
Abstract
Aim:
The activation of microglia triggers an inflammatory response, which is frequently associated with an imbalance of iron metabolism. This study aimed to determine whether inflammation-associated iron dyshomeostasis contributes to impaired poststroke recovery and to explore the underlying mechanisms.
Results:
Ferroportin 1 (FPN1) deficiency in neurons and glial cells delayed sensorimotor function recovery following cerebral ischemia. FPN1 deficiency was associated with aggravated neuronal injury, enhanced apoptosis- and necroptosis-associated signaling, impaired myelin- and synapse-related repair, and reduced dendritic spine density in the ischemic cortex. Histological analyses, including hematoxylin and eosin staining and Nissl staining, further supported more severe peri-infarct pathological damage in Fpn1Nestin-CKO mice. In addition, increased IgG extravasation indicated aggravated blood–brain barrier (BBB) disruption and secondary neurovascular injury after stroke. These pathological changes were accompanied by increased iron accumulation in the ischemic cortex and altered expression of iron metabolism-related molecules. Elevated inflammatory cytokine expression and increased hepcidin levels were associated with disrupted brain iron homeostasis in Fpn1Nestin-CKO mice. Inhibition of JAK-STAT signaling with AG490 reduced p-STAT3 and hepcidin levels and was associated with modulation of iron-related and repair-associated responses, with more pronounced effects observed in Fpn1-deficient mice.
Innovation and Conclusion:
These findings highlight a close association between inflammatory signaling, BBB dysfunction, and iron dyshomeostasis during poststroke recovery. Our results suggest that delayed sensorimotor recovery in mice with neuronal and glial FPN1 deficiency may be linked to inflammation-associated BBB disruption and subsequent iron accumulation in the ischemic brain. Antioxid. Redox Signal. 00, 000–000.
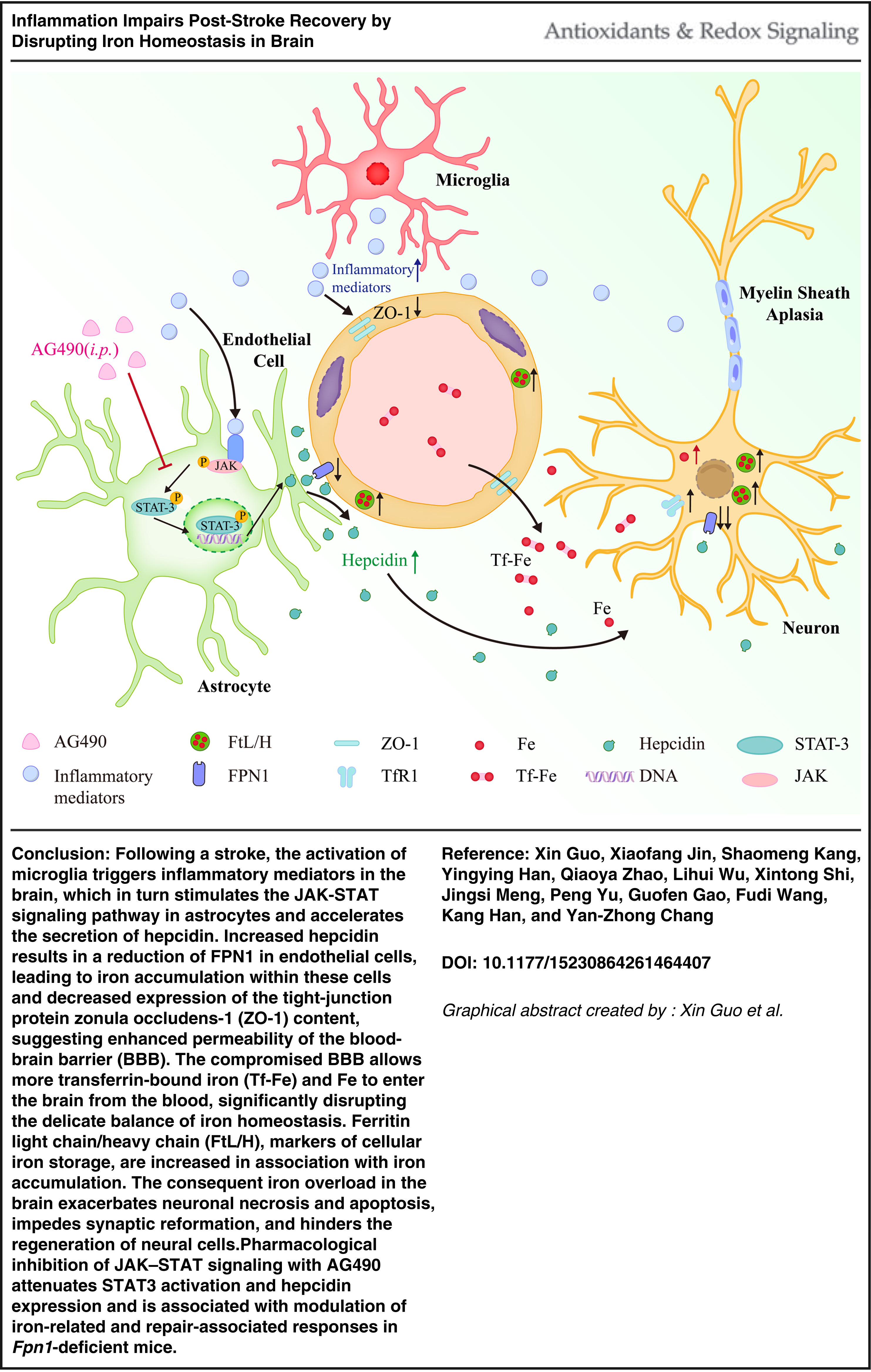
Introduction
Stroke remains a leading cause of mortality and long-term disability worldwide, with ischemic stroke accounting for the majority of cases (Hilkens et al., 2024). Despite advances in reperfusion therapies, a substantial proportion of patients continue to experience persistent neurological deficits, underscoring the need to better understand the mechanisms governing poststroke recovery (Campbell et al., 2019; Feigin et al., 2021). Recovery is not solely determined by the extent of the primary ischemic insult but is critically shaped by the dynamic interplay between secondary injury processes and endogenous repair mechanisms within the peri-infarct region (Cassidy and Cramer, 2017; Murphy and Corbett, 2009).
Poststroke pathology evolves in a time-dependent manner. The acute phase (hours to days) is characterized by excitotoxicity, oxidative stress, and rapid activation of innate immune responses, whereas the subacute and chronic phases involve sustained neuroinflammation, blood–brain barrier (BBB) dysfunction, and tissue remodeling, all of which critically influence functional recovery (Iadecola and Anrather, 2011; Wei et al., 2023). Among these processes, neuroinflammation has emerged as a key determinant with dual roles, capable of promoting support repair or exacerbating injury depending on its magnitude and duration (Hu et al., 2015; Jayaraj et al., 2019). Activated microglia, astrocytes, infiltrating immune cells, and injured neurons release inflammatory mediators that shape neuronal survival and the local tissue microenvironment (Candelario-Jalil et al., 2022). However, persistent or excessive inflammation can exacerbate secondary brain injury, disrupt BBB integrity, and impair neurorepair.
Innovation
Although inflammation and iron dysregulation have been independently associated with ischemic stroke, their interaction during poststroke recovery remains incompletely understood. In the present study, Fpn1 deficiency was associated with enhanced activation of JAK-STAT signaling, increased hepcidin expression, disrupted iron homeostasis, aggravated BBB damage, and impaired neurological recovery after cerebral ischemia. Pharmacological inhibition of JAK-STAT signaling with AG490 produced genotype-dependent effects, with more pronounced modulation of hepcidin signaling, inflammatory responses, and repair-associated processes observed in Fpn1-deficient mice. Collectively, these findings support an interaction between inflammatory signaling and iron metabolism during stroke recovery and suggest that the JAK-STAT3-hepcidin axis may represent a context-dependent regulator of poststroke pathology under conditions of disrupted iron homeostasis.
Emerging evidence highlights a close interplay between inflammatory signaling and iron metabolism. Inflammation regulates systemic and cellular iron homeostasis primarily through the JAK-STAT-hepcidin axis (Zhang et al., 2025). Hepcidin, a central iron-regulatory hormone, induces internalization and degradation of ferroportin 1 (FPN1), the only known cellular iron exporter, thereby limiting iron efflux and promoting intracellular iron retention (You et al., 2022). Disruption of brain iron homeostasis is increasingly recognized as a key contributor to poststroke pathology. During the acute phase, elevated levels of labile iron promote reactive oxygen species (ROS) generation via Fenton chemistry, leading to lipid peroxidation and neuronal injury (Ward et al., 2014; Yoshida et al., 2019). Consistently, iron chelation with deferoxamine has been shown to reduce neuronal iron accumulation and attenuate neuroinflammation-associated injury during cerebral infarction (Nemeth et al., 2004).
Importantly, inflammation-driven iron sequestration may further exacerbate neuronal vulnerability after stroke (Tuo et al., 2017). Although conditional deletion of Fpn1 disrupts brain iron homeostasis (Wu et al., 2021), the functional consequences of impaired neuronal iron export during poststroke recovery remain unclear. In particular, whether inflammation-induced alterations in the hepcidin-FPN1 axis contribute to iron dyshomeostasis and thereby impede recovery has not been fully elucidated.
In the present study, we employed Nestin-Cre-mediated conditional knockout mice to investigate how disruption of iron export affects poststroke recovery. We further examined the interaction between neuroinflammation and iron metabolism, with a focus on the JAK-STAT-hepcidin pathway. Our findings demonstrate that loss of Fpn1 is associated with increased iron accumulation, exacerbated inflammatory responses, and impaired functional recovery following ischemic stroke, underscoring the critical role of iron homeostasis as a potential therapeutic target.
