
Abstract
Background:
Approval of medical devices is typically based on data from relatively small clinical studies with a highly selected patient population. Postmarket surveillance is required by regulatory bodies after approval to collect and evaluate experience gained from real world use in larger and unselected populations. Terumo Aortic is a manufacturer of off-the-shelf and custom-made stent-grafts for endovascular repair of thoracic and abdominal aortic pathologies and is assessing device performance in a large registry.
Methods:
A multiarm, multicenter, open label, prospective observational registry designed to obtain both short- and long-term safety and performance data on the use of standard and custom-made Terumo Aortic endovascular devices in patients with thoracic and abdominal aortic pathologies. Eligibility requirements are minimal, and a standard-of-care protocol will ensure real-world evidence is collected as far as 10 years.
Discussion:
Challenges to this research reflect its real-world nature such as differences in standard of care between centers and geographies, varying levels of experience and expertise with the devices or techniques, all-comer populations that may not always be comparable, and a design specifically limited to a single manufacturer. Advantages of this registry design include long-term follow-up, different modules to collect standardized outcomes across pathologies and global reach to reflect practice in many different geographies with a wide range of latest-generation endovascular devices.
Conclusion:
This protocol is a large endovascular registry of all aortic pathologies that are treated by both off-the-shelf and custom-made Terumo Aortic products. It is ambitious in scope and projection and will be part of an overall response involving patients, physicians, and manufacturers to answer the remaining questions of endovascular aortic repair, contribute to continuing improvement of the techniques and technologies, and present an accurate picture of outcomes with latest generation stent-graft devices.
Clinical Impact
This large, long-term registry will generate robust real-world evidence on the safety and performance of both standard and custom-made Terumo Aortic endovascular devices in treating thoracic and abdominal aortic pathologies. By including a broad, minimally selected patient population across diverse global centers, the study mirrors everyday clinical practice and helps bridge the gap between clinical trials and real-world outcomes. Its findings will inform clinical decision-making, support regulatory compliance, and guide ongoing device development. Ultimately, the registry aims to enhance patient care by improving the understanding of endovascular treatment effectiveness and long-term durability in heterogeneous populations.
Introduction
Since the early 1990s, endovascular aortic repair (EVAR) has become the standard treatment for abdominal aortic aneurysms (AAA) with a suitable anatomy, and later as thoracic EVAR for thoracic pathologies such as aneurysm, dissection, and blunt traumatic aortic injury. 1 The success of the technology has led to more complex techniques such as fenestrated and branched EVAR and even to treatment of the ascending aorta and aortic valve. 2 A number of grafts and models exist for the various types of endovascular repair of aortic pathologies, and all may have different instructions for use and availability. Given that this technique is still in development with an uncertain long-term effectiveness, lifelong imaging surveillance is required. 3
Postmarket surveillance is the process of continuing assessment of safety and effectiveness after a medical device is authorized for commercial use. 4 The European Union’s Medical Device Regulation 2017/745 defines it as a “systematic procedure to proactively collect and review experience gained from devices on the market for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions.” 5 The US Food and Drug Administration (FDA) describes it as an “active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device.” This requirement has often been met by the conduct of postmarket clinical follow-up studies, often referred to as registries, to distinguish them from trials with investigational devices and confirmatory studies. 6
Some characteristics specific to medical devices mean that registries (which may provide a larger perspective) are particularly appropriate as a study design compared to other clinical research methodologies which may face challenges including uncertainty about long-term outcomes; substantial design variation within a class; the potential for clinically significant variation in outcomes across populations; high cost; difficulty in blinding (making randomization impossible or unethical); confounding factors such as operator experience and preference with regard to device oversizing; patient and device selection, and continuous minor device changes/developments.6,7
The Terumo Aortic Global Endovascular Registry (TiGER) is a multiarm, multicenter, open label, prospective observational registry of Terumo Aortic endovascular grafts that will provide long-term data on device performance in real-world populations with thoracic and abdominal aortic pathologies and insight into both off-the-shelf and custom-made devices based on the RELAY, TREO, and ANACONDA, Terumo Aortic, Inchinnan, United Kingdom platforms. The size and range of TiGER will reflect and allow for the diverse reality of aortic repair which depends on many varied factors from patient-specific (pathology and anatomy), to the physician-specific (skills and experience), to the environment (institution and regulatory situation, culture) and the manufacturer (availability and specifications). Such a wide-ranging undertaking will go some way to addressing the challenges of researching medical devices.
