
Abstract
Background:
Several studies have demonstrated the efficacy of paclitaxel drug-coated balloons (DCBs) for the treatment of femoropopliteal (FP) lesions in patients with peripheral artery disease (PAD). Despite the available evidence, long-term real-world efficacy data are limited.
Objectives:
To report the 3-year outcomes of the prospective, multicenter, single-arm real-world registry conducted in the United States, assessing the clinical use of the Lutonix 035 DCB in arteries of the superficial femoral artery (SFA) and popliteal artery (PA).
Methods:
In the SAFE-DCB US Registry, a total of 1005 subjects at 74 investigational sites were treated with the Lutonix 035 DCB and were followed up to 36 months. The per protocol included 966 patients (mean age 69.1 years; 56.7% male). A total of 1237 target lesions were treated, in which 93.3% were de novo, 85.2% were located in the SFA, and 88% had mild-to-severe calcification. Forty-five percent of the patients were Rutherford 3, and 35.1% had critical limb-threatening ischemia. The primary efficacy endpoint is target lesion revascularization (TLR) at 12 months. Secondary endpoints included rate of primary patency at 12 months and freedom from TLR and freedom from target vessel revascularization (TVR) evaluated through 36 months. The safety endpoints evaluated through 36 months were freedom from the composite of device- and/or procedure-related perioperative (≤30 day) death, TVR, and freedom from major amputation of the target limb.
Results:
Primary patency at 12 months post-index procedure by Kaplan-Meier estimates was 83.7%. Freedom from TLR at 12 months was 88.6%, 75.7% at 24 months, and 74.4% at 36 months. At 36 months, freedom from TVR post-index procedure by Kaplan-Meier estimates was 69.5%. Freedom from primary safety events at 30 days was 98.2%. Freedom from composite of all-cause perioperative (≤30 day) death and from the following: index limb amputation, index limb reintervention, and index-limb-related death by Kaplan-Meier estimates at 12 months was 80.8%, at 24 months 67.4% and 58.4% at 36 months. Freedom from major amputation of the target limb at 36 months was 95.6%.
Conclusion:
The results from the SAFE-DCB US Registry for femoral popliteal (FP) disease demonstrated sustained safety and efficacy over 3 years following the Lutonix 035 DCB intervention.
Clinical Impact
This prospective real-world population study supports long-term clinical outcomes of Lutonix and provides meaningful insights into the sustained efficacy of drug-coated balloon (DCB) therapy in real-world femoropopliteal population. The low-bailout stenting rate (1.7%), and the 36-month sustained Freedom from target lesion revascularization rate (74.4%) supporting DCB usage when a “leave nothing behind” approach is favored. Furthermore, these results reinforce the effectiveness of DCBs in the endovascular treatment peripheral artery disease.
Keywords
Background
Drug-coated balloons (DCBs) deliver anti-proliferative drugs to the arterial wall to reduce restenosis while preserving a “leave-nothing-behind” approach to endovascular treatment. Paclitaxel-based DCBs for femoropopliteal (FP) disease have demonstrated efficacy across several randomized controlled trials (RCTs), including Lutonix Levant 1 1 and Levant 2, 2 which established the Lutonix DCB as superior to standard percutaneous transluminal angioplasty (PTA). Similar findings were reported in other contemporary DCB trials such as the IN.PACT SFA Trial 3 and Ranger II SFA Randomized Trial. 4
Although RCTs remain the gold standard for establishing device efficacy, their strict inclusion criteria, often excluding patients with complex comorbidities and calcified lesions, limit generalizability to routine clinical practice. Real-world registries have provided broader insight into DCB performance across more diverse patient populations. For example, the real-world Global SFA Registry 5 demonstrated favorable outcomes in patients with long lesions and in-stent restenosis (ISR), and the LANDMARK Japanese registry showed comparable performance between low-dose (Lutonix) and high-dose (IN.PACT) DCBs in matched real-world FP lesions. 6 Smaller prospective and retrospective studies have further demonstrated long-term durability of the Lutonix DCB in specific subgroups, though limited by cohort size.7-9
Despite these contributions, there remains a lack of large-scale, prospective, multicenter (>2 years) real-world data evaluating the long-term effectiveness and safety of the Lutonix DCB. The Lutonix SAFE-DCB US Registry is the largest prospective multicenter study of the device to date, designed to capture outcomes in an unrestricted real-world population over 3 years of follow-up. While only the 6-month results have previously been reported, 10 the long-term findings have not yet been described.