Results
Nestin-Cre-mediated Fpn1 knockout impairs sensorimotor functional recovery after cerebral ischemia
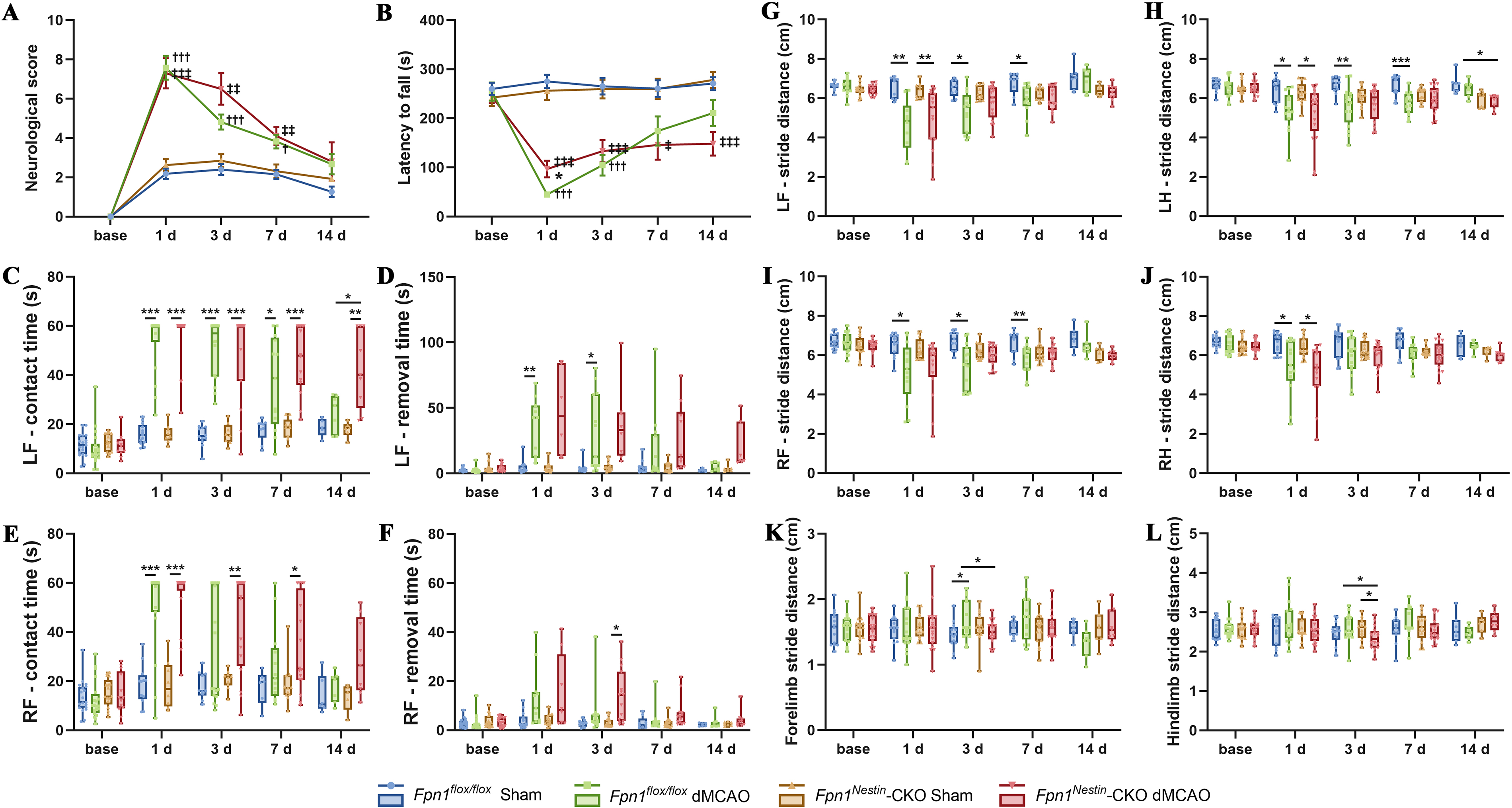
To evaluate functional recovery after stroke, we conducted modified neurological severity score (mNSS), rotarod tests, adhesive removal tests, and gait analysis tests in mice with or without conditional Fpn1 knockout before distal middle cerebral artery occlusion (dMCAO) and on days 1, 3, 7, and 14 after the operation (Supplementary Fig. S1C). At day 1 after dMCAO, mNSS scores did not differ significantly between groups. However, during the recovery phase, Fpn1Nestin-CKO dMCAO mice exhibited significantly higher mNSS scores than Fpn1flox/flox dMCAO group on days 3 and 7. On day 14, mNSS scores in both groups approached sham levels (Fig. 1A). The rotarod test showed the residence time of Fpn1flox/flox dMCAO mice was significantly shorter than that of the Fpn1Nestin-CKO dMCAO group on day 1. However, on day 14, the residence time of Fpn1Nestin-CKO dMCAO group was significantly shorter compared with the Fpn1flox/flox dMCAO group (p < 0.05, Fig. 1B). The adhesive removal test showed that on day 14, both the contact time and removal time for the left and right limbs in the Fpn1flox/flox dMCAO group returned to a control level, while these the parameters of left forelimb (LF)/right forelimb (RF)-contact time and LF-removal time in the Fpn1Nestin-CKO dMCAO group remained significantly higher than the control level (p < 0.05, Fig. 1C–F). The results of gait analysis experiments reveal that on day 14, the step amplitude of LF, RF, left hindlimb and right hindlimb, and the standing width of forelimb standing and hind limb standing in the Fpn1Nestin-CKO dMCAO group were all different from those of the Fpn1flox/flox dMCAO group, with shorter stride, increased forelimb and hindlimb standing width (p < 0.05, Fig. 1G–L). In general, these results demonstrate that Nestin-Cre-mediated Fpn1 knockout delays sensorimotor functional recovery following cerebral ischemia.
Fpn1 knockout aggravates neuronal injury, BBB disruption, and enhances apoptosis- and necroptosis-associated signaling after cerebral ischemia
Neuronal injury after ischemic stroke involves multiple forms of cell death-related signaling. In the present study, we performed hematoxylin and eosin (H&E) and Nissl staining in the peri-infarct cortex to further validate tissue injury histologically. H&E staining revealed more severe histopathological damage in the peri-infarct cortex of Fpn1Nestin-CKO dMCAO mice compared with Fpn1flox/flox dMCAO mice. Quantitative analysis further confirmed a significant increase in tissue injury in Fpn1Nestin-CKO dMCAO mice (Fig. 2A–B). Nissl staining showed a marked reduction in surviving neurons in the peri-infarct cortex, which was significantly more pronounced in Fpn1Nestin-CKO dMCAO mice than in Fpn1flox/flox dMCAO mice (Fig. 2C–D). In addition, BBB integrity was further evaluated by IgG staining. Increased IgG leakage was observed in the injured cortex of Fpn1Nestin-CKO dMCAO mice, indicating exacerbated BBB disruption and secondary neurovascular injury following cerebral ischemia (Fig. 2E–F). To investigate the underlying mechanisms, we mainly evaluated markers related to apoptosis- and necrosis-associated signaling. The expression of receptor-interacting protein kinases RIP1 and RIP3 was significantly increased in the ipsilateral cortex of Fpn1Nestin-CKO dMCAO group compared with the Fpn1flox/flox dMCAO mice. Both RIP1 and RIP3 content on the infarct side in each group were significantly higher than that on the contralateral side (CS) (Fig. 2G–H). Notably, phosphorylation of mixed lineage kinase domain-like protein, a key executioner of necroptosis, was increased in Fpn1Nestin-CKO dMCAO mice, further supporting enhanced necroptosis-associated signaling after cerebral ischemia (Fig. 2G–H).
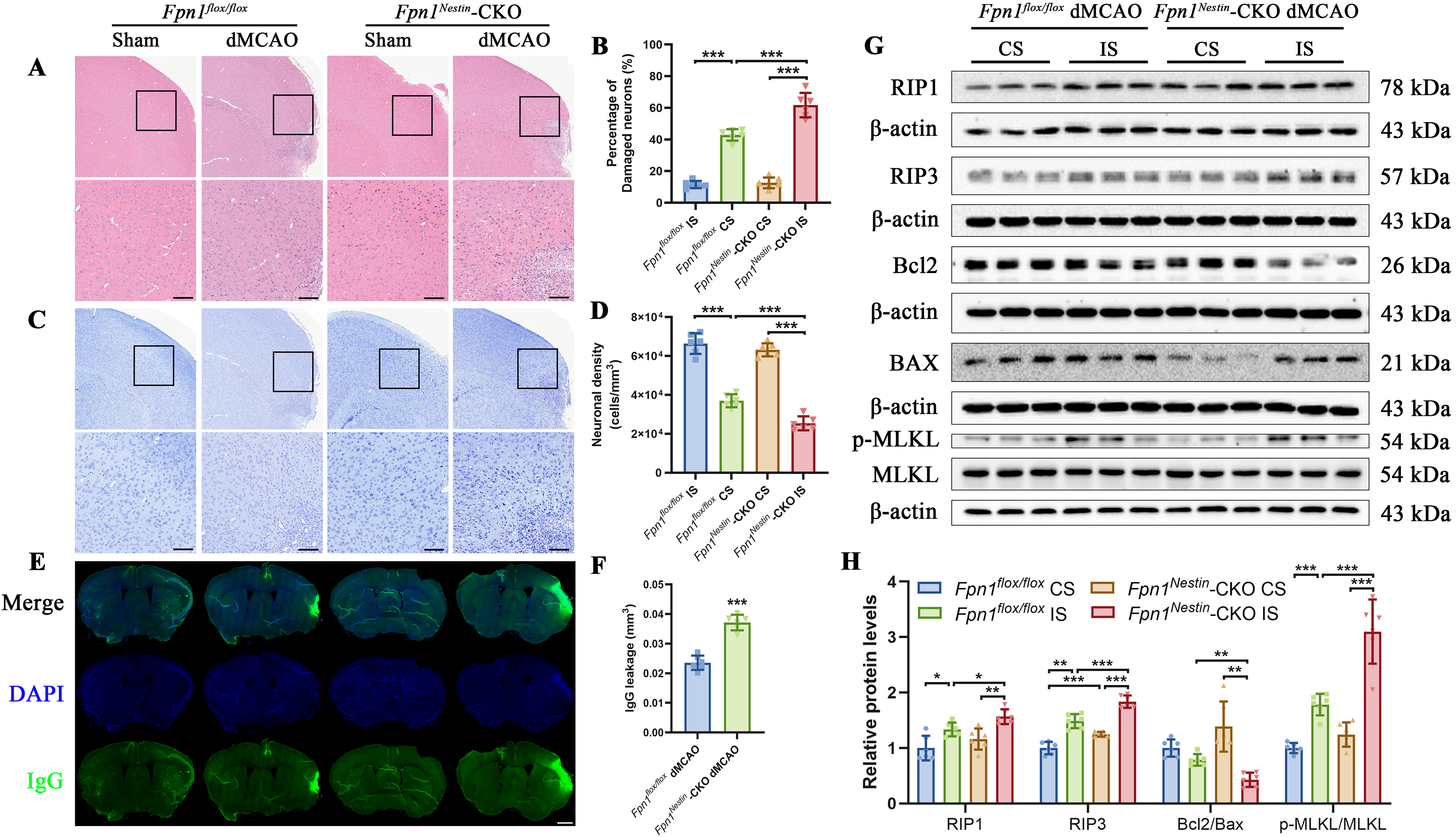
Apoptosis-related signaling was also assessed. The Bcl2/BAX ratio on the infarct side in the Fpn1Nestin-CKO dMCAO group was significantly more reduced than that in the Fpn1flox/flox dMCAO group (Fig. 2G–H). However, cleaved caspase3 levels did not differ significantly between groups (Supplementary Fig. S2A–S2B), suggesting that classical caspase-dependent apoptosis may not be prominently altered.