Materials and Methods
The objective of the TiGER study is to collect real-world, post-approval safety, performance, patient-reported health outcomes, and health economic data on patients treated with Terumo Aortic endovascular stent-grafts in standard clinical practice. This is a multiarm, multicenter, open label, prospective observational registry designed to obtain safety and performance data on the use of Conformité Européenne marked and custom Terumo Aortic endovascular stent-grafts in standard clinical practice. Table 1 details the enrollment schedule, interventions, and assessments for the study.
Participant Timeline Showing Schedule of Enrollment, Interventions, and Assessments.


t1: Per SOC, after implantation, each patient will be evaluated prior to (or at) hospital discharge/30 days to 6 months; t2: 12 months postimplant (±30 days); t3: 24 months postimplant (±180 days) and annually thereafter; tn: the expected duration of each subject’s involvement in the study is from date of implant until lifetime of subject or the subject is withdrawn or LTFU.
Abbreviation: AEs, adverse events; EC, Ethics Committee; EVAR, endovascular aortic repair; FEVAR, fenestrated endovascular aortic repair; IIAA, isolated iliac artery aneurysm; IRB, Institutional Review Board; LTFU, lost to follow-up; SOC, standard of care; TEVAR: thoracic endovascular aortic repair.
May be performed over multiple clinic visits.
Where a patient is deemed emergent, or per conditions for retrospective informed consent subject to EC/IRB approval may apply.
Study completion where patient withdraws or is LTFU.
Where a patient is deemed emergent, informed consent can be done retrospectively.
Routine assessments include vital signs, physical examination, blood collection and analysis, blood pressures, and pregnancy tests (where required).
Indicates that task is optional depending on local practice.
Thoracic group—imaging is required for a minimum of 5 years.
The AEs recorded will be limited to condition/procedure and device-related events only, treatment/concomitant/antibiotics will be captured for any AEs.
Study completion where patient withdraws or is LTFU.
Depending on pathology and treatment, patients enrolled in the study are allocated to one of the 4 modules (Figure 1). The study will include a minimum of 1000 subjects, without upper limit, and involve up to 80 sites globally. Currently enrolling sites are in Belgium, France, Germany, Italy, The Netherlands, Portugal, Spain, Switzerland, United Kingdom, and the United States. A current list of study sites is available on ClinicalTrials.gov (www.clinicaltrials.gov/ct2/show/NCT04246463).
