuclear translocation → NOX1 transcription → ferroptosis.
Impact on the field
Current mechanistic studies on DC predominantly focus on classical oxidative or endoplasmic reticulum stress (Li et al., 2024; Zhang et al., 2023). Although ferroptosis is increasingly recognized in diabetic complications (Liu et al., 2024), it is traditionally viewed in DC as a passive lipid peroxidation outcome rather than an actively regulated epigenetic process. Moreover, although histone lactylation is increasingly recognized as a bridge between metabolic reprogramming and epigenetics (Zhang et al., 2024) and actively drives ferroptosis in other pathological contexts (Wu et al., 2024), its role in diabetic ocular complications remains entirely unexplored. Our study bridges this gap by revealing an unprecedented “glycolysis–lactylation–fucosylation” cascade. Unlike models treating metabolic reprogramming and cell death separately, our study demonstrates that glycolytic lactate directly drives H3K18 lactylation, subsequently orchestrating TSTA3-mediated protein fucosylation to trigger ferroptosis. This shifts the mechanistic understanding of DC from isolated biochemical accumulation to an integrated metabolic–epigenetic network, thereby redefining how hyperglycemia dictates the fate of LECs.
Clinical implications
The clinical implications are substantial. Current management of DC depends solely on surgical intervention at advanced stages (Chung et al., 2009; Pollreisz and Schmidt-Erfurth, 2010). Our findings suggest that targeting the upstream metabolic trigger (glycolysis with agents such as 2-DG) or the key molecular node TSTA3 could represent a novel pharmacological strategy to delay or prevent cataract onset in patients with diabetes. The conservation of this pathway in HLEs further supports its translational significance. Moreover, the components of this axis—such as H3K18la levels, TSTA3 expression, and fucosylated NF-κB p50—could serve as potential biomarkers for early detection or patient stratification, thus paving the way for precision medicine approaches in DC management.
Limitations
This study has limitations. Our primary evidence derives from rodent models and cultured cells; hence, validation in human diabetic lens tissues is required to confirm clinical applicability. Although we focused on H3K18la, other lactylation sites or concurrent epigenetic modifications may also contribute to the regulatory network. In addition, the long-term safety of systemic glycolysis inhibition for ocular applications needs careful evaluation to avoid off-target effects on other energy-dependent tissues such as the retina.
Future directions
Future investigations should aim at mapping the complete landscape of lactylation-driven gene regulation in LECs using multiomics approaches. Clinical cohort studies correlating the proposed biomarkers with cataract progression rates in patients with diabetes are warranted. Finally, the development of specific TSTA3 inhibitors or targeted delivery systems holds significant promise and merits rigorous preclinical testing for preventing this common and debilitating complication of diabetes.
Methods
Animal experimental methods
All procedures involving animals were conducted strictly according to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of Kunming Children’s Hospital (Approval No. 2023-03-050-K01). This study was reported in compliance with the ARRIVE 2.0 guidelines.
A total of 60 male Sprague–Dawley (SD) rats (aged 8 weeks, body weight 200–220 g) were obtained from Jasper Laboratories (Shanghai, China). Upon arrival, the animals were acclimatized for 1 week under controlled environmental conditions, namely, 12-h light/dark cycle (lights on at 7:00 AM), ambient temperature of 22°C ± 1°C, and relative humidity of 50%–60%. Standard rodent chow and autoclaved water were provided ad libitum. All rats were monitored daily for general health, and predefined humane endpoints (e.g., >20% body weight loss, severe lethargy, and signs of infection) were established to minimize suffering.
Rats were randomly assigned using a computer-generated randomization sequence into two groups, NC group (n = 15) that received intraperitoneal injection of 0.1 M citric acid–sodium citrate buffer (pH 4.2–4.5) and diabetes mellitus (DM) group (n = 45) that received a single intraperitoneal injection of STZ (60 mg/kg, Sigma-Aldrich, USA), freshly dissolved in the same citrate buffer on ice and administered within 15 min to prevent degradation.
At 3 days postinjection, nonfasting blood glucose levels were measured from tail vein samples using a calibrated Accu-Chek® Aviva glucometer (Roche Diagnostics, Germany). Rats with blood glucose levels of ≥16.7 mmol/L on two consecutive measurements were considered diabetic. Of the 45 STZ-injected rats, 36 met this criterion and were included in the DM cohort, and the remaining 9 rats were excluded due to failure to achieve sustained hyperglycemia.
At 2 weeks after diabetes confirmation—allowing sufficient time for the establishment of stable hyperglycemia and early metabolic dysregulation—10 rats from the NC group and 30 rats from the DM group were randomly selected for experimental interventions. The sample size was determined a priori based on pilot data exhibiting a 30% difference in retinal ganglion cell density between groups (standard deviation = 15%), with a statistical power of 80% and α = 0.05, requiring a minimum of n = 8 per subgroup. Accounting for an estimated 20% attrition due to STZ-related morbidity, initial group sizes were inflated accordingly.
The 40 selected rats were further randomized into four subgroups (n = 10 each) as follows: NC + vehicle; DM + vehicle; DM + AAV-mediated TSTA3 shRNA (intravitreal injection of 1 × 1010 viral genomes per eye); and DM + 2-DG (intraperitoneal injection, 100 mg/kg/day, dissolved in saline).
Interventions commenced on day 17 post-STZ administration and continued for 4 weeks. All outcome assessments—including blood glucose monitoring and histological analyses—were conducted by investigators blinded to group allocation.
At the end of the study, the animals were euthanized via an intraperitoneal overdose of sodium pentobarbital (150 mg/kg), followed by cervical dislocation as a secondary method, according to American Veterinary Medical Association guidelines.