To further explore whether ferroptosis-related pathways were involved, we examined several ferroptosis-related proteins, including acyl-CoA synthetase long-chain family member 4 (ACSL4), glutathione peroxidase 4 (GPX4), and nuclear receptor co-activator 4 (NCOA4) (Doll et al., 2019). ACSL4 content on the infarct side was significantly increased compared with the CS in both groups, whereas no significant change was observed between groups. The other protein content of GPX4 and NCOA4 on the infarct side did not differ significantly from that on the CS in either group, and no significant difference was observed between the infarct sides of Fpn1Nestin-CKO dMCAO and Fpn1flox/flox dMCAO mice (Supplementary Fig. S2C-S2F). Taken together, these results indicate that the present data do not provide evidence for enhanced ferroptosis in Nestin-Cre-mediated Fpn1 knockout mice after cerebral ischemia.
Fpn1 knockout impairs myelin-associated repair, synaptic remodeling, and endogenous repair-related responses after cerebral ischemia
To evaluate myelin-related repair histologically, we performed immunofluorescence staining for Olig2 and myelin basic protein (MBP) in the peri-infarct cortex. Olig2-positive area increased on the injured side after dMCAO in both groups, but the increase was less pronounced in Fpn1Nestin-CKO dMCAO than in Fpn1flox/flox mice (Fig. 3A,C). In contrast, MBP-positive area was reduced after dMCAO, and this reduction was more pronounced in Fpn1Nestin-CKO dMCAO (Fig. 3B,D).
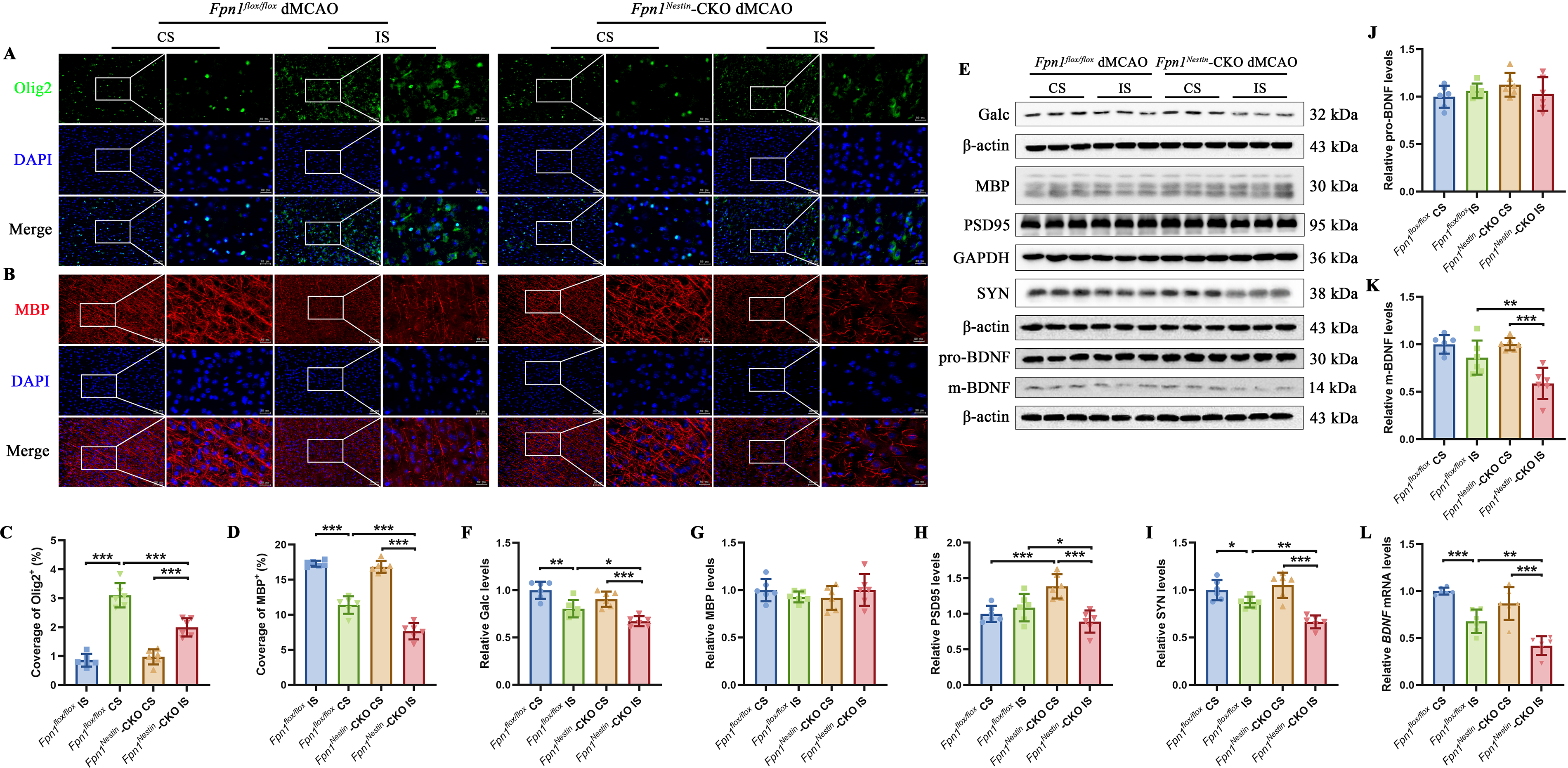
To further assess myelin and synaptic remodeling, we examined the expression of myelin sheath- and synapse-related proteins in the cerebral cortex on day 14 after cerebral ischemia by Western blot analysis (Fig. 3E). Galactosyl ceramidase (Galc) is an enzyme involved in the metabolism of myelin lipids, whereas MBP is a major structural component of the myelin sheath. Galc content was significantly reduced in the injured side of Fpn1Nestin-CKO dMCAO group compared with the Fpn1flox/flox dMCAO group (Fig. 3F), whereas MBP content did not differ significantly between groups (Fig. 3G). Synaptic integrity was evaluated by postsynaptic density protein-95 (PSD95) and synaptophysin (SYN). PSD95 content was significantly decreased in the injured cortex of Fpn1Nestin-CKO dMCAO group compared with both the CS and the Fpn1flox/flox dMCAO group (Fig. 3H). Similarly, the SYN content was reduced after dMCAO in both groups, and significantly lower in Fpn1Nestin-CKO dMCAO group than in Fpn1flox/flox dMCAO group (Fig. 3I). Although pro-brain-derived neurotrophic factor content did not differ significantly between groups (Fig. 3J), mature BDNF content was significantly lower in the injured cortex of Fpn1Nestin-CKO dMCAO mice than in the Fpn1flox/flox dMCAO group (Fig. 3K). Consistently, BDNF messenger ribonucleic acid (mRNA) abundance was reduced after dMCAO and showed a greater decrease in the Fpn1Nestin-CKO dMCAO group (Fig. 3L).
To investigate endogenous repair-related cellular responses, we examined cell proliferation marker protein Ki-67 (Ki67) and 5-bromodeoxyuridine (BrdU) and neurogenesis-associated marker doublecortin (DCX) level by immunofluorescence and Western blot. Immunofluorescence staining showed reduced Ki67- and DCX-positive areas in the Fpn1Nestin-CKO dMCAO group compared with Fpn1flox/flox dMCAO group (Fig. 4A–B, 4D–E). BrdU incorporation, reflecting DNA synthesis in proliferating cells, was also decreased in the Fpn1Nestin-CKO dMCAO (Fig. 4C,F). Western blot analysis further showed that Ki67 and DCX content were significantly lower in the Fpn1Nestin-CKO dMCAO group than in the Fpn1flox/flox dMCAO group (Fig. 4G–I). Finally, Golgi staining showed that a significant reduction in dendritic spine density on the injured side after dMCAO, with a further decrease observed in Fpn1Nestin-CKO dMCAO group compared with Fpn1flox/flox dMCAO group (Fig. 4J–K). Taken together, these findings indicate that Nestin-Cre-mediated Fpn1 knockout is associated with impaired cellular proliferation, reduced neurogenesis-related responses, and attenuated dendritic spine remodeling after cerebral ischemia. These alterations may contribute to compromised structural plasticity and delayed functional recovery following stroke.

Fpn1 knockout disrupts brain iron homeostasis and aggravates iron-related changes in the ischemic cortex
To explore whether Fpn1 deficiency alters iron homeostasis after stroke, we measured the total iron content in the cerebral cortex by inductively coupled plasma mass spectrometry (ICP-MS). Under baseline conditions, Fpn1 knockout significantly decreased the total cortical iron content (Supplementary Fig. S3A). However, after dMCAO, the total iron content on the injured side increased significantly in both Fpn1flox/flox and Fpn1Nestin-CKO groups, indicating disturbed local iron homeostasis in the ischemic cortex (Fig. 5A).
