
Abstract
Purpose:
This study investigated the 2-year safety and effectiveness of the PQ Bypass DETOUR system as a percutaneous femoropopliteal bypass.
Materials and Methods:
Seventy-eight patients with 82 long-segment femoropopliteal lesions were enrolled in this prospective, single-arm, multicenter study. The DETOUR system deployed Torus stent grafts directed through a transvenous route. Eligible patients included those with lesions of >10 cm and average of 371±55 mm. Key safety endpoints included major adverse events (MAEs) and symptomatic deep venous thrombosis in the target limb. Effectiveness endpoints included primary patency defined as freedom from ≥50% stenosis, occlusion, or clinically-driven target vessel revascularization (CD-TVR), primary assisted, and secondary patency.
Results:
Chronic total occlusions and severe calcium occurred in 96% and 67% of lesions, respectively. Core laboratory-assessed total lesion length averaged 371±51 mm with a mean occlusion length of 159±88 mm. The rates of technical and procedural success were 96%, with satisfactory delivery and deployment of the device without in-hospital MAEs in 79/82 limbs. The MAE rate was 22.0%, with 3 unrelated deaths (4%), 12 CD-TVRs (16%), and 1 major amputation (1%). Deep venous thrombosis developed in 2.8% of target limbs, and there were no reported pulmonary emboli. Primary, assisted primary, and secondary patency rates by the Kaplan–Meier analysis were 79±5%, 79±5%, and 86±4%, respectively.
Conclusions:
The PQ Bypass DETOUR system is a safe and effective percutaneous alternative to femoropopliteal open bypass with favorable results through 2 years. The DETOUR system provides a durable alternative to conventional endovascular modalities and open surgery for patients with long, severely calcified, or occluded femoropopliteal lesions.
Introduction
Peripheral arterial disease (PAD) affected more than 200 million people worldwide in 2010, with an increase of over 23% from the previous decade. 1 Beginning in the midportion of the 20th century, open surgical bypass procedures evolved as a definitive treatment for symptomatic PAD. Outcome was better with autogenous vein grafts compared with prosthetic grafts, but both were associated with high rates of success in treating claudication and limb-threatening symptoms. Open surgical revascularization, while successful in restoring flow, is associated with significant perioperative morbidity/wound complications, especially in the older and medically compromised patients who present with PAD. 2
On this landscape, percutaneous arterial revascularization procedures were innovated, beginning with the first angioplasty procedure performed over 50 years ago, followed by balloons, stent placement, and atherectomy in later years. 3 Drug-eluting stents and drug-coated balloons followed, but the long-term safety of the drugs, primarily paclitaxel, has come into question. 4 Covered stents have been used for longer femoropopliteal lesions, and recently heparin-coasted endoprostheses have been studied.5-8 In general, however, the outcome of percutaneous technologies has been inferior for the treatment of longer, heavily calcific, and chronically occluded lesions. 9
The PQ Bypass DETOUR system was designed as a technique for femoropopliteal bypass via a minimally invasive, percutaneous revascularization method. This system has the potential to avoid the morbidity of open surgical revascularization. The objective of the DETOUR study was to investigate the safety and effectiveness of the PQ Bypass DETOUR system in treating long, often severely calcified and chronically occluded femoropopliteal lesions. Herein, we report the 2-year results of the study.
Materials and Methods
Study Design
The DETOUR study is a prospective, single-arm, multi-center, international, pre-market investigation that examined the safety and effectiveness of the PQ Bypass DETOUR system for the treatment of long-segment femoropopliteal disease (NCT02471638). Over a 2-year period beginning in the first quarter of 2015 through the fourth quarter of 2017, 78 patients were enrolled at 8 investigational sites in Poland, Germany, Latvia, Italy, Chile, and New Zealand. Institutional Review Board (IRB) approval was obtained for the study protocol, and all patients provided written informed consent before undergoing any study procedures. Follow-up visits were scheduled at 1, 3, and 6 months, and then every 6 months thereafter through 36 month. Assessments at the follow-up visits included Rutherford category, ankle-brachial index (ABI), lower-extremity arterial and venous duplex ultrasound imaging, and venous quality of life measures [venous clinical severity score (VCSS) and Villalta score]. Adverse events were identified and recorded at each visit and at other times when investigational sites became aware of an event.
Patient Eligibility
Eligible patients were between the age of 18 and 90 with body mass index (BMI) less than 40 and a resting ABI less than 0.9 or, in those with noncompressible vessels, a toe-brachial index (TBI) of less than 0.7. Patients were required to have symptoms of severe claudication, rest pain, or ischemic ulceration (Rutherford categories 3–5), but those with tissue loss extending proximal to the toes were excluded. Screening procedures to determine eligibility included deep vein duplex ultrasonography and either computed tomographic angiography (CTA) or magnetic resonance angiography (MRA) demonstrating patent femoral veins measuring ≥10 mm in diameter or presence of a duplicate femoral vein. Femoropopliteal target lesions were required to be at least 10 cm in length and could include diffuse stenotic disease, chronic total occlusions (CTOs), or in-stent restenosis. Iliac and femoral arterial and venous systems were required to be of sufficient size and morphology, including tortuosity, to allow endovascular access with an 8-F sheath.
Patients were ineligible to participate in the DETOUR study if they had a history of comorbidities including but not limited to deep vein thrombosis (DVT), renal failure, dementia, congestive heart failure, chronic obstructive pulmonary disease (COPD), or metastatic malignancy. In addition, patients were excluded from the study if they had untreated flow-limiting aortoiliac occlusive disease (while this could be treated >30 days before the index intervention), had previous bypass surgery, thrombolysis in the target vessel within 72 hours prior to the index procedure, or were receiving immunosuppressant therapy. A complete list of inclusion and exclusion criteria is included in Supplemental Material Table S1.