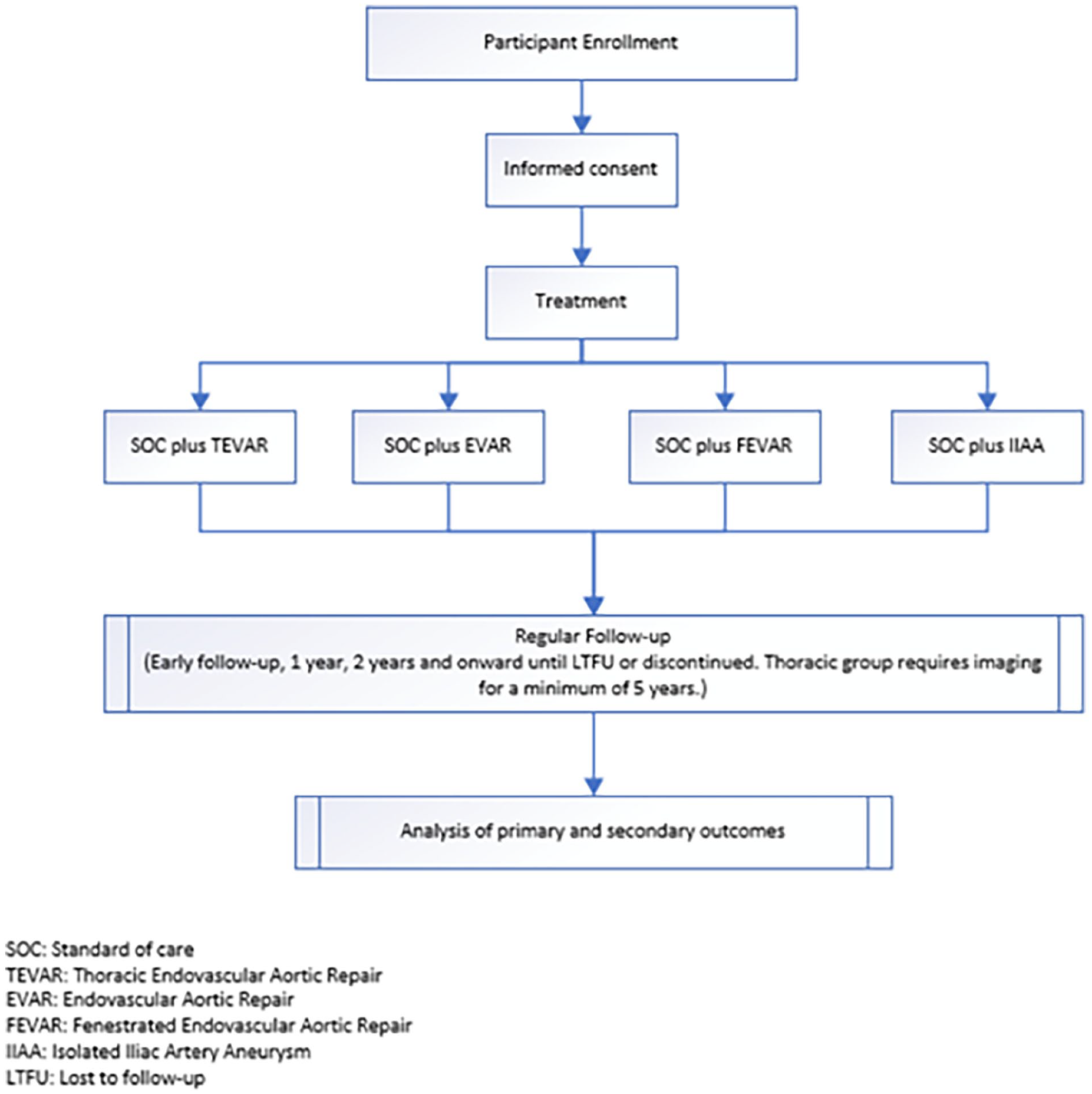
Allocation flowchart by patient treatment.
This is a noninterventional study and therefore only patients who would receive treatment with a Terumo Aortic device as part of standard of care treatment can be enrolled. Patients will be enrolled under one of the following modules:
• Thoracic module: Patients requiring urgent or elective endovascular treatment of thoracic aortic aneurysm, pseudoaneurysm, dissection, penetrating atherosclerotic ulcer (PAU), or intramural hematoma (IMH).
• Abdominal module: Patients requiring urgent or elective endovascular treatment of infrarenal AAA.
• Custom module: Patients with a documented thoracic, thoracoabdominal, or abdominal pathology suitable for elective treatment using a Terumo Aortic custom-made device for one of the following: ○ Abdominal/thoracoabdominal custom—fenestrated devices and fenestrated combination devices, scalloped devices. ○ Thoracic custom—branched, fenestrated, or scalloped devices or otherwise customized devices.
• Other module: Patients requiring urgent or elective endovascular treatment of isolated iliac artery aneurysm.
A novel element of TiGER is that the FDA has agreed to embedding 2 postapproval studies into TiGER as part of its respective conditions of approval of the aneurysm and dissections indications of the RelayPro, Terumo Aortic, Inchinnan, United Kingdom device: a minimum of 177 aneurysm or PAU subjects treated with the RelayPro device, of which, a minimum 88 will be enrolled in the United States and a minimum of 110 will be evaluable at 5 years; a minimum of 120 dissection subjects treated with the RelayPro device, of which, 60 will be chronic dissections and 60 will be acute dissections, where a minimum of 30 patients in each dissection classification will be enrolled in the United States. To note, patients in the United States will be enrolled in the thoracic module only.
As new indications and devices are approved, those subjects may also be captured within expanded modules of the registry as part of their postmarket follow-up plan. Sites are selected to reflect a range of user experience and clinical situations with both high- and low-volume institutions; however, all will have previous experience with at least one Terumo Aortic product included in the registry at the time of selection.