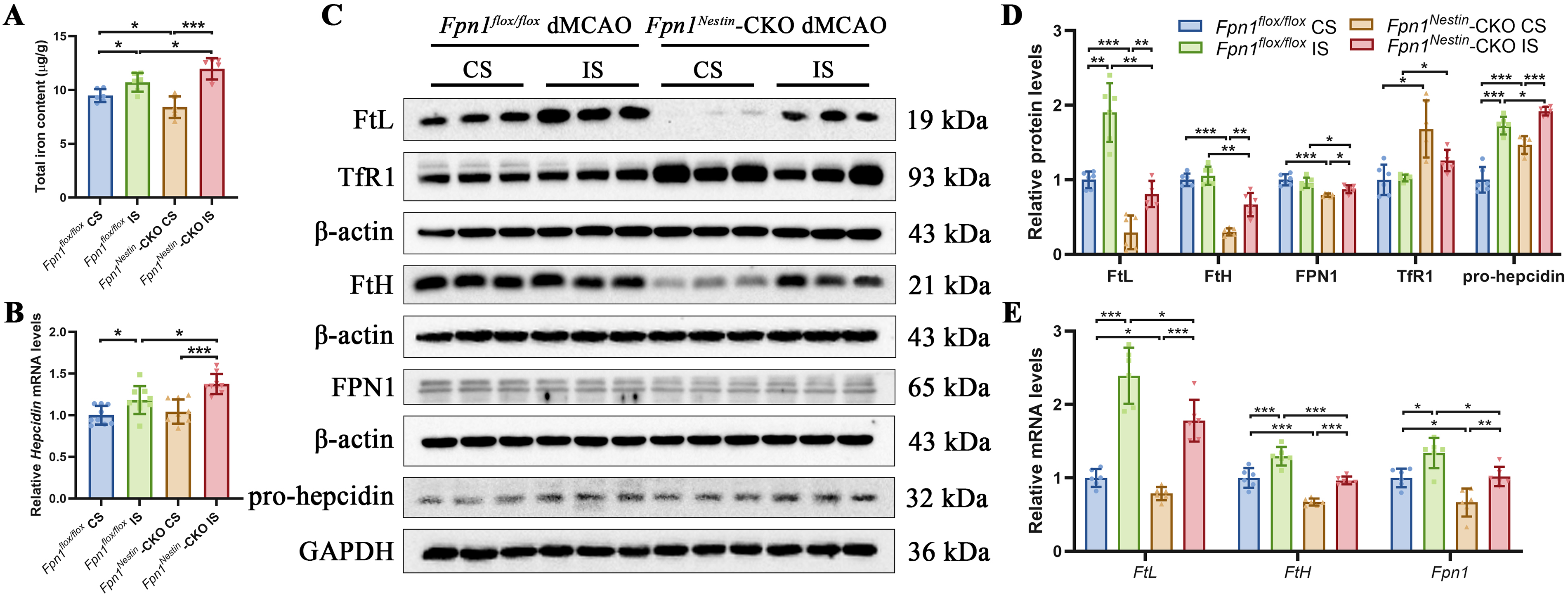
Consistent with the increased cortical iron content, ferritin light chain (FtL) and ferritin heavy chain (FtH) content were elevated in the injured side compared with the CSs in both groups. Although absolute FtL and FtH levels in Fpn1Nestin-CKO dMCAO group were lower than those in Fpn1flox/flox dMCAO group, the relative increase between the injured and CS in the Fpn1Nestin-CKO dMCAO group was more pronounced in the Fpn1flox/flox dMCAO group (Fig. 5C–E). At the transcriptional level, the Fpn1 mRNA abundance on the injured side was upregulated, with FPN1 content on the injured side in Fpn1Nestin-CKO dMCAO mice showing a lower level than that in the Fpn1flox/flox dMCAO group (Fig. 5C–E). Transferrin receptor (TfR1) content, a key mediator of cellular iron uptake, on the injured side of Fpn1Nestin-CKO dMCAO group was lower than that on the opposite side, and there was an increase compared with the Fpn1flox/flox dMCAO group (Fig. 5C–D). Hepcidin, a central regulator of iron metabolism produced by the liver and glial cells, controls iron efflux by promoting FPN1 internalization and degradation (Gao et al., 2019). The relative mRNA abundance of hepcidin on the infarcted side was higher than that on the CS and was further elevated in the Fpn1Nestin-CKO dMCAO group compared with the Fpn1flox/flox dMCAO group (Fig. 5B). Nestin-Cre-mediated Fpn1 knockout led to an increase in pro-hepcidin content on the injured side after cerebral ischemia (Fig. 5C–D). Finally, we examined divalent metal transporter 1 (DMT1), including both +IRE (iron-responsive element) and −IRE isoforms. No significant differences were observed in DMT1 content between groups (Supplementary Fig. S3B–S3D). Overall, these findings indicate that Nestin-Cre-mediated Fpn1 knockout is associated with dysregulated iron homeostasis during the postischemic phase, characterized by enhanced iron accumulation and altered iron-handling responses in the ischemic cortex.
Increased endothelial iron content is associated with aggravated BBB disruption after cerebral ischemia
Disruption of iron homeostasis has been implicated in the regulation of BBB integrity (You et al., 2022). To determine whether altered iron levels contribute to BBB dysfunction after stroke, we first assessed the content of tight-junction proteins by Western blot analysis (Fig. 6A). Compared with the Fpn1flox/flox dMCAO group, the zonula occludens-1 (ZO-1), occludin, and claudin-5 content were all diminished on the injured side of Fpn1Nestin-CKO dMCAO group (Fig. 6B–D). Immunofluorescence analysis further demonstrated that the ZO-1 positive cell numbers were a marked reduction in the injury side of Fpn1Nestin-CKO dMCAO mice (Fig. 6E–F). Ultrastructural examination by transmission electron microscopy (TEM) revealed damaged cerebral microvascular endothelial cells after dMCAO, with more severe endothelial damage observed in Fpn1Nestin-CKO dMCAO, including increased intercellular gaps between endothelial cells and surrounding structures (Fig. 6G).

In addition, double immunofluorescence staining revealed that reduced CD31/FPN1 co-localization and increased CD31/FtL co-localization in the injury side of Fpn1Nestin-CKO dMCAO mice (Fig. 6H–K), suggesting altered endothelial iron handling following cerebral ischemia. Thus, these findings indicate that Fpn1 deficiency is associated with increased endothelial iron content and exacerbated BBB disruption following cerebral ischemia.
BBB disruption is associated with aggravated gliosis and inflammatory responses after cerebral ischemia
Astrocytes and microglia play central roles in coordinating neuroinflammatory responses after brain injury. To assess glial activation, we examined the expression of glial fibrillary acidic protein (GFAP) and ionized calcium-binding adaptor molecule 1 (Iba1) in the cortex after dMCAO by Western blot analysis (Fig. 7A). Compared with the CS, GFAP content was significantly increased in the injured side in both groups, and was further elevated in the Fpn1Nestin-CKO dMCAO group relative to the Fpn1flox/flox dMCAO group (Fig. 7B). Similarly, Iba1 content was significantly higher in the Fpn1Nestin-CKO dMCAO group than in the Fpn1flox/flox dMCAO group (Fig. 7C). Immunofluorescence staining further demonstrated an increased GFAP-positive areas in the peri-infarct region of Fpn1Nestin-CKO dMCAO group compared with the Fpn1flox/flox dMCAO group (Fig. 7D–E). The results of immunohistochemical staining showed that, compared with the Fpn1flox/flox dMCAO group, an increased number of Iba1-positive cells in the peri-infarct region of the Fpn1Nestin-CKO dMCAO mice. (Fig. 7F–G). These results suggest that Nestin-Cre-mediated Fpn1 knock out aggravated the gliosis associated with cerebral ischemia recovery.
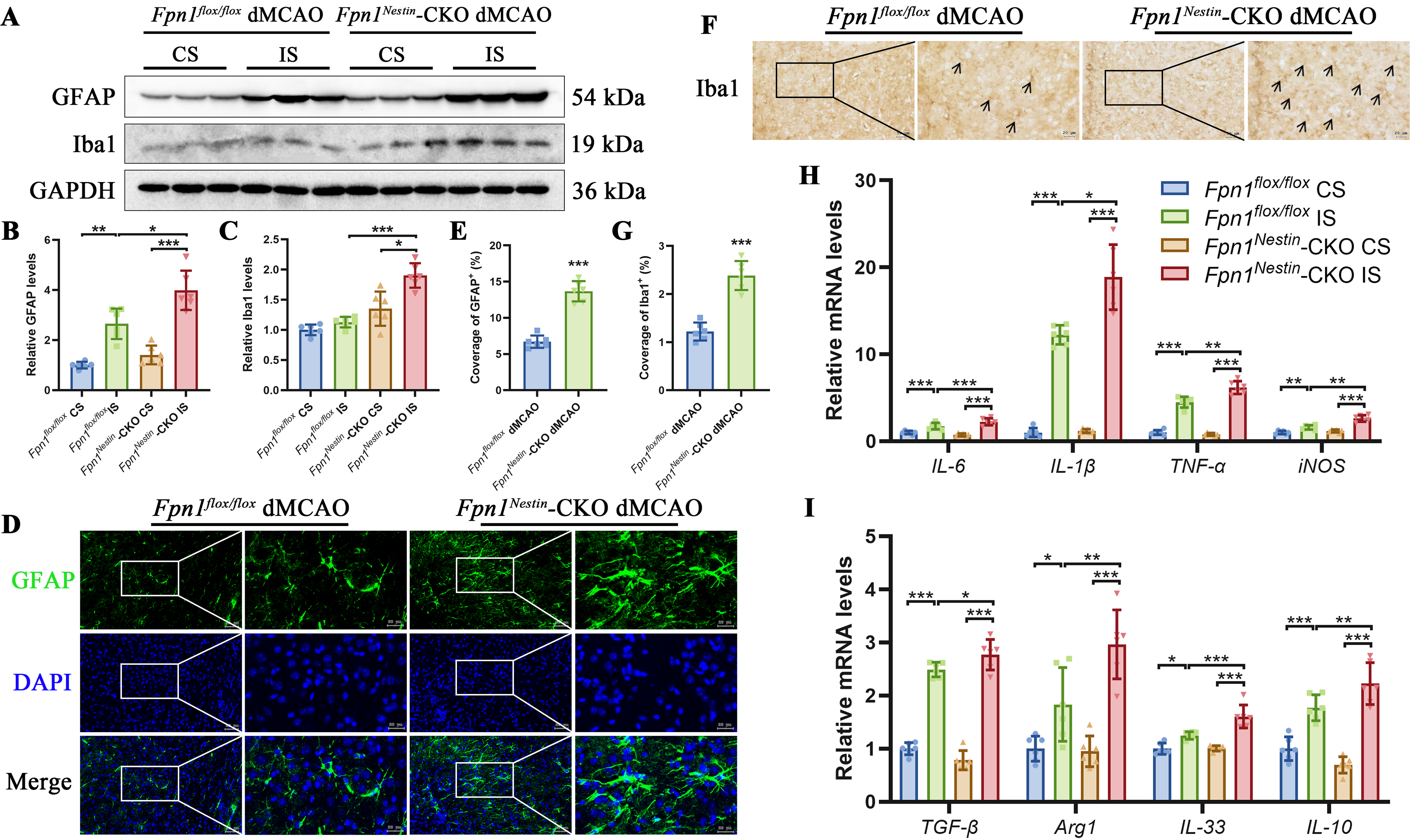
To further characterize the inflammatory milieu, we quantified the mRNA abundance of cytokines and immune-related markers in the cortex at day 14 post-dMCAO using real-time quantitative polymerase chain reaction (PCR). Pro-inflammatory mediators, including interleukin-6 (IL-6), interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and inducible nitric oxide synthase, were significantly upregulated in the injured side compared with the CS in both groups, with a further increase observed in Fpn1Nestin-CKO dMCAO mice compared with Fpn1flox/flox dMCAO group (Fig. 7H). We also assessed markers associated with anti-inflammatory and reparative responses. The mRNA levels of transforming growth factor-β, arginase 1 (Arg1), interleukin-33, and interleukin-10 were elevated in the injured side after dMCAO and were further increased in Fpn1Nestin-CKO dMCAO mice (Fig. 7I). Arg1, a key enzyme in arginine metabolism, is commonly associated with alternatively activated macrophages/microglia and is regulated, at least in part, by the JAK-STAT3 signaling pathway. Together, these findings indicate that Nestin-Cre-mediated Fpn1 knockout is associated with enhanced reactive gliosis and an overall amplification of inflammatory responses in the ischemic cortex, characterized by concurrent upregulation of both pro-inflammatory and reparative mediators, suggesting a heightened but potentially dysregulated immune response following cerebral ischemia.