Description of the Device and Procedure
The PQ Bypass DETOUR system consisted of the PQ Snare, PQ Crossing Device, and Torus stent graft. A description of the device and the procedure appeared in a prior publication. 10 Briefly, 8-F arterial access was via the contralateral femoral artery (with subsequent crossing of the aortic bifurcation). Venous access was gained through an ipsilateral posterior tibial vein in most cases. Using the PQ Crossing Device, the femoral vein was accessed through the ipsilateral superficial femoral artery (SFA) at the level 4 cm distally to SFA orifice. Patency beyond the first 1 cm was not required. Where the proximal SFA is diseased or occluded beyond 1 cm, crossing into the vein entry was performed through the recanalized artery. The diseased arterial segment was fully reconstituted after deployment of the device at the SFA orifice. Entry to the popliteal artery from the deep venous system was at a level just beyond the distal aspect of the arterial obstruction. The Torus stent grafts (100 mm, 150 mm, or 200 mm in length) are self-expanding nitinol stents encapsulated in expanded polytetrafluoroethylene (ePTFE), which are deployed distally to proximally with the first Torus stent graft bridging the popliteal artery to the vein, and the next stent grafts are serially deployed re-entering the SFA, with the proximal uncovered metal struts of the Torus stent graft extending into the distal common femoral artery. The Torus grafts are overlapped by at least 6 cm to avoid component separation. This is followed by post-dilation angioplasty to nominal diameter. Study protocol requirements included dual antiplatelet therapy (eg, 100 mg aspirin and 75 mg clopidogrel) throughout the duration of the study.
Endpoint Assessment
CTA, duplex ultrasound, and plain film X-ray images were assessed by an independent core laboratory (Cleveland Clinic, Cleveland, OH, USA) and evaluated for arterial and venous anatomic endpoints. Each adverse event was reviewed by a medical monitor who is a licensed physician (Syntactx, New York, NY, USA) to determine whether adjudication was necessary. The safety endpoints, including all components of the composite major adverse event (MAE) and major adverse vascular event (MAVE) endpoints, were adjudicated by an independent Clinical Events Committee (CEC, Syntactx, New York, NY, USA). The same group determined whether any revascularization procedures met the definition of clinically-driven target vessel revascularization (CD-TVR).
The primary safety endpoint was the MAE rate at 1 month (defined as a composite endpoint of death, CD-TVR, or major target limb amputation). CD-TVR was defined as endovascular or open surgical revascularization of a target vessel for a symptomatic, core laboratory reported ≥50% stenosis or occlusion. 11 Secondary composite endpoints included MAVEs, triggered by stent-graft thrombosis, target limb amputation, clinically apparent distal embolization with tissue loss, procedure-related arterial rupture, acute limb ischemia, or bleeding events that required transfusion of at least 1 unit of blood. Other safety endpoints included stent fractures, symptomatic target DVT, and changes in VCSS and Villalta scale. Major bleeding was defined as bleeding requiring transfusion of ≥2 units of blood.
The primary effectiveness endpoint was primary patency, defined as freedom from ≥50% diameter stent-graft stenosis, occlusion, or CD-TVR. A ≥50% stenosis was defined when the peak systolic velocity ratio exceeded 2.5 on core laboratory-assessed duplex ultrasound images or as observed on core laboratory-assessed angiographic imaging. Primary assisted patency was defined as freedom from ≥50% stenosis or occlusion, irrespective of interventions. Secondary patency was defined as freedom from permanent or occlusion (last follow-up), irrespective of interventions or remediated occlusions. Other effectiveness endpoints included technical success, defined as successful delivery of the investigational devices to the identified area and removal of delivery system, procedural success defined as technical success in the absence of in-hospital MAEs, and clinical success defined by improvement in the Rutherford category compared with baseline.
Statistical Analysis
The study sample size calculation was performed using a literature-derived, protocol-specified performance goal of 70% for the primary effectiveness endpoint of stent-graft patency at 2 year. An anticipated 85% rate for the primary effectiveness endpoint was based upon early investigations. With these assumptions, accounting for a 60-subject sample size provided 80% power with a 1-sided alpha level of 0.05 for detecting a difference compared with the performance goal, allowing for attrition of 10 patients over 2 years.
All analyses were performed on an intent-to-treat (ITT) basis. Continuous variables were expressed as mean and standard deviation. Categorical variables were expressed as the number of events divided by the number of observations with the 95% 1-sided exact (Clopper–Pearson) confidence intervals (CIs) for the primary safety and effectiveness endpoints. The Kaplan–Meier methodology was used to calculate estimates with standard errors for primary, assisted-primary, and secondary patency, as well as for freedom from CD-TVR, MAE, MAVE, and stent thrombosis. Statistical analyses were performed with SAS version 9.4 (SAS Institute).
Results
Demographic and Baseline Characteristics
Among the 78 patients enrolled in the ITT cohort, treatment was attempted in 82 limbs. The procedure was unable to be completed in 1 patient (1%), leaving 77 patients and 81 limbs in the per-protocol cohort. The per-protocol cohort will not be considered further; all analyses were conducted on the ITT population. The 65 men (83%) and 13 women (17%) had a mean age of 65±7 years. Patient clinical characteristics, including comorbidities and lesion characteristics, are listed in Table 1. A history of smoking was reported in 87% (68/78) of patients, hypertension in 83% (65/78), coronary artery disease (CAD) or myocardial infarction (MI) in 44% (34/78), and diabetes in 26% (20/78). 10
Demographic and Baseline Characteristics.