Imaging
Imaging will be performed as per standard practice and reported locally at site level into the database. Where an adverse event (AE) occurs (defined as any untoward medical occurrence, unintended disease or injury, or untoward clinical signs (including abnormal laboratory findings) in subjects, users or other persons), whether or not related to the investigational medical device, imaging analysis may be further reviewed by the sponsor. Any site-reported device-related/possibly device-related events will be submitted to an independent third-party reader (Core Laboratory; NAMSA, Minneapolis, MN) for imaging analysis. Furthermore, the Medical Monitor will perform independent MedDRA (medical dictionary for regulatory activities) coding for all AEs.
Characteristics to Participate
To ensure that patients reflect real-world use, eligibility for participation is as simple as possible while meeting regulatory requirements. Patients must be consenting adults (minimum age as per local regulations) and have an indication for aortic endovascular repair with a Terumo Aortic, Terumo Aortic, Inchinnan, United Kingdom approved (marketed) off-the-shelf device or custom-made devices and be willing to comply with the registry protocol and adhere to follow-up visits. In cases requiring urgent treatment (typically younger patients with traumatic injuries), participation is possible if there is an Institutional Review Board (IRB)/Ethics Committee (EC) agreement to allow consent after intervention (ie, consent to study participation, not to treatment).
As a real-world registry that follows standard-of-care protocols, there is a reliance on sites to encourage follow-up with all patients; however, this is continually tracked by the sponsor. Several practices are/will be implemented to minimize the number of patients lost-to-follow-up (LTFU) including: the option for remote study visits, routine calls and check-ins with sites to discuss compliance/maintenance of compliance; encouragement of annual visit reminders from sites; and one-to-one calls with investigators for sites with poor follow-up compliance. Furthermore, the statistical analysis plan details methods to account for the impact of LTFU on the interpretability of study outcomes.
Ethics Approval and Consent to Participate
Prior to starting the registry, each investigator will provide the sponsor with documented evidence that an appropriate EC or IRB (and any other relevant approval board) has approved the study protocol, patient information sheet (PIS), and informed consent form (ICF). The consenting process for the registry was aligned with the requirements necessary for ethical and IRB review and approvals, and well as General Data Protection Regulations specific to each country, and site, included. Written confirmation of the informed consent process shall be obtained from each patient prior to any study activities. This is a noninterventional protocol and the standard of care at each institute should always be followed. The sponsor recognizes that standard of care can differ between hospitals and therefore if any of the procedures specified in this protocol are not in line with standard of care at a particular institute, then the principal investigator/institute must inform the sponsor.
Retrospective consent to study participation is permitted under the following limited circumstances subject to EC/IRB approval:
For U.S. sites only, if a patient has visited before the study starts (when IRB outcome is favorable, but the site was pending study activation) and is not due to return to the site for some time, consent may be obtained via telephone. The PIS and ICF should be sent via post or e-mail for the patient to read and consider the study. A telephone call should be used to discuss the study with the patient (including risks and benefits), if the patient is happy to participate in the study, they should be asked to sign a copy of the ICF and send back via post (the patient should also retain a copy). The following information should be recorded in the patient’s medical notes at a minimum; time and date of telephone call, name and role of personnel who conducted the call, version of PIS and ICF provided to patient and date sent, document any questions asked by the patient and record date patient verbally confirmed consented to the study. Once a copy of the signed ICF is received, this should be signed by appropriate study personnel. No study data will be entered into the study database until the signed ICF is fully signed.
In the event the patient has not consented prior to procedure (eg, due to admission type or study site personnel availability at the time of pre-procedure) consent may be collected retrospectively immediately after procedure and no later than discharge/early follow-up (up to 6 months). The written informed consent documentation process will be followed as per study protocol and will be documented clearly in the patients’ medical source notes. Where retrospective consent is used, no study data will be collected prior to consent being obtained.
Where approved by site IRB, informed consent requirement will be waived. Documentation of IRB decision and approval to waive ICF will be collected prior to data collection.
The sponsor will provide a generic ICF and PIS for the study in English. These will be translated into appropriate country-specific languages by the sponsor. Any changes to PIS or ICF requested by local EC/IRB must be notified to the sponsor. Once approved by the EC/IRB, no changes should be made to these documents without first seeking approval from the relevant EC/IRB and the Sponsor.