AG490 exerts effects on JAK/STAT3 signaling and promotes repair-related processes after cerebral ischemia
To examine the role of JAK/STAT signaling in the poststroke pathology, we first assessed pathway activation by examining phosphorylation of STAT3 and JAK in the cortex after dMCAO. Consistent with activation of inflammatory signaling after ischemia, the p-STAT3 and p-JAK content were significantly increased in the infarcted side compared with the CS in both groups. Moreover, both p-STAT3 and p-JAK content were further elevated in Fpn1Nestin-CKO mice relative to Fpn1flox/flox mice, indicating enhanced activation of the JAK/STAT3 signaling pathway in the context of Fpn1 deficiency (Fig. 8A–C).
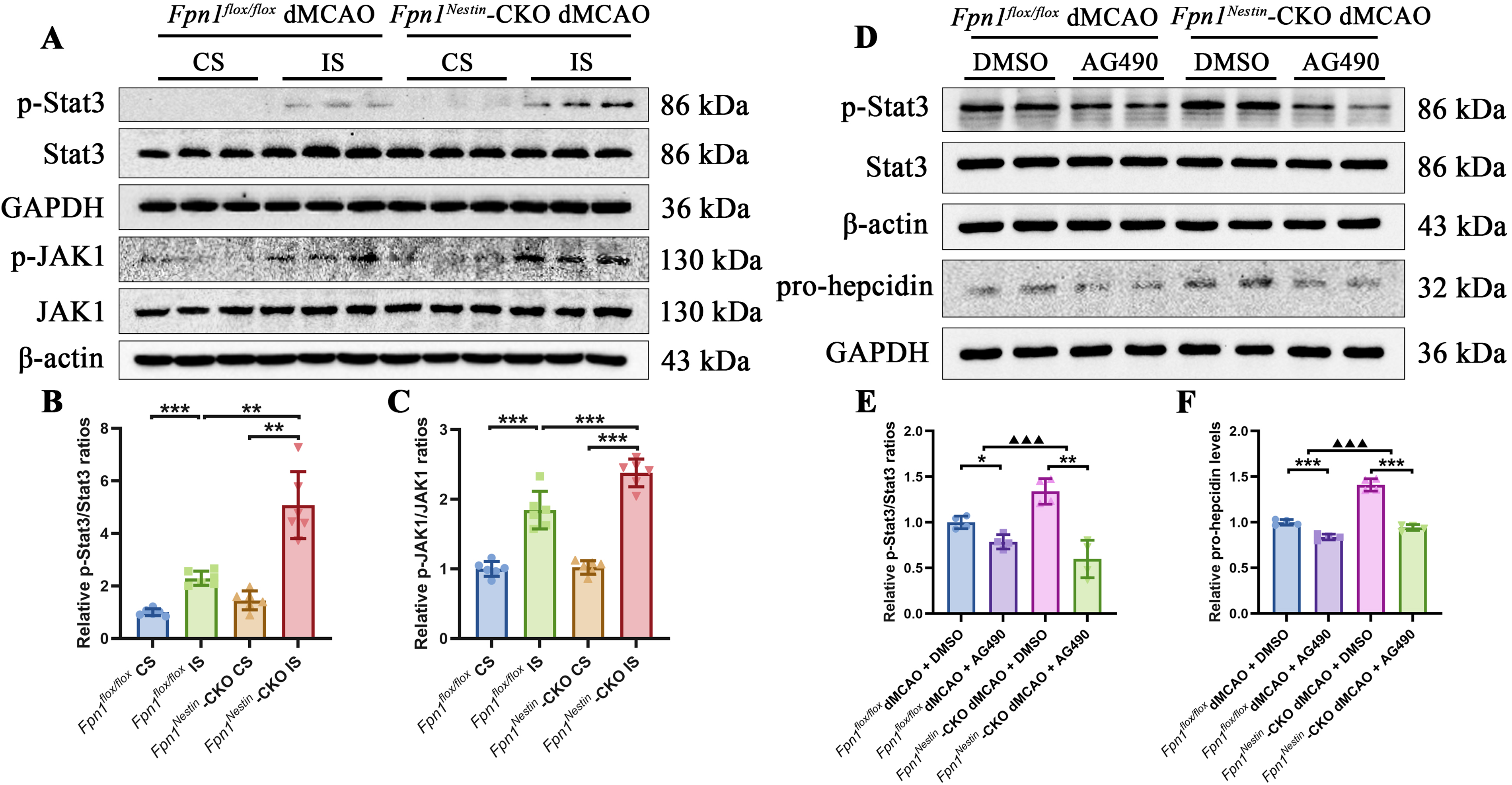
To further investigate the functional significance of JAK/STAT3 signaling, we administered AG490, a well-characterized tyrosine kinase inhibitor that suppresses JAK/STAT signaling by inhibiting STAT3 phosphorylation. Behavioral assessments were performed before dMCAO and on days 1, 3, 7, and 14 after surgery to evaluate the effects of AG490 treatment on poststroke functional recovery. AG490 treatment was associated with improved neurological performance, including reduced mNSS scores, increased rotarod performance, and improved sensorimotor function in the adhesive removal test. No significant differences were observed in gait parameters (Supplementary Fig. S4A-S4L). At the molecular level, AG490 effectively reduced p-STAT3 levels after dMCAO (Supplementary Fig. S5A-S5B). This was accompanied by a decrease in pro-hepcidin, with a more pronounced reduction observed in Fpn1Nestin-CKO mice (Fig. 8D–F).
Under pathological conditions, pro-inflammatory cytokines such as IL-6 can activate the JAK-STAT3 pathway, thereby amplifying inflammatory signaling cascades (Johnson et al., 2018). AG490, a tyrosine kinase inhibitor, suppresses JAK-STAT3 signaling by inhibiting STAT3 phosphorylation and has been shown to attenuate inflammatory responses (Chen et al., 2017). In Fpn1flox/flox mice, AG490 treatment significantly increased Arg1, FTL, and FTH content. However, in Fpn1Nestin-CKO mice, AG490 treatment significantly decreased Arg1 content, whereas FTL and FTH did not change significantly. No significant changes in IL-1β, TNF-α, and FPN1 content were observed in either genotype. Notably, when comparing AG490 treated-to-untreated ratios, the reductions in p-STAT3 and pro-hepcidin content were more pronounced in Fpn1Nestin-CKO mice than in Fpn1flox/flox mice, whereas the increases in TNF-α, Arg1, FTL, and FTH content were greater in Fpn1flox/flox mice (Supplementary Fig. S6A-S6H).
AG490 treatment exerted differential effects on myelin- and synapse-related proteins between genotypes. In Fpn1flox/flox mice, AG490 treatment reduced Galc and MBP content. However, in Fpn1Nestin-CKO mice, Galc content was decreased, whereas MBP content remained unchanged. Regarding synaptic markers, PSD95 content in Fpn1Nestin-CKO mice did not differ from that in control mice following AG490 treatment. However, SYN content increased in Fpn1Nestin-CKO mice but decreased in Fpn1flox/flox mice. Further comparison between genotypes showed that the reduction in Galc and MBP content was more pronounced in Fpn1flox/flox mice than in Fpn1Nestin-CKO mice, whereas the increase in SYN content was greater in Fpn1Nestin-CKO mice (Supplementary Fig. S7A-S7F).
AG490 also induced changes in proliferation- and gliosis-related markers. In Fpn1flox/flox mice, AG490 treatment was associated with decreased Ki67 and NeuN, accompanied by increased GFAP and Iba1 content. In contrast, in Fpn1Nestin-CKO mice, AG490 treatment was associated with increased Ki67 and NeuN content and decreased Iba1 content, with no significant change in GFAP content. Consistently, genotype comparison showed more pronounced increases in Ki67 and NeuN content and greater reductions in GFAP and Iba1 content in Fpn1Nestin-CKO mice than those in Fpn1flox/flox mice (Supplementary Fig. S7G-S7L). Taken together, inhibition of JAK-STAT signaling with AG490 reduced p-STAT3 and hepcidin levels and was associated with changes in iron-related and repair-associated responses. These effects were more evident in Fpn1-deficient mice than in control mice, indicating that the impact of JAK-STAT signaling may depend on the underlying state of iron homeostasis.
Discussion
Iron is an essential component in numerous physiological processes; however, its levels must be tightly regulated to ensure an adequate supply while preventing the accumulation of uncommitted iron, which catalyzes the production of highly toxic radical species (Anderson and Wang, 2012) through the Fenton reaction- the primary source of ROS. Elevated ROS can damage lipids, carbohydrates, proteins, and nucleic acids (Qian and Wang, 1998). Increased iron levels are commonly observed in neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease (Han et al., 2021; Ndayisaba et al., 2019). The accumulation of iron in the brain promotes lipid peroxidation and neuronal damage by catalyzing the conversion of superoxide and hydrogen peroxide into highly active toxic hydroxyl radicals (·OH) (Jomova and Valko, 2011). Consistently, multiple studies have reported increased brain iron levels following cerebral infarction (Clark et al., 2011; Guo et al., 2022; Zheng et al., 2023), resulting in harmful iron accumulation that not only disrupts brain iron homeostasis but also results in iron-mediated neurotoxicity (Long et al., 2023).