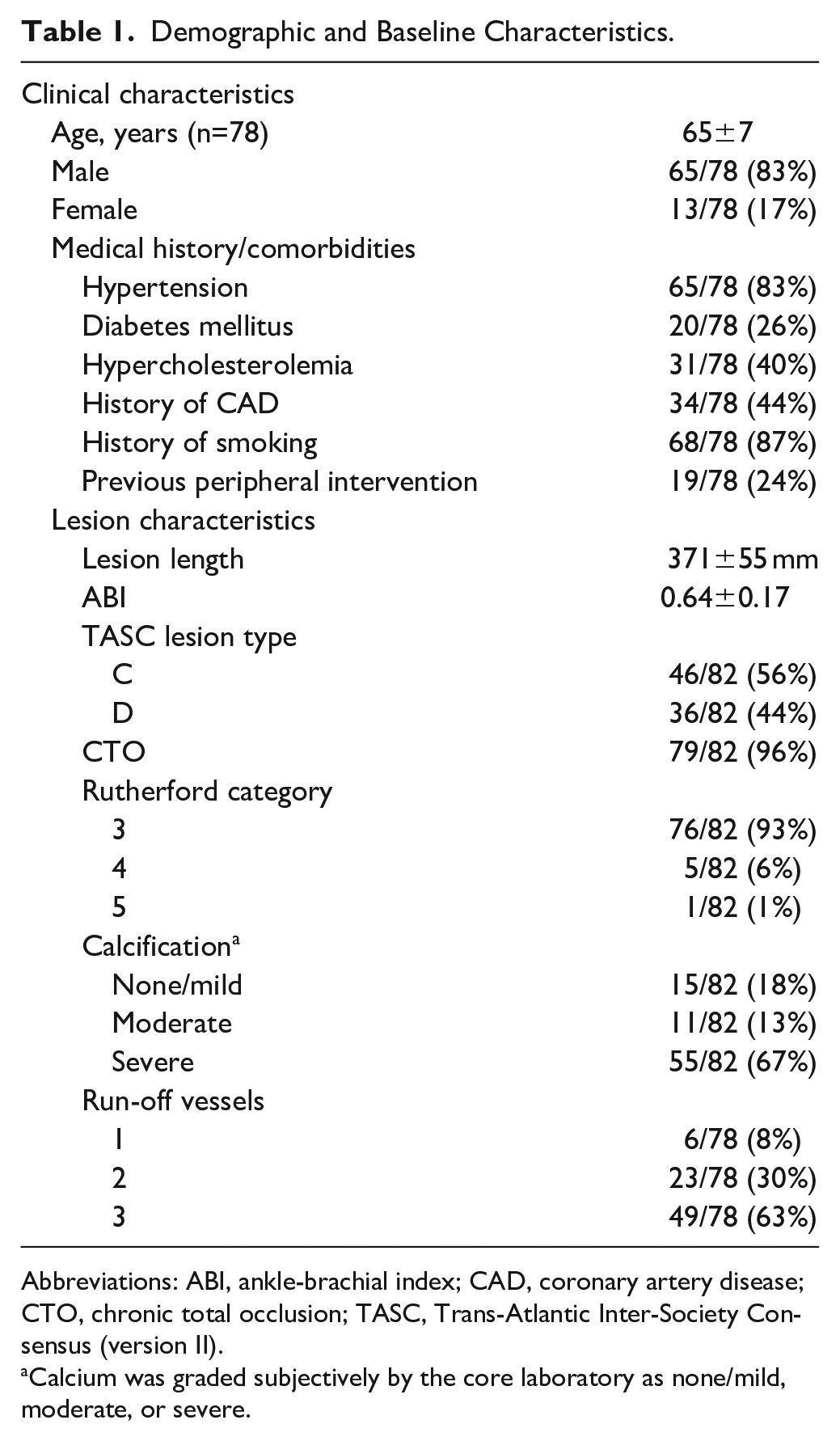
Abbreviations: ABI, ankle-brachial index; CAD, coronary artery disease; CTO, chronic total occlusion; TASC, Trans-Atlantic Inter-Society Consensus (version II).
Calcium was graded subjectively by the core laboratory as none/mild, moderate, or severe.
The mean ABI was 0.64±0.17 at baseline, with a mean core laboratory-reported pretreatment target lesion length of 371±55 mm and a range of 222 to 472 mm (Table 1). All but 3 of the lesions (96%) were CTOs, and the length of the occlusions averaged 150±88 mm. Most patients (93%) had presenting symptoms of lifestyle-limiting claudication. All lesions were either Trans-Atlantic Inter-Society Consensus (TASC II) C (56%) or TASC D (44%). Arterial calcification was graded by the core laboratory as severe in 67% (55/82) of limbs. Most limbs (63%) had 3-vessel runoff, whereas 30% had 2-vessel runoff and 8% had single vessel runoff. There were 4 limbs with unknown run-off status.
Periprocedural Outcomes
The rate of technical success and procedural success was 96% (79/82). There were 3 limbs in which the investigator reported unsatisfactory initial delivery and deployment of the device; however, the final procedural result was satisfactory in 2 of these limbs, whereas the procedure was unable to be completed in the third. 10 The mean number of devices was 2.4 (standard deviation, 0.5) with a range of 2 to 3.
Early Outcomes
The primary safety endpoint of freedom from 1-month MAE was 97% (79/81 limbs 95% CI 92%–100%); CD-TVRs were performed in 2 limbs on postoperative days 6 and 29, respectively (Table 2). Through 1 month, CEC-adjudicated stent-graft thrombosis occurred in 4% (3/81) limbs, 1 patient had bleeding requiring transfusion (1%), and 1 patient developed an ipsilateral DVT (1%; Table 2). The 1-month MAVE rate was 5% (4/81) accounted for by a stent-graft thrombosis (3/81) and a bleeding episode with a 2-unit transfusion at the posterior tibial vein access site (1/81). No patient experienced arterial rupture or distal embolization through the 1-month timepoint. Clinical success was reported in 94% (76/81) of limbs at the 1-month follow-up visit (Table 3). At the 1-month visit, 74% (60/81) of limbs were asymptomatic and 17% (14/81) of limbs had symptoms limited to mild claudication (Rutherford category 1; Table 3). The mean ABI was 0.94±0.20 at the 1-month visit, significantly higher than the preprocedure baseline ABI (p<0.001).
Safety Outcomes.