Patients that meet the inclusion criteria (and none of the exclusion criteria) for this study may require emergency surgery to repair the aorta. In these cases, there may not be time to perform the consent procedure as described in the protocol, or the patient may not be able to give consent due to their medical condition. Where possible, preoperative consent should be obtained from the patient or legally authorized representative (LAR); however, retrospective written informed consent to participate in the study is acceptable for emergency patients only (eg, because of trauma or imminent rupture). Where retrospective consent is used, no patient identifiable study data will be collected prior to consent being obtained. Where the patient/LAR is unwilling or unable to give retrospective consent, no study data will be collected.
Clinical Outcomes
All operative, postoperative and follow-up data will be entered into electronic case report forms (eCRFs). Reporting standards, including the European Society for Vascular Surgery and European Society of Cardiology were reviewed and utilized for development of eCRFs.8,9
The primary outcome will be aortic-related mortality, which is defined as any death due to aortic rupture, malperfusion, or aortic dissection. All-cause mortality (any death occurring during the study period, regardless of cause) is a secondary endpoint and subdivided by: aortic-related death (any death occurring within 30 days of implant, due to rupture or following any procedure intended to treat the target lesion), cardiac-related death (death due to arrhythmia, heart failure including cardiogenic shock, or myocardial infarction), pulmonary-related death (death due to due to pulmonary edema, respiratory failure, or pulmonary embolism), vascular-related death (death due to stroke, cerebral hemorrhage, or another clear vascular event that is not categorized as cardiac-related or pulmonary-related), other (death due to any event that cannot be clearly categorized as above, but where some information is available), unknown (a death where no information is available). All deaths within 30 days of the procedure will be automatically considered to be procedure-related or device-related unless obviously unrelated or aortic-related.
Other secondary endpoints include performance measurements such as technical success which is a composite of: successful delivery of the device through the vasculature (ie, ability to deliver the implant to the intended location without the need for unanticipated corrective intervention related to delivery), deployment of the endovascular stent graft in the planned location with coverage of the target lesion (or entry tear in the case of dissection), absence of unplanned coverage or aortic branch vessels, patency of the endovascular stent graft, absence of device deformations (eg, kinks, stent eversion, mal-deployment, misaligned deployment) requiring unplanned placement of an additional device within the endovascular stent graft, successful withdrawal (ie, successful withdrawal of the delivery system, without the need for unanticipated corrective intervention related to withdrawal), absence of Type Ia, Type Ib, Type IIIa, and Type IIIb endoleak that extends beyond 30 days of confirmatory imaging (eg, computed tomography angiography, magnetic resonance angiography, or duplex ultrasound).
Composite clinical success is defined as the absence of target lesion-related mortality, occurrence of endoleaks (Type Ia, Ib, IIIa, and IIIb), graft infection, loss of stent-graft patency (>50%), target lesion aortic rupture, conversion to open repair, arrest of the original pathological process (eg, total aortic diameter expansion >5 mm in the aorta that has a study endograft, embolization from penetrating ulcer, extension of dissection, false lumen perfusion) or a new aortic pathology as a result of the intervention (eg, pseudoaneurysm, dissection, IMH, unintentional adverse dissection septum rupture, stent-graft induced aortic wall injury, new aortobronchial/tracheal, or aortoenteric fistula formation). Additional endpoints include all-cause mortality, target lesion reintervention, all secondary interventions, device performance based on imaging assessments of all morphological variables (aortic remodeling, all endoleaks, any loss of patency, stent-graft migration, device integrity, etc).
Safety endpoints will be measured as the incidence of AEs classified by seriousness (serious and nonserious), incidence of serious AEs by relationship to device, procedure, and preexisting condition, events of interest (such as major AEs) which includes, death during the study period, myocardial infarction, stroke [modified Rankin Scale (mRS) score due to neurological deficit >2 at 90 days postevent and an increase in at last 1 mRS category from an individual’s prestroke baseline], new onset renal failure requiring dialysis, renal failure, respiratory failure, grade 3 spinal cord ischemia (Society for Vascular Surgery reporting standards), bowel ischemia, and procedural blood loss >1000 cc.