Isolation and culture of primary LECs
Eyeballs retrieved from 1-month-old SD rats were enucleated and rinsed with phosphate-buffered saline (PBS) containing antibiotics. After removing the cornea and iris, the lens capsule with adherent epithelial cells was excised. The capsular membrane was dissected into 1 × 1 mm2 fragments and cultured in minimum essential medium supplemented with 20% fetal bovine serum (FBS), nonessential amino acids, and 50 µg/mL gentamicin.
Cells and culture
The HLE line HLE-B3 (ATCC CRL-11421) was maintained in Dulbecco’s modified Eagle medium containing 10% FBS and 15 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). The cells were incubated at 37°C in a humidified 5% CO2 atmosphere.
Histology and IF
Enucleated eyes were snap-frozen in chilled methanol/acetic acid (M-AA). For hematoxylin and eosin staining, tissues were embedded in paraffin, sectioned (5 µm), and stained using standard protocols. For immunohistochemistry and IF, M-AA-fixed tissues were embedded in optimal cutting temperature compound and sectioned (5–8 µm). The sections were blocked with 5% bovine serum albumin in PBS containing 0.2% Triton X-100 before incubation with primary antibodies. For IF, after secondary antibody incubation and 4′,6-diamidino-2-phenylindole nuclear counterstaining, slides were mounted and imaged using a Nikon A1R confocal microscope.
ROS detection
ROS and superoxide levels in lens capsule sections were quantified using 2ʹ,7ʹ-dichlorofluorescein diacetate (DCF) and dihydroethidium (DHE). Capsular flaps were incubated with 1 µM DHE or DCF at 37°C for 20 min in the dark. Fluorescence signals were captured using a Zeiss LSM microscope.
Western blotting
Protein lysates were extracted using radioimmunoprecipitation assay buffer and separated by sodium dodecyl sulfate–polyacrylamide gel (SDS–PAGE). Resolved proteins were transferred to PVDF membranes. The membranes were blocked and incubated overnight with primary antibodies at 4°C, followed by horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized using chemiluminescent reagents.
Transfection and infection
For gene knockdown, siRNA transfections were conducted using Lipofectamine RNAiMAX. For gene overexpression, plasmids were transfected using Lipofectamine 3000. For stable knockdown or overexpression, lentiviruses were produced by cotransfecting HEK293T cells and used to infect target cells with polybrene. AAV-mediated shRNA knockdown of TSTA3 was achieved via intravitreal injection in vivo.
Plasmids, siRNA, shRNA, and viruses
FLAG/HA-tagged eukaryotic expression plasmids were cloned into pcDNA3. For lentiviral overexpression, genes were cloned into pCDH vectors. shRNA constructs for lentivirus or AAV were inserted into appropriate vectors (e.g., pSIH-H1-Puro). The primer sequences are detailed in Supplementary Table S1.
RNA isolation and qPCR analysis
Total RNA was isolated using TRIzol reagent. cDNA synthesis and qPCR were performed according to the manufacturer’s guidelines. β-Actin and GAPDH served as endogenous reference genes, with relative expression levels calculated using the 2−ΔΔCt method. The primer sequences are provided in Supplementary Table S2.
Flow cytometry and lectin affinity assays for fucosylation
Cell surface fucosylation was examined by flow cytometry using fluorescein isothiocyanate-conjugated Ulex europaeus agglutinin 1. For the enrichment of fucosylated proteins, lectin affinity chromatography was performed using biotinylated LCA and streptavidin agarose beads, followed by elution with SDS–PAGE loading buffer or 100 mM α-
Metabolic assays
Glucose uptake and lactate, ATP, and key glycolytic enzyme activities (HK, PFK, ALDO, PK, and lactate dehydrogenase) were quantified using colorimetric kits according to the manufacturer’s instructions.
Chromatin immunoprecipitation
ChIP was performed using an anti-H3K18la antibody. Cross-linked chromatin was sheared by sonication and immunoprecipitated, and the enriched DNA was quantified by qPCR.
Dual luciferase reporter assay
HLE-B3 cells were cotransfected with luciferase reporter plasmids and a Renilla luciferase control plasmid. Firefly luciferase activity was normalized to Renilla luciferase activity to determine promoter activity.
Detection of lipid peroxidation and antioxidant activity
Lipid peroxidation was determined using the C11-BODIPY probe and by quantifying MDA levels. GSH peroxidase activity was measured to assess antioxidant capacity.
Statistical analysis
Data are expressed as mean ± standard deviation from at least three independent replicates. Comparisons between two groups were performed using Student’s t-test, and comparisons among multiple groups were performed using one-way ANOVA with Bonferroni post hoc correction. p < 0.05 was considered statistically significant.
Authors’ Contributions
D.T., H.W., and Z.L. made contributions to the conception and design; Y.W., L.L., S.L., and R.L. analyzed and interpreted data; D.T. drafted the article; N.N. revised it critically for important intellectual content. All authors approved the final version to be published.
Footnotes
Funding Information
This study is funded by the National Natural Science Foundation of China—Regional Science Foundation Project (Project Number: 82360203), the Joint Special Project of Kunming Medical University (Project Number: 202401AY070001-294), and the Medical and Health Talent Special Project of Yunnan Province under the “Support Program for Talents of Developing Yunnan.”
Ethical and Legal Declarations
This study and included experimental procedures were approved by Kunming Children’s Hospital. All animal experiments were approved by the Animal Care and Use Committee of the Ethical Institution of Kunming Children’s Hospital (No. 2023-03-050-K01).
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Disclosure Statement
The authors confirm that there are no conflicts of interest.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.