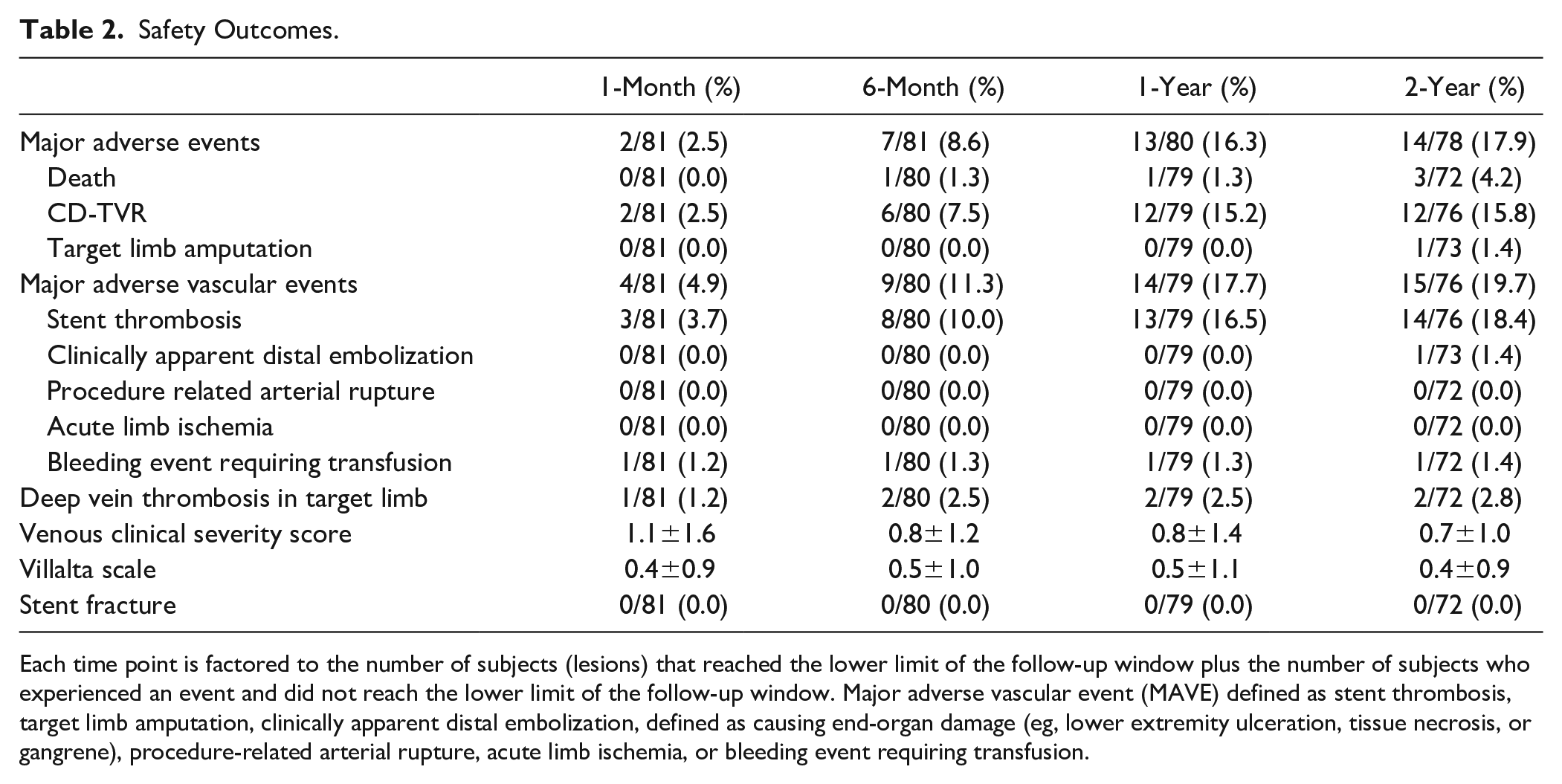
Each time point is factored to the number of subjects (lesions) that reached the lower limit of the follow-up window plus the number of subjects who experienced an event and did not reach the lower limit of the follow-up window. Major adverse vascular event (MAVE) defined as stent thrombosis, target limb amputation, clinically apparent distal embolization, defined as causing end-organ damage (eg, lower extremity ulceration, tissue necrosis, or gangrene), procedure-related arterial rupture, acute limb ischemia, or bleeding event requiring transfusion.
Effectiveness Outcomes.
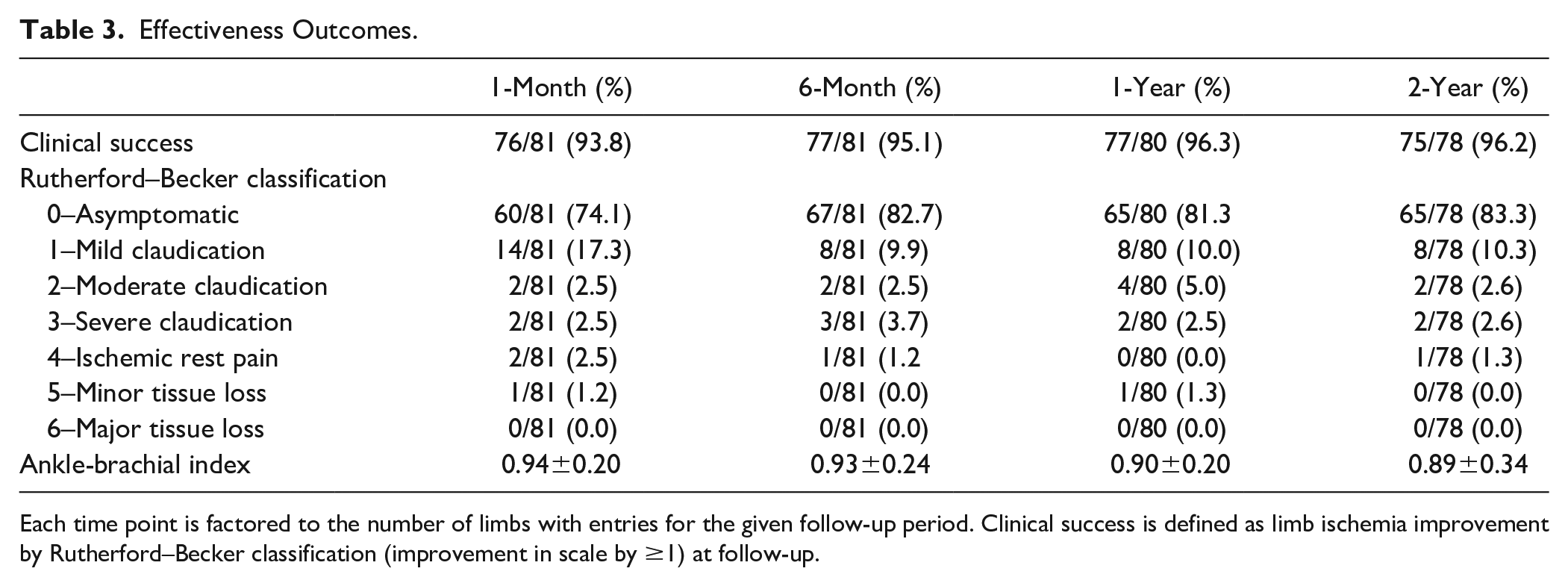
Each time point is factored to the number of limbs with entries for the given follow-up period. Clinical success is defined as limb ischemia improvement by Rutherford–Becker classification (improvement in scale by ≥1) at follow-up.