Patient outcomes will be measured by postoperative return to normal activities—employment, household activities, social life, and hobbies—at 1 year follow-up. Procedure-related health economics will be described using the following parameters: total procedure time (from the first break of skin to final closure, ie, skin to skin time, percutaneous, first access to last access closure), arterial access route and type, volume of contrast media used, fluoroscopy time, image fusion technology used, magnet system used, device re-positioned, blood loss, blood transfusion required, lower limb ischemia, time in intensive care unit (ICU)/maximum care unit (from the first administration of anesthesia to release from intensive care or postanesthesia care unit providing ICU-level care), time to hospital discharge (from initiation of the procedure to physical discharge from the hospital), complications requiring reintervention/re-hospitalization.
Statistical Methods
The intention is to use the registry data for a variety of subanalyses which may utilize data collected on a single device, indication, or data from several devices implanted for the same clinical indication. Each subanalysis will have a separate statistical analysis plan and sample size calculation. Poolability across sites will be assessed for the primary outcome which will be calculated for each individual site to evaluate the consistency of the primary outcome estimate or to detect outcomes that differ significantly (outliers) across sites. A binary logistic regression will determine whether the effect of site is statistically significant. If a significant effect of site is found, estimate of the primary endpoint will be calculated by excluding the extremely low or high primary outcome estimate one site at a time and the effect of excluding individual sites on the overall proportion will be examined. If after considering the totality of evidence from these evaluations, it appears that the primary outcome result is not consistent across sites, the final proportion and confidence interval will be constructed as a weighted average by site with weights proportional to the sample size within each site. Statistical analysis will be carried out using SAS 9.4, Cary, NC or later. Generation of descriptive statistics will be an ongoing process. Survival analysis and modeling will be performed at the discretion of the biostatistician and the lead investigators to address specific issues as they occur.
Discussion
The objective of postmarket research is to determine whether the results of clinical trials with investigational devices can be replicated in real-world practice over the short- and long-term. 10 This fundamental objective is usually referred to as “generalizability” or “external validity” between research that evaluates efficacy in a well-defined and controlled setting and effectiveness in standard clinical practice. 11
The importance of follow-up after EVAR is to confirm that the early benefits (significant reduction in perioperative mortality and morbidity) are maintained despite, for example, late secondary interventions and mortality. 12 Famously, the early EVAR 1 trial showed significant late (>5 years) complications in the endovascular arm that reversed the initial survival benefit at 8 years. 11 Stent-graft design has evolved and improved—as has physician expertise—since those early days and resulted in contemporary series with fewer long-term complications, increased reintervention-free survival, and lower all-cause mortality. 13
While it is accepted that longer-term follow-up is essential, how that should be conducted is less clear. A systematic review of 147 articles reporting on 27,058 endovascular repairs recommended noninferiority studies (better performance than the worst performing 25% stent-grafts; with at least 525 patients) and endpoints of cumulative endoleak rate (excluding type II) and reintervention rate at 2 years. 14 Others argue that postapproval device evaluation using real-world data from registries can be done much more economically and faster than traditional independent industry studies. 15
TiGER is an ambitious project prioritizing longer-term follow-up of a large patient population treated with EVAR in several different configurations and thus aims to address some of the limitations of some product registries. Investing in large-scale registries that can facilitate international collaboration is important to generate high-quality data efficiently that can inform policy and change clinical and public health practices. As clinical trials of custom-made devices may be too small to provide conclusive safety and performance evidence, this registry focuses on consolidating resources (at both a site and sponsor level) to allow the inclusion of a larger number of study sites and provide a greater chance of collecting data on these devices (although data are collected in different study arms and results will be analyzed separately). Despite its size and scope, a guiding principle has been simplicity because it is well reported that research complexity adversely effects quality and performance. 16 We have attempted to limit the number of endpoints, eligibility criteria, and procedures per visit, despite the fact that the number of countries and investigative sites positively correlates with amendment frequency, longer screening and study duration, and study participant dropout rates. Since initial registration on clinicaltrials.gov, minor improvements have been made to the protocol to further clarify the inclusion/exclusion criteria as well as definitions for the secondary outcome measures of technical success and composite clinical success.
The chosen primary endpoint (aortic-related mortality) speaks to this principle and the need to provide a relevant and easily understood, collected, and reported data point that does not require special training or interpretation. It is also relevant to the wide range of aortic pathologies that will be included in the registry. Finally, it is the only clinically relevant and long-term clinical parameter for patients with aortic pathologies. Secondary endpoints will of course capture many other details—all-cause mortality and secondary interventions, most especially—that are relevant to patients, physicians, regulators, and manufacturers alike.