To better interpret these findings, it is important to consider the baseline iron status in Fpn1Nestin-CKO mice. Our previous observations indicate that iron levels in the cortex and hippocampus are reduced in young Fpn1Nestin-CKO mice, accompanied by decreased ferritin expression in neurons and astrocytes, suggesting baseline alterations in iron handling. Given the dynamic and age-dependent nature of brain iron metabolism, these changes may evolve over time and under pathological conditions. FPN1, the only known cellular iron export protein (Ganz, 2005), plays a critical role in maintaining brain iron homeostasis. Nestin-Cre-mediated deletion of Fpn1 impairs iron efflux in neurons and astrocytes, leading to intracellular iron retention and compensatory upregulation of hepcidin, a key negative regulator of iron export. Elevated hepcidin can act on brain microvascular endothelial cells to induce internalization and degradation of endothelial FPN1, which represents a major pathway for iron transfer from the circulation into the brain. Consequently, suppression of endothelial FPN1 may limit iron influx across the BBB, thereby contributing to reduced brain iron content under baseline conditions.
However, the pathological context following cerebral ischemia differs substantially. In the present study, total brain iron content was assessed by ICP-MS at day 14 after dMCAO. Consistent with previous reports, iron content was elevated in the dMCAO groups (Guo et al., 2022; Wang et al., 2021b), with a more pronounced elevation observed in the Fpn1Nestin-CKO mice. This apparent discrepancy can be explained by ischemia-induced BBB disruption, which permits the entry of circulating iron into the brain parenchyma. Indeed, IgG staining demonstrated more severe BBB leakage in Fpn1-deficient mice compared with controls, indicating increased permeability. Under these conditions, enhanced influx of peripheral iron likely overrides the baseline restriction of iron entry, resulting in iron accumulation in the injured cortex. Such iron overload may further promote lipid peroxidation and exacerbate neuronal injury after stroke.
We also observed exacerbated BBB disruption in stroke mice, indicated by reduced expression of tight junction proteins, and impaired microvascular function. This allows blood-borne substances, including inflammatory factors produced by various immune cells, to infiltrate the brain. This phenomenon is more pronounced in Fpn1-deficient mice, where FtL protein expression level is increased and FPN1 content is decreased in endothelial cells. These findings suggest that altered endothelial iron-related content is associated with aggravated BBB damage, which may facilitate abnormal iron influx into brain tissue after stroke.
We next examined glial and inflammatory changes associated with aggravated BBB damage following stroke. Inflammation is often associated with the disruption of the BBB and imbalance of iron metabolism (Urrutia et al., 2013), and it also induces hepcidin expression. In the Fpn1-deficient mice, microglial activation triggered a neuroinflammatory response that stimulated the JAK-STAT signaling pathway in astrocytes and accelerated the secretion of hepcidin. In addition to microglia-derived inflammatory mediators such as IL-6 and IL-1β, peripheral inflammatory factors entering through a compromised BBB (Jin et al., 2013) exacerbate brain injury (Chen et al., 2017; Liu et al., 2020). These cytokines can further damage the BBB, promote angiogenic edema, and facilitate neutrophil adhesion and infiltration into affected areas (Amantea et al., 2015; Barthels and Das, 2020). Notably, IL-6 enhances the transcriptional expression of hepcidin via the JAK-STAT signaling pathway activation in astrocytes (Varga et al., 2021), where p-Stat3 binds to its cognate promotor sequence in the hepcidin gene to activate transcription (Fleming, 2007). We propose that the increased hepcidin in Fpn1Nestin-CKO mice after cerebral infarction is in response to an inflammatory response in the brain, which further exacerbates brain iron deposition. In addition, after stroke injury, astrocyte proliferation in the injured area can form a glial scar, which is to protect neurons and limit inflammation and infection around the affected area in central nervous system injury (Barthels and Das, 2020; Sims and Yew, 2017). However, persistent reactive astrogliosis and scar-associated extracellular matrix may create a nonpermissive peri-infarct microenvironment that limits tissue remodeling, including axonal sprouting and the migration of repair-related cells (Choudhury and Ding, 2016).
Iron accumulation in neurons promotes apoptosis (Jomova et al., 2010; Ke and Qian, 2003,2007; You et al., 2017), and blocking the expression of hepcidin in astrocytes has been shown to prevent iron overload and neuronal death (You et al., 2017). In addition, the energy supply of neurons after stroke is interrupted (Hilkens et al., 2024), which can result in irreversible necrosis (Jomova et al., 2010). Apoptosis- and necrosis-related signaling was enhanced in the infarct-side cortex of the Fpn1Nestin-CKO dMCAO group, which may be related to disturbed iron homeostasis and increased intracellular iron retention in injured neural cells after Fpn1 knockout.
The recovery process of nerve injury in ischemic cerebral infarction involves myelination, synaptogenesis, and neural stem cell differentiation in functional areas (Guo et al., 2022). On day 14 after the stroke, Fpn1 knockout was associated with altered oligodendrocyte lineage responses and impaired myelin-related repair in the ischemic cortex. Synapse formation and dendritic spine density were also reduced, indicating that brain iron imbalance disrupted synaptic and neural function restoration. In Fpn1Nestin-CKO mice, inflammation exacerbated microglial phagocytosis of synapses and myelin sheaths, promoted neuronal necrosis, and further compromised BBB integrity, collectively impeding behavioral recovery. Immature or newly proliferated neurons migrate from the subventricular zone area to the peri-infarct region and differentiate into new neurons or glial cells, which then participate in the process of brain injury repair in the peri-infarct region (Guo et al., 2022; Wang et al., 2021a). The brain iron imbalance in the brain also prevented the regeneration of neural stem cells during cerebral ischemia injury, as evidenced by lower levels of Ki67, DCX, and BrdU in cerebral infarction mice, and the number of mature neurons in the injury area was also lower after Fpn1 knockout. Thus, FPN1 deficiency slows neural stem cell migration and differentiation during stroke recovery. A stable iron environment is essential for neurological restoration, and Fpn1 knockout induces iron-overloaded that is detrimental to tissue repair, underscoring the critical role of FPN1 in neuronal and glial recovery after stroke.
Is it possible to improve endogenous repair by targeting a key node within this pathological network? Previous studies have found AG490 can block epidermal growth factor receptor, Stat3, and JAK by inhibiting tyrosine kinase activity. Inhibiting the activity of Stat3 in microglia under cerebral ischemia has been reported to reduce the production of pro-inflammatory cytokines such as TNF-α and IL-6 (Chen et al., 2017). In the present study, enhanced activation of the JAK-STAT pathway under conditions of Fpn1 deficiency. This suggests that dysregulated iron handling may be associated with overactivation of STAT3 signaling following cerebral ischemia. Consistent with this, AG490 treatment exerted differential effects depending on genotype. In Fpn1flox/flox mice, AG490 did not attenuate pro-inflammatory cytokine expression and was associated with increased ferritin-related responses and altered expression of several repair-associated markers. These findings suggest that inhibition of JAK-STAT signaling does not necessarily confer benefit under baseline conditions and may produce complex downstream effects. By contrast, in Fpn1-deficient mice, where STAT3 signaling appears to be overactivated, AG490 more effectively reduced p-STAT3 and pro-hepcidin levels, accompanied by decreased microglial activation and partial normalization of several repair-associated markers. This is consistent with the notion that AG490 may attenuate pathological JAK-STAT3 overactivation and thereby modulate downstream inflammatory and repair-associated responses in the context of Fpn1 deficiency. Importantly, the effects of AG490 were largely restricted to Fpn1-deficient mice, suggesting that the contribution of the JAK-STAT3-hepcidin axis to poststroke pathology is context-dependent and may be particularly relevant in the setting of disrupted iron homeostasis.
Beyond the mechanistic findings, the temporal evolution of BBB disruption, inflammation, iron dysregulation, and ferroptosis is critical for therapeutic efficacy after stroke. In the dMCAO model, BBB disruption occurs early and persists, followed by sustained inflammation and altered iron handling. As our analyses were performed at day 14, representing the subacute phase, ferroptosis-related pathways were not prominently activated, suggesting a limited role at this stage. These dynamics may differ in ischemia-reperfusion models, where early oxidative stress may favor ferroptosis. Accordingly, therapeutic strategies may need to be phase-specific, targeting BBB integrity, oxidative stress, and ferroptosis early, and focusing on inflammation and iron homeostasis at later stages. However, these implications should be interpreted cautiously, as early time points were not assessed. Given the lack of early time-point data in the present study, future time-course analyses, including comparisons with reperfusion models, are warranted. In addition, the use of only male mice is a limitation of the present study, as sex differences may influence stroke pathology, inflammation, and iron metabolism. Future studies including both sexes are warranted.
In summary, FPN1 deficiency in neurons and glial cells was associated with delayed sensorimotor function recovery after cerebral ischemia. Fpn1 deficiency exacerbated BBB disruption, altered iron homeostasis, enhanced neuronal injury, and impaired endogenous repair-related responses, including synaptic remodeling and neurogenesis-associated processes. These changes were accompanied by increased gliosis, elevated inflammatory signaling, and enhanced activation of the JAK-STAT3 pathway. Importantly, the effects of AG490 were genotype-dependent, with more pronounced modulation of JAK-STAT3/hepcidin signaling and repair-associated responses observed in Fpn1-deficient mice than in control mice. These findings suggest that dysregulated iron metabolism may interact with inflammatory signaling to influence poststroke recovery.