Mid-Term Outcomes
There were no stent fractures reported by the core laboratory through 2 years. MAE rates at 6 months, 1 year, and 2 years were 9% (7/81), 16% (13/80), and 18% (14/78), respectively (Table 2). The estimates of freedom from MAE by Kaplan–Meier analysis were 91±3%, 84±4%, and 83±4% at 6 months, 1 year, and 2 years, respectively (Figure 1). The 6-month Kaplan–Meier estimate for freedom from MAVE was 89±4%, the 1-year estimate was 83±4%, and the 2-year estimate was 83±4% (Figure 1). Through 2 years of follow-up, there were 3 deaths (4%), due to stroke, intracranial bleeding during a contralateral (non-index limb) thrombolytic procedure, and bowel ischemia following lung tumor resection. Through 2 years of follow-up, 1 patient (1%) had a major target limb amputation. This patient had a PQ Bypass stent-graft thrombosis with thrombi extending into the profunda femoral artery and the tibial arteries. Pharmacologic thrombolysis was successful in restoring blood flow through the graft, but multiple attempts at aspiration thrombectomy of the outflow vessels were not.
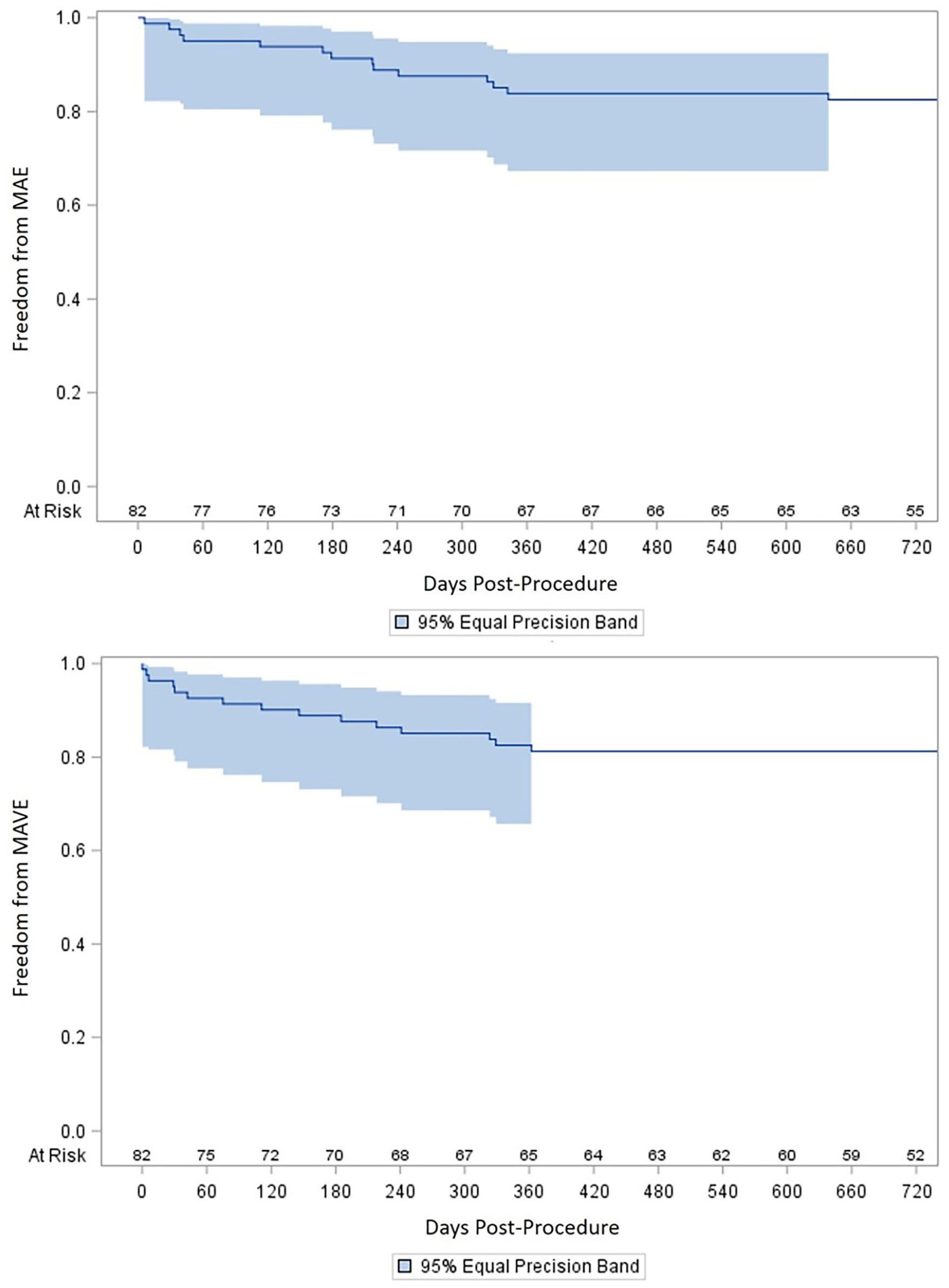
Kaplan–Meier estimates of MAE and MAVE.
The 6-month primary effectiveness endpoint of primary patency was 89% (72/81 of limbs, 95% CI 81%–94%), exceeding the 70% prespecified performance goal for the study (Figure 2). Among the 9 limbs that lost primary patency, there were 6 stent-graft occlusions (5 with CD-TVRs) and 3 stent-graft stenoses ≥50% diameter reduction (all 3 with CD-TVRs; Figure 3). Through 2 years, there were 17 limbs that lost primary patency: 13 with stent-graft occlusion (8 with CD-TVRs) and 4 with ≥50% stent-graft stenoses (all 4 with CD-TVRs). No one pattern of restenosis predominated; lesions were located at the proximal stent, distal graft, and in 1 case, there was a diffuse lesion of the entire stent graft.