Although wide-ranging and extensive, the registry nevertheless focuses on the product (as it is required to do by the regulatory authorities), not the pathology or the patient. As we witness the emergence of an aortic specialty, therefore, the current design is limited because it will not adequately capture the probable interaction of different endovascular devices and the subtle but significant differences between secondary interventions and staged procedures, for example.17,18 Furthermore, as the registry includes Terumo Aortic devices only, there will be no direct comparison possible with other commercially available endovascular devices; however, the data generated will be used to compare outcomes between devices as part of postmarket activities required to maintain medical device approval in Europe. Additional device-specific analyses of patient outcomes may support the expansion of indications for commercially available devices, thereby driving innovation and advancing the standard of care.
Similarly, the use of Core Laboratory assessment of imaging-based outcomes is often held up as a gold standard in quality. We have sought to have some independent oversight by having Core Laboratory review of imaging associated with events—but not uniform review of all imaging, which would be impractical for so many patients and sites.
We recognize the delay in publication of this registry due, in part, to the global shutdown as a result of the Coronavirus Disease 2019 (COVID-19) pandemic in March 2020 resulting in unprecedented challenges and significant disruption in clinical trial research management and reporting due to redirected resource in both clinical and nonclinical settings. However, this study remains relevant and addresses ongoing, current issues in the approval and postmarket surveillance of medical devices. The COVID-19 pandemic highlighted the need for more coordination and collaboration in clinical trial research, 19 and we believe these data have the potential to provide important information to other researchers on the design and conduct of real-world research, ultimately informing clinical practice and improving patient outcomes.
Conclusion
In summary, this protocol is a large endovascular registry of all aortic pathologies that are treated by both off-the-shelf and custom-made Terumo Aortic products. It is ambitious in scope and projection and will be part of an overall response involving patients, physicians, and manufacturers to answer the remaining questions of EVAR, contribute to continuing improvement of the techniques and technologies, and present an accurate picture of outcomes with latest generation stent-graft devices.
Footnotes
Acknowledgements
Jill Bryson, Terumo Aortic, Head of Clinical Affairs. Valerie Merkle, Ph.D. Terumo Aortic, Medical and Scientific Affairs Senior Director. Michelle Durnan, Terumo Aortic, Clinical Research Manager. Saminderjit Dhaliwal, Terumo Aortic, Clinical Study Manager. Katie Taggart, Terumo Aortic, Clinical Study Manager, Guillermo Rosero, Terumo Aortic, Senior Medical Affairs Associate.
Trial Status and Registration
Protocol version 5.0, 17 August 2023. Actual study start date: 17 December 2019. Estimated primary completion date: November 2029. Estimated study completion date: November 2030. Clinical trial name: A Global Post Market Evaluation of Terumo Aortic Endovascular Grafts (TiGER-001). ClinicalTrials.gov identifier: NCT04246463. URL: ![]() . Date of registration: 26 November 2019.
. Date of registration: 26 November 2019.
Author Contributions
V.R. is the Chief Investigator; they conceived the study, led the proposal and protocol development. S.D. contributed to study design and to development of the proposal. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: There is no competing interest, but there are principal investigators sponsored by Terumo Aortic.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Vascutek Ltd, Bolton Medical (trading as Terumo Aortic).
Ethical Considerations
Prior to starting the registry, each investigator will provide the sponsor with documented evidence that an appropriate EC or IRB (and any other relevant approval board) has approved the study protocol, patient information sheet (PIS), and informed consent form (ICF).
Consent for Publication
Authorship and manuscript composition will reflect cooperation between multiple investigators and sites, and the sponsor. A publication committee will be established chaired by the Global Coordinating Investigator. Authorship will be established by the publication committee prior to writing the manuscript. Primary publications will be published by the publication committee under a group name on behalf of the wider group, with the names of all participating investigators and institutes acknowledged. Secondary publications with individual authors should be agreed to by the publication committee. Authorship in these cases can be the full group, or a sub-set as agreed. The sponsor must receive any proposed publication and/or presentation materials at least 30 days prior to the proposed date of the presentation or the initial submission of the proposed publication for the materials to be reviewed by the sponsor. The sponsor may provide comments regarding any publication, particularly in relation to technical details of the device; however, the publication committee and the publishing investigators retain the right to make final editorial decisions for all publications.