Experimental Section
Animals
Mice were kept under 12-h light and 12-h dark cycles at 22 ± 1°C and 45%−55% relative humidity and were provided feed and drink ad libitum. All procedures were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals issued by the National Institutes of Health and approved by the Animal Care and Use Committee of Hebei Science and Technology Bureau, China. All experiments in the study were approved by the Animal Care and Use Committee of Hebei Normal University (authorization number: 2023LLSC027). We generated Nestin-Cre recombinase mice on a C57BL/6 genetic background and Fpn1flox/flox mice on a 129/SvEvTac genetic background. Progeny of Fpn1flox/W with Nestin-Cre recombinase were obtained and mated to produce the next generation, from which genotypes were identified by PCR amplification. Fpn1flox/flox mice showed only a band around 500 bp bands (Supplementary Fig. S1A), while only a band around 100 bp bands was found in Nestin-Cre homozygous mice (Supplementary Fig. S1B), and the primer sequences used were shown in Supplementary Table S1. Three-month-old male Fpn1flox/flox mice and Fpn1flox/flox mice with Nestin-Cre recombinase (Fpn1Nestin-CKO mice) were selected for experiments. A total of 190 male mice were used in this study. Mice were allocated to the Sham, dMCAO, and AG490 intervention groups as described below: Fpn1flox/flox Sham (n = 15): Fpn1flox/flox mice subjected to sham surgery, Fpn1Nestin-CKO Sham (n = 15): conditional Fpn1 knockout mice subjected to sham surgery, Fpn1flox/flox dMCAO (n = 30): Fpn1flox/flox mice subjected to dMCAO surgery, Fpn1Nestin-CKO dMCAO (n = 30): conditional Fpn1 knockout mice subjected to dMCAO surgery. Mice were allocated to the Sham, dMCAO, and AG490 intervention groups as follows: Sham (n = 9), dMCAO (n = 14), Sham + AG490 (n = 7), dMCAO + AG490 (n = 10). For AG490 intervention, dMCAO groups were further subdivided into: Fpn1flox/flox dMCAO + DMSO (n = 15): Vehicle control (intraperitoneal injection of DMSO), Fpn1Nestin-CKO dMCAO + DMSO (n = 15): Vehicle control, Fpn1flox/flox dMCAO + AG490 (n = 15): AG490 treatment (3 mg/kg, i.p.), Fpn1Nestin-CKO dMCAO + AG490 (n = 15): AG490 treatment. Sample sizes were determined based on power analysis (α = 0.05, power = 0.8) using historical data from pilot studies.
All animal experiments were conducted using randomized and blinded methods.
dMCAO model
The dMCAO model was generated by electrocoagulation under microscopy (Guo et al., 2022), where the right focal cortex was infarcted. The specific steps were as follows: (i) The mice were fasted for 18 h before surgery and anesthetized with pentobarbital sodium (20 mg/kg, intraperitoneally). To minimize pain and distress, animals received peri- and postoperative analgesia (buprenorphine, 0.05–0.1 mg/kg, subcutaneously). Before surgery, the surgical area was prepared and disinfected with povidone-iodine. (ii) After conventional skin preparation and disinfection, the mice were fixed on the operating table in a supine position. A ∼1 cm incision was cut at the center of the neck, then the neck subcutaneous muscle and fascia were separated to fully expose the neck triangle on the right side, the right common carotid artery (CCA) was carefully separated and permanently ligated, and the neck skin incision was then closed. (iii) The mice were immobilized on their left side, a 1 cm skin incision was made between the external canthus and the auditory meatus of the right eye to expose the temporalis muscle, which was carefully separated to expose skull, then the right middle cerebral artery cortical branches were identified, a miniature cranial drill was used to create a ∼1.5 mm diameter hole, and the cortical branch of the right middle cerebral artery was exposed. (iv) The cortical branch of the right middle cerebral artery was cauterized with a coagulation pen, minimizing damage to the surrounding brain tissue as much as possible. (v) After confirming under the microscope that no reperfusion occurred in the arterial branch, the skin was sutured. (vi) The mice were maintained in a clean, sterile cage at 37°C with controlled humidity until they recovered from anesthesia, after which they were provided with soft food and allowed free access to food and water. Animals were monitored daily for general condition, mobility, and signs of pain or distress. All surgical procedures were performed under aseptic conditions. No prophylactic antibiotics were routinely administered in accordance with institutional guidelines. dMCAO group: the dMCAO mouse model was established by the above methods. Sham group: the model establishment procedures were the same as the dMCAO group, but the right CCA was separated without ligation, the skull was exposed and drilled, but the cortical branches of the right middle cerebral artery were not cauterized. Exclusion criteria: (i) Intraoperative subarachnoid hemorrhage. (ii) Blood flows after two rounds of electrocoagulation. For experiments, the mice in each group were graded and euthanized, and any tissue or organs were collected at the corresponding time points.
Neuroethological tests
The following four behavioral tests were performed on mice 1 day before dMCAO and 1, 3, 7, and 14 days after dMCAO, by researchers who were unaware of the experimental groups.
Modified neurological severity score
The mNSS was used to detect a series of behavioral activities in mice in terms of motor, sensory, balance, and reflex. The scores ranged from 0 to 18. The higher the score, the more serious the neurological deficit.
Rotarod test
The motor and limb coordination of the mice was evaluated after operation. Before surgery, the mice were placed on a clean rotary rod and run at a speed of 4 rpm. The mice that did not drop within 60 s were selected for follow-up experiments. Before surgery, the mice in each group were trained for 5 consecutive days at 4 rpm, three times a day, 5 min each time, and the two intervals between training runs were at least 15 min. In the experiment, the trained mice were placed on the rotating rod, and the rotation speed increased uniformly from 4 to 40 rpm within 90 s. The duration until the mice fell off the rotating rod was recorded, with the longest time being 300 s. The test was conducted three times, with the average values calculated as the results.
Adhesive removal test
To evaluate the motor sensory ability of mice, rectangular 3M medical tapes of different colors (3 mm × 4 mm) were pasted onto the two forelimbs of the mice. The order of attaching to the left and right limbs of each mouse varied, and the adhesive angle and strength were approximately the same. After the adhesive tape was attached, the mice were placed in a large transparent box for observation for 120 s. The contact time and end time of the LF and RF tape attachment were recorded. Baseline sensorimotor performance was evaluated before surgery, and only mice that were able to contact and remove the adhesive tapes from both forelimbs within 120 s after pretraining were included in the subsequent experiments. Mice failing to meet this criterion were excluded. Afterwards, the mice were trained for 5 days, three times a day, so that the mice could reach horizontally and remove the tape at the same time. Additionally, each mouse was acclimated to the transparent box before the test.
Gait analysis
To evaluate the motor coordination of the mice, the front and rear feet were painted with different colors of nontoxic ink, and the mice were placed on a 50 cm × 7 cm track. Before each test, a new piece of wrinkle-free white paper of appropriate size was placed on the track, upon which paw prints were left by the mice on the paper. All mice were trained three times to walk on the track before the formal testing. The stride length and the standing width of forelimbs and hindlimbs of three consecutive steps were measured, and the average value was calculated as the result.
Staining of brain tissue sections
Frozen section: After thoroughly perfusing the brain with saline, the tissue was fixed in 4% paraformaldehyde, dehydrated in 30% sucrose, and sectioned coronally with an ultrathin frozen slicer to a thickness of 15 μm.
Paraffin sections: After euthanizing the mice, brain tissue was extracted and placed in a 4% (w/v) paraformaldehyde solution for fixation at 4°C for 24 h. Following fixation, the tissue underwent routine ethanol gradient dehydration, followed by clearing, paraffin embedding, and sectioning. Continuous coronal sections with a thickness of 4 μm were prepared.
H&E staining and Nissl staining
To validate tissue injury histologically, brain sections were stained with H&E (Solarbio, G1125) for H&E staining and cresyl violet (Solarbio, G1430) for Nissl staining according to the manufacturer’s instructions. The sections were then dehydrated sequentially in 70%, 80%, 90%, and 95% ethanol for 5 min each, followed by absolute ethanol for 10 min, mounted with coverslips, and imaged under a bright-field microscope (Tissue FAXS Plus 5).
Immunohistochemistry
At room temperature, the brain sections were put into 3% H2O2 to eliminate endogenous peroxidase for 10 min and then were blocked with 10% goat serum prepared in PBS for 60 min at room temperature. The blocking solution was then removed, and primary antibodies diluted in PBS were applied to the sections for overnight incubation at 4°C. The working dilutions were as follows: on the following day, sections were washed three times with PBS and incubated with biotinylated goat-antirabbit or antimouse IgG secondary antibody diluted (1:500) in PBS for 50 min at 37°C. After washing with PBS, horseradish peroxidase-labeled streptavidin working solution was added, and the sections were incubated for 50 min at 37°C, followed by DAB under microscopic observation, and were counterstained with hematoxylin. The sections were then dehydrated sequentially in 70%, 80%, 90%, and 95% ethanol for 5 min each, followed by absolute ethanol for 10 min, mounted with coverslips, and imaged under a bright-field microscope (Tissue FAXS Plus 5).
Immunofluorescence
Brain slices were washed three times in 0.01 M PBS, and placed into sodium citrate antigen repair solution (dissolve 1.206 g sodium citrate and 0.189 g citric acid to 500 mL) at 90°C for 10 min in a water bath. After cooling to room temperature, the sections were washed with 0.01 M PBS and then incubated with 10% goat serum prepared in PBS at 37°C for 50 min. The serum was replaced with primary antibodies diluted in PBS, and the samples were incubated overnight at 4°C. The next day, the sections were washed with 0.01 M PBS at room temperature three times. In the dark, DyLight 488 goat antimouse IgG and DyLight 549 goat antirabbit IgG, diluted in PBS was added, after which the samples were incubated at 37°C for 50 min. The sections were then washed with 0.01 M PBS and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 4 min. Finally, the sections were washed by 0.01 M PBS, fixed with antifluorescence quenching agent (Solarbio, S2100), and imaged by a fluorescence microscopy (Tissue FAXS Plus 5). The antibodies’ information and working dilutions were presented in Supplementary Table S3.
Histological analyses
For all histological analyses, images were acquired from the peri-infarct cortex adjacent to the ischemic core. For each animal, six anatomically matched coronal sections at comparable rostrocaudal levels were analyzed. Whole-slide images of each section were acquired using a Tissue FAXS Plus 5 imaging system (Tissue Gnostics, Vienna, Austria) under identical acquisition settings across all experimental groups. For H&E staining quantification, the percentage of damaged neurons was calculated as the number of damaged neurons divided by the total number of neurons × 100%. For Nissl staining quantification, five consecutive 4-μm-thick coronal sections from the peri-infarct cortex were analyzed for each animal. Quantification was performed using ImageJ software. The number of viable Nissl-positive neurons was counted within standardized regions of interest in the peri-infarct cortex and expressed as neuronal density (cells/mm3). For immunofluorescence and immunohistochemical staining, quantitative analysis was performed using ImageJ software. Images from the same experiment were analyzed using identical threshold settings. The integrated density (IntDen) and corresponding stained area were measured, and signal intensity was calculated as IntDen divided by the total stained area.