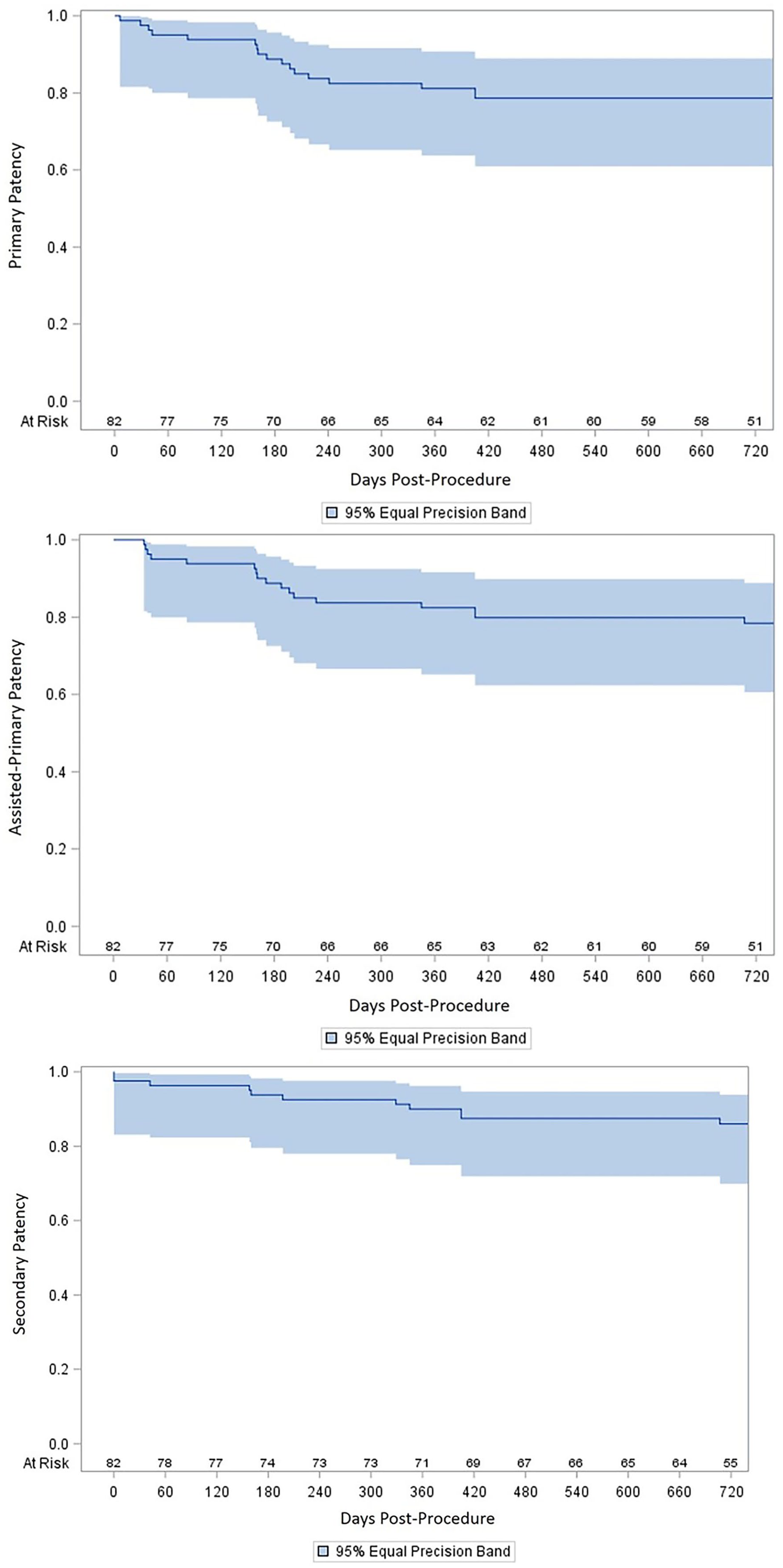
Kaplan–Meier estimates of primary, assisted-primary, and secondary patency.
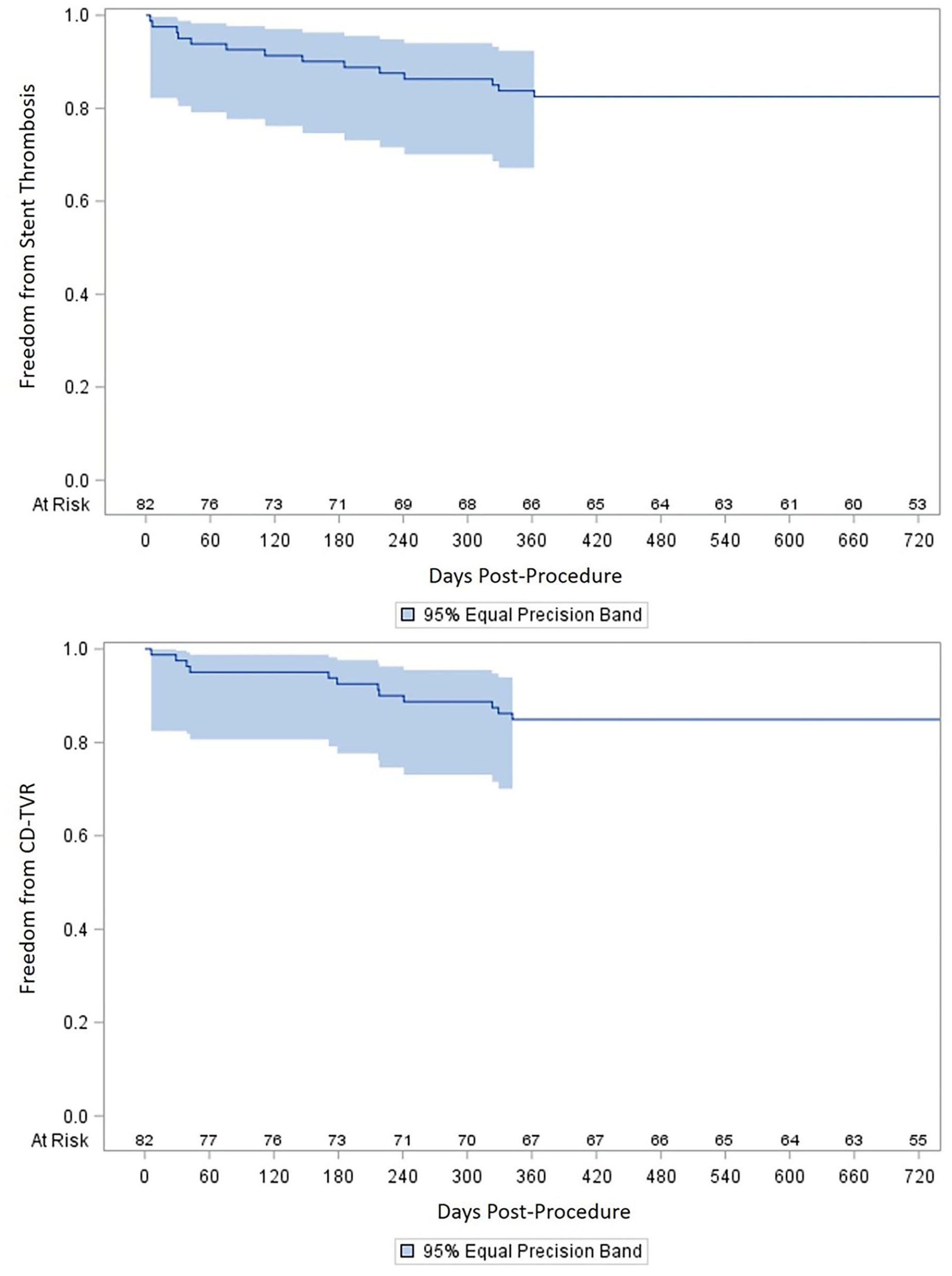
Kaplan–Meier estimates of freedom from stent thrombosis and CD-TVR.
The 1-year Kaplan–Meier patency rates for protocol-defined primary, assisted-primary, and secondary patency were 81±4%, 82±4%, and 90±4%, respectively (Figure 2). The 2-year Kaplan–Meier patency rates for primary, assisted-primary, and secondary patency were 79±5%, 78±5%, and 86±4%, respectively (assisted-primary patency was 0.2% higher than primary patency, 78.6% vs 78.4%, due to an artifact of the Kaplan–Meier analysis from differential timing of censored patients). Expressed as a binary endpoint, primary patency was 65/80 (81%) at 12 months and 61/78 (78%) at 24 months. Primary patency as a binary endpoint in very long (>360 mm) lesions was 44/52 (85%) at 12 months and 40/50 (80%) at 24 months. Freedom from CD-TVR at 1 year and 2 years was 85±4% and 85±4%, respectively (Kaplan–Meier estimates, Figure 3), with 12 CD-TVRs (12/76, 15%) through the first 2 years after the index procedure. Through 1 year and 2 years, there were 13/79 (16%) stent-graft thrombosis and 14/76 (18%) stent-graft thrombosis, respectively, accounting for Kaplan–Meier estimates of freedom from stent-graft thrombosis of 84±4% at 1 year, and 84±4% at 2 years (Figure 3).
The ABI was 0.90±0.20 (p<0.001 vs baseline) and 0.89±0.34 (p<0.001 vs baseline) at 1 year and 2 years, respectively (Table 3). The 2-year Rutherford category improved from baseline; 83% (67/81) of limbs were asymptomatic, and 10% (8/81) had symptoms limited to mild claudication (Rutherford category 1, p<0.001 vs baseline). The Rutherford category improved in 96% (77/80) of limbs with observations at 1 year; 81% (65/80) were asymptomatic, and 10% (8/80) had mild claudication (p<0.001 vs baseline). The Rutherford category improved in 96% (75/78) of limbs with observations at 2 years; 83% (65/78) were asymptomatic, and 10% (8/78) had mild claudication (p<0.001 vs baseline).
Venous Status Over Follow-Up
Through 2 years, DVTs occurred in 3% (2/72) of target limbs (Table 2). The DVT involved the femoral and popliteal veins in each case. Both patients were treated with local standard of care which included anticoagulation and duplex ultrasound, in addition to the protocol-specified follow-up visits, imaging, VCSS, and Villalta scale. One presented at post-op day 27 and the other one at post-op day 178. The DVT involved the femoral and popliteal veins in each case. Overall, there were no significant changes in the VCSS or Villalta scales during follow-up. The VCSS scores were 1.1±1.6, 0.8±1.2, 0.8±1.4, and 0.7±1.0 at 1 month, 6 months, 1 year, and 2 years, respectively, compared to the mean VCSS of 0.8±1.3 at baseline. The corresponding Villalta scale values were 0.4±0.9, 0.5±1.0, 0.5±1.1, and 0.4±0.9 at 1 month, 6 months, 1 year, and 2 years respectively, compared to the mean Villalta scale value of 0.4±0.9 at baseline.
Discussion
The DETOUR system was designed to be an endovascular therapy (EVT) created femoral popliteal bypass with an acceptable safety profile. The published 1-year results demonstrated short-term safety and effectiveness of the device, despite a challenging patient population. 10 The 2-year results document a favorable safety and effectiveness profile, with patency rates similar to open surgical bypass. Furthermore, the morbidity of the procedure is reduced compared with traditional open revascularization. There was no venous morbidity as measured by VCSS and Villalta scales with two DVTs. There were no wound infections as opposed to up to 30% of lower extremity bypasses.12-14
Traditional treatment of long segment femoropopliteal PAD frequently involved open surgical bypass via autogenous vein or prosthetic grafts, both of which are associated with acceptable long-term patency rates.2,15 However, such procedures are invasive and involve long recovery times. The morbidity associated with open revascularization is an important consideration when making treatment decisions for the frequently elderly and medically compromised PAD population. Hospital stays after open bypass can approximate 5 to 10 days and in complicated cases can take months, adding significant financial burden. 16 The prolonged rehabilitation and risk of post-operative complications such as occurrence of post-procedure wound complications in 10% to 20% of patients also result in a significant financial burden.2,17,18 On this landscape, EVT has become common place for the treatment of PAD. Endovascular methods for treating femoropopliteal disease are less invasive, associated with decreased perioperative morbidity, length of hospital stays, and recovery time. However, first-generation interventions, such as percutaneous transluminal angioplasty (PTA), have suboptimal long-term outcomes, especially with lesions that are >100 mm in length.19-23
Newer EVT interventions such as stents, covered stents, and drug-coated balloons have resulted in varying degrees of improvement over PTA.24-27 The 2-year ILLUMENATE trial results showed significant higher primary patency for paclitaxel drug-coated balloons versus Plain balloons (76% vs 61%, respectively). 28 Additionally, provisional stents remain necessary in 25% of CTOs for residual stenosis following drug-coated balloon angioplasty. 27 Moreover, the rate of reintervention at 2 years for plain balloons, drug-coated balloons, bare metal stents, and drug-coated stents (DCSs) was 36%, 18%, 27%, and 19%, respectively. 29
With lesion lengths averaging 371 mm and CTO lengths of 159 mm, the primary patency rates in the DETOUR study were 81% at 12 months and 79% at 24 months. Taken in the context of a mean lesion length of 371 mm and 96% CTO with occlusion lengths averaging 159 mm, the DETOUR results compare favorably to those of other endovascular modalities used for long-lesion femoropopliteal disease, including covered stents and drug-coated balloons. For long femoropopliteal lesions, the VIASTAR study of heparin-coated covered stents reported 2-year patency rates of 65%, compared with 27% for bare metal stents. 30 In the IN.PACT Global study, the 3-year CD-TLR rate was 23.1% overall, with a 2-fold increased risk of CD-TLR in lesions lengths exceeding 200 mm. 31