Golgi staining
Golgi staining was performed using GolgiStain Kit (Shanghai Jiemei Gene, GMS80020.2) according to the manufacturer’s instructions. Mice were anesthetized with pentobarbital sodium, and the brains were removed and immersed in 4% paraformaldehyde at 4°C for 24 h. In the dark, brain tissues were immersed in a mixture of 7 mL GENMED fixative solution A and 7 mL GENMED fixative solution B and incubated at room temperature for 14 days. The tissue was then transferred to 30% sucrose solution C and kept at 4°C in the dark for 48 h. The brains were sectioned at 50 μm using a microtome (VT1000S, Leica, Wetzlar, Germany). Sections were imaged using a bright-field microscope (Axio Imager, Zeiss). Dendritic spine density was quantified using ImageJ software by counting the number of spines per unit length of dendrite.
Transmission electron microscopy
Brain tissue samples from the peri-infarct cortex were rapidly dissected and cut into small blocks (∼1 mm3), then fixed in 2.5% glutaraldehyde prepared in 0.1M phosphate buffer (PB, pH 7.4) at 4°C overnight. After primary fixation, the samples were rinsed three times with 0.1M PB for 15 min each. The tissues were then postfixed in 1% osmium tetroxide for 4 h in the dark under a fume hood, followed by rinsing twice with 0.1M PB for 20 min each. Samples were dehydrated through a graded acetone series, including 50%, 70%, 90%, and 95% acetone for 20 min each at 4°C, followed by 100% acetone for 20 min twice at room temperature. For embedding, samples were sequentially infiltrated with acetone and embedding medium mixtures at room temperature as follows: acetone: embedding medium = 3:1 for 3–4 h, 1:1 for 5–6 h, and 1:3 overnight. The samples were then transferred into pure embedding medium and incubated at 37°C overnight, followed by embedding in flat molds and polymerization at 65°C for 48 h. After polymerization, the samples were sectioned at 100 nm, lifted onto 3-mm copper grids, and stained for 30 min in 1.5% aqueous uranyl acetate and for 10 min in lead citrate. The sections were finally dried and viewed on a Hitachi H7000 TEM (Bruker, Germany).
RNA isolation and real-time quantitative PCR
Total RNA was extracted from brain tissues using TRIzol-based method (Thermo Fisher, 15596018) following a standard multistep protocol. Briefly, tissue samples were homogenized in TRIzol reagent, followed by phase separation with chloroform. After centrifugation, the aqueous phase containing RNA was transferred to a new tube, and RNA was precipitated using isopropanol. The RNA pellet was washed with 75% ethanol, air-dried, and resuspended in RNase-free water. RNA concentration and purity were assessed using NanoDrop. RNA purity was evaluated by measuring the A260/A280 ratio, and only samples meeting the quality criteria were used for reverse transcription before downstream analysis. Using a Prime Script RT Kit (Takara, rr047a), total RNA (1 μg) was reverse transcribed into cDNA. Using a Bio Rad CFX Connect Real-Time system, SYBR Green PCR Master Mix (CWBIO, CW0957) was used for PCR amplification. The cycle parameters were as follows: 95°C for 10 min, then 40 cycles of 95°C for 15 s and 60°C for 1 min. Relative mRNA abundance was calculated using the 2−ΔΔCT method, with β-actin as the internal reference gene and the Fpn1flox/flox CS group as the calibrator, and primers used were shown in Supplementary Table S2.
Determination of iron content in tissues by ICP-MS
After perfusion with saline, the mouse brain tissue was extracted, weighed, and transferred into 1.5 mL metal-free digestion tubes (NEST, 615601). No predrying or ashing step was performed, and wet tissue weight was used directly. Tissue samples were digested in 500 μL of 65% nitric acid (Thermo Fisher, HPLC grade, 010984.AK) overnight at room temperature. On the following day, the tube caps were opened in a fume hood, and the remaining acid was evaporated in a 90°C metal bath for 20 min. Subsequently, 500 μL of H2O2 (Sigma, #323381) was added, and the samples were heated in a metal bath at 70°C for 15 min and then at 100°C until the remaining acid had evaporated. In the blank control, only nitric acid was added to the tube. Finally, the digested residue was reconstituted to a final volume of 1 mL with deionized water (resistivity ≥18.2 MΩ·cm).
Iron concentrations were measured by ICP-MS using an iCAP RQplus system (Thermo Fisher Scientific). An internal standard (Indium, SCP Science, 140-051-491) was added to all standards and samples to correct for instrumental drift and matrix effects. Calibration standards were prepared by serial dilution of a certified iron standard solution (SCP Science, 700-101-121) in the same acid matrix as the samples to generate a calibration curve covering (concentration range, 0, 5, 10, 50, 100, 200 μg/L). Calibration blanks and standards were measured together with the samples under the same conditions. Iron concentrations were calculated from the calibration curve and normalized to [wet tissue weight/dry tissue weight].
Western blot analysis
Protein lysis buffer was prepared by adding 10 μL phenylmethanesulfonylfluoride to 1 mL radioimmunoprecipitation assay buffer (50.0 mM Tris–HCl, pH 7.4, 150.0 mM NaCl, 1% NP40, 0.1% SDS) at 4°C, which was then immediately added to mouse brain tissue at a ratio of 1 mg tissue to 15 μL buffer. The cerebral cortex tissue was cut into pieces on ice, and ultrasonicated (power: 560 W, 10 s of sonication and 10 s of rest, repeated six times) until the liquid became clear without any lumps. The mixture was centrifuged at 12,000 × g at 4°C for 20 min, and the supernatant was collected. The protein concentration in the supernatant was determined using a BCA protein concentration determination kit (Yeasen, 20201ES76). Protein samples of 25 µg were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred to nitrocellulose membranes. After blocking with 5% nonfat milk in TBST for 90 min at room temperature, membranes were incubated overnight at 4°C with primary antibodies diluted in TBST. Next, the membranes were washed four times for 15 min with TBST (pH 7.6, 137.0 mM NaCl, 0.05% Tween-20), after which HRP-conjugated goat antimouse IgG or HRP-conjugated goat antirabbit IgG diluted in TBST was added for 90 min at room temperature. The membranes were again washed four times for 15 min with TBST. The protein bands were visualized by chemiluminescence imaging, and the relative content was normalized to β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content. The Antibodies information and working dilutions were presented in Supplementary Table S3.
Drug injection
On days 1–5 after the operation, 20 mg/mL BrdU solution (Sigma, B9285) was injected intraperitoneally at a dose of 50 mg/kg, and the control group was injected with the same dose of normal saline. 20 mM AG490 (MCE, HY-12000) or dimethyl sulfoxide (DMSO) was injected intraperitoneally at 3 mg/kg 1 h before the operation and on days 1, 3, 5, 7, 9, 11, and 13 after the operation. The Fpn1flox/flox mice and Fpn1Nestin-CKO mice were divided into four groups: Fpn1flox/flox dMCAO + DMSO group, Fpn1flox/flox dMCAO + AG490 group, Fpn1Nestin-CKO dMCAO + DMSO group, and Fpn1Nestin-CKO dMCAO + AG490 group (Supplementary Fig. S1C–S1D).
Statistical analysis
The densities of Western blot image bands were quantified using Image J software, and the protein content was normalized to their respective values of β-actin or GAPDH bands. The positive signals of the target proteins in the images of the cortex obtained through immunofluorescence/immunohistochemical staining were quantified using Image J software based on the coverage area and quantity of the same molecule in the same area size of the region of interest in different groups. The proportions obtained were then analyzed. The Graph Pad Prism 8.0 was used to generate graphs.
The experimental data were processed and analyzed using SPSS 26.0 software. Normality and homogeneity of variance were assessed using the Shapiro-Wilk and Levene’s tests, respectively. Depending on data distribution and variance equality, appropriate parametric or nonparametric tests were applied, with Welch’s correction used when variances were unequal. For behavioral experiments with repeated measurements over time, we first applied linear mixed-effects models to evaluate overall group × time interactions. Subsequently, two-way analysis of variance (ANOVA) was performed at each individual time point to further examine group differences. Homogeneity of variance was assessed using Levene’s test at each time point, followed by appropriate post hoc comparisons: Tukey’s test was used when variances were equal, whereas Games–Howell correction was applied when variances were unequal. The data of >2 variables were analyzed using two-way ANOVA, with time as a within-subject factor and treatment/genotype as between-subject factors. Student’s t-test was used to analyze the difference between the two groups. The quantitative data are presented as the mean ± standard deviation. The difference of p < 0.05 was considered to be statistically significant. Electronic laboratory notebook was not used.
Authors’ Contributions
Y.C.and K.H. designed the project, obtained funding, and provided overall guidance for the study; X.G. and X.J. performed most of the experiments, data analysis, and wrote the article; S.M.K. performed behavioral testing experiments and data analysis; Y.H., Q.Z., L.W., X.S., and J.M. performed behavioral testing experiments; Y.P., G.G., and F.W. participated in the experimental discussions and provided directions for the study. All authors read and approved the final article.
Availability of Data and Materials
The data that support the findings of this study are included in this published article and its additional file. Other materials are available from the corresponding authors on reasonable request.
Supplemental Material
sj-pdf-1-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-pdf-1-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-2-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-2-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-3-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-3-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-4-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-4-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-5-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-5-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-6-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-6-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-7-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-7-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-8-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-8-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Supplemental Material
sj-docx-9-are-10.1177_15230864261464407 — Supplemental material for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain
Supplemental material, sj-docx-9-are-10.1177_15230864261464407 for Inflammation Impairs Poststroke Recovery by Disrupting Iron Homeostasis in Brain by Xin Guo, Xiaofang Jin, Shaomeng Kang, Yingying Han, Qiaoya Zhao, Lihui Wu, Xintong Shi, Jingsi Meng, Peng Yu, Guofen Gao, Fudi Wang, Kang Han, and Yan-Zhong Chang
Footnotes
Acknowledgments
The authors are especially grateful to Dr. Du Jiulin (Shanghai Institute of Neuroscience) for providing Nestin-Cre recombinase mice with the C57BL/6 genetic background. They would like to thank all the participants in the study.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
The work was supported by the National Natural Science Foundation of China (Nos. 82301458 and U23A20169), the Nature Science Foundation of Hebei Province (Nos. H2024206372 and C2025205065), Hebei Province Government-funded Excellent Talents Project in Clinical Medicine (No. ZF2024147), and the Key Project of the Natural Science Foundation of Hebei Province, China (No. E2021205003).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.