The IN.PACT Admiral DCB Long Lesion Sub-Cohort study is a series with some of the most favorable results for percutaneous treatment of long lesion (>180 mm) femoropopliteal disease. The lesions lengths in the IN.PACT Long Lesion study population were somewhat shorter than those in DETOUR. The mean total lesion and occlusion lengths were 287±71 and 117±113 mm, respectively in IN.PACT compared with 371±55 and 159 ±88 mm, respectively in DETOUR. The 12-month binary primary patency rates in IN.PACT and DETOUR were 65% and 81%, respectively. In the subgroup of lesions longer than 360 mm, the 12-month primary patency rate fell to 44% in IN.PACT but remained at 85% in DETOUR. 32
Although open bypass is generally recommended for complex femoropopliteal lesions, a minimally invasive intervention with comparable effectiveness outcomes remains an unmet need and the goal for the next-generation EVT. An area of initial concern with the PQ Bypass procedure was regarding DVT risk, given the path of the stent graft through the femoral vein. However, only 2 limbs (3%) receiving the DETOUR system experienced DVT through to 2-year follow-up. This DVT rate was not significantly greater than what is seen following open bypass, despite the presence of a stent graft coursing through the deep venous system. 33 Additionally, no symptoms of venous outflow obstruction or worsening of VCSS and Villalta indices were evident. There were no symptomatic pulmonary emboli over the 2-year follow-up period. Whether this reflects of the low DVT rate or intrinsically less-embolic nature of the PQ stent graft-associated thrombus is an area worthy of further study.
Claudication symptoms were remediated by the DETOUR device system effectively through 2 years, with improved Rutherford in 96% of patients and 83% of limbs were asymptomatic (Rutherford class 0). Outpatient evaluation found 62% and 50% in Rutherford class 0 for open bypass and EVT groups, respectively. 34 Similar to the mid-term follow-up, ABI for the open bypass and EVT groups, consistent ABI normalization through 2 years, was also found to be a hemodynamic benefit. 34
Potential limitations of the DETOUR study include the inability to find a single, fair comparator device, since the DETOUR system is the first of its kind. Given the mechanics of the device, it is expected to carry a very different effectiveness profile as compared to standard EVTs such as plain balloon angioplasty or even arterial stents in the treatment of more complex lesions. Additionally, noting the minimally invasive mechanism of deployment, it is difficult to compare safety outcomes of the DETOUR system to the highly invasive open bypass. Comparisons of the DETOUR system to alternative therapies are limited to literature-reported data generated from distinct patient groups. An additional limitation of the study was the inclusion of 4 patients with bilateral treatment; a design feature that might confound the analysis to the extent that outcome in the 2 sides was correlated. Finally, clinical considerations of how the device affects the venous circulation, including complications of the arteriovenous connection, symptomatic DVT versus non-occlusive venous material associated with the graft, and the venous luminal preservation will need more thorough assessment.
Conclusions
The 2-year data from the DETOUR study suggest that the technology offers a promising percutaneous option for patients with symptomatic long-segment femoropopliteal occlusive disease. Mid-term patency rates are favorable, and complications including venous thromboembolic disease are relatively infrequent. These mid-term results confirm the safety and effectiveness of the system as a viable solution for patients with complex femoropopliteal disease who would otherwise be poor candidates for open surgical revascularization.
Supplemental Material
sj-docx-1-jet-10.1177_15266028211034862 – Supplemental material for Percutaneous Femoropopliteal Bypass: 2-Year Results of the DETOUR System
Supplemental material, sj-docx-1-jet-10.1177_15266028211034862 for Percutaneous Femoropopliteal Bypass: 2-Year Results of the DETOUR System by Grzegorz Halena, Dainis K. Krievins, Dierk Scheinert, Janis Savlovskis, Piotr Szopiński, Albrecht Krämer, Kenneth Ouriel, Andrej Schmidt, Michal Zdunek and Sean P. Lyden in Journal of Endovascular Therapy
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DKK, GH, JS, and PS have no conflicts of interest. DS is on the advisory board/is a consultant for Abbott, Bayer, Boston Scientific, Cook Medical, Cardionovum, CR Bard, Gardia Medical/Allium, Medtronic, Philips, and Upstream Peripheral Technologies. AK’s hospital received payments from PQ Bypass for expenses incurred during admission and procedure. The professional staff also received honoraria; however, AK does not have an employment arrangement or royalty/stock agreement with PQ Bypass. AH is Clinical Investigator and Advisory Board Member for Gore, Medtronic, and Boston Scientific as well as a Clinical Investigator for Cook, BD-Bard, Cagent, Endologix, PQ Bypass, Merit, and Surmodics. KO is an employee of Syntactx and has equity in Syntactx. AH has no royalty or stock involvement. AS is a consultant for Abbott, Bard/BD, Cook, Cordis/Cardinal Health, Reflow Medical, and Upstream Peripherality in Syntactx. AH has no royalty or stock involvement. AS is a consultant for Abbott, Bard/BD, Cook, Cordis/Cardinal Health, Reflow Medical, and Upstream Peripheral.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study sponsor (PQ Bypass, Milpitas, CA, USA), while not involved in the data analysis or interpretation, was involved in the study design and data acquisition. Andrew Hill, MBChB (Auckland City Hospital, Auckland, New Zealand), and Yamume Tshomba, MD (Fondazione Policlinico Universitario Agostino Gemelli, Milan, Italy) were involved in data collection. Paul Bishop, MSEE (Cleveland Clinic, Cleveland, OH, USA) assisted with the data interpretation and analysis. Victoria Lee, MD and Minyi Hu, PhD (Syntactx, New York, NY, USA) provided technical services.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.