The aim of the present analysis is to report the 3-year real-world effectiveness and safety outcomes from the Lutonix SAFE-DCB Registry, thereby addressing the evidence gap between controlled RCT data and long-term clinical performance in routine clinical practice.
Materials and Methods
Study Design
The study was designed as a prospective, multicenter real-world registry in the U.S. to assess the safety and efficacy of Lutonix 035 DCB Catheter in a real-world heterogeneous patient population. Subjects with stenotic or obstructive vascular lesions in the superficial femoral artery (SFA) and/or popliteal artery (PA) at 74 investigational sites in the United States were followed for up for 36 months post-index procedure. The inclusion criteria were intentionally broad to reflect the current clinical practice and included male or nonpregnant female subjects ≥21 years of age with an expected lifespan sufficient to allow for completion of all patient registry procedures. Subjects signed informed consent form and agreed to comply with the protocol-mandated follow-up procedures and visits. Exclusions criteria included had another medical condition or is currently participating in an investigational drug or an investigational device study that, which, in the opinion of the investigator, may cause him/her to be noncompliant with the protocol, confounds the data interpretation, or is associated with a life expectancy insufficient. The study was approved by the institutional review board at each site, before commencement.
Device Description and Procedure
The Lutonix 035 DCB Catheter is an over-the-wire (OTW) drug-coated PTA dilatation catheter with a semi-compliant balloon coated with a specialized immediate release nonpolymer based formulation that includes the anti-proliferative drug paclitaxel at a surface concentration of 2 μg/mm2 and polysorbate and sorbitol, which facilitate drug release and tissue deposition.
All procedural preparation, angiography treatment, and hospital discharge procedures were conducted per the investigational site’s standard of care. Treatment with Lutonix DCB was per the investigational sites’ standard of care and adhering to the instruction for use (IFU), which states that dual antiplatelet therapy should be administered according to current medical standards per-procedure and for a minimum of 4 weeks after the intervention.
The Lutonix Catheter is indicated for PTA, after pre-dilation. Several IFU changes occurred over the course of this registry including (1) the target time for the Lutonix Catheter to advance to the target site (shortest as possible ~30 seconds), (2) target inflation pressure to ensure full-wall apposition (balloon to artery ratio ≥1:1), and (3) target inflation time of the DCB (2 minutes), which occurred in September 2015. The requirement for pre-dilation changed to “vessel preparation of the target lesion, using the appropriate vessel preparation method as determined by the treating physician, is required prior to the use of the Lutonix 035 DCB Catheter,” was added to the IFU in May 2016, roughly 1 year after the start of this registry.
Endpoints and Definitions
Primary effectiveness was freedom from target lesion revascularization (TLR) at 12 months. TLR was defined as the first revascularization procedure (eg, PTA and stenting) of the target lesion after the index procedure. Primary safety was freedom from composite of device and/or procedure-related perioperative (≤30 days) death, target limb major amputation (above the ankle), and target vessel revascularization (TVR).
Secondary endpoints included TLR and TVR at 6, 12, 24, and 36 months. Target vessel revascularization was defined as the first revascularization procedure (eg, PTA, stenting, and surgical bypass) in the target vessel after the index procedure. Primary patency at 12 months, was defined as the absence of target lesion restenosis (as adjudicated by Independent Core Lab for duplex ultrasound [DUS] peak-systolic velocity ratio<2.5) and freedom from TLR. Acute device and procedural success, as defined by attainment of <30% residual stenosis of the target lesion after the index procedure using any percutaneous method and/or noninvestigational device and no periprocedural complications (death, stroke, myocardial infarction, emergent surgical revascularization, significant distal embolization in target limb, and thrombosis of target vessel) prior to hospital discharge. Freedom from major amputation of the target limb defined as above the ankle amputation, and composite of all-cause perioperative (≤30 days) death, and from the following: index limb amputation, index limb reintervention, and index-limb-related death at 30 days, 6, 12, 24, and 36 months post-index procedure.
Statistical Analysis
All analyses were based on the per-protocol population, which excludes subjects with major protocol deviation from all enrolled patients. Baseline characteristics, patient registry endpoint and outcomes are summarized using descriptive statistics. Summary statistics for categorical variables include frequency counts and percentages; for continuous variables, the mean, standard deviation, minimum, median, and maximum values are used.
The Kaplan-Meier method was used to evaluate time to event (days), with 95% confidence intervals (CIs), for freedom from TLR, TVR, and patency for the duration of the registry (ie, 36 months). The calculation of rates at each time point were based on the available data at the time point. The rates at each time point take into account information from registry subjects who discontinued from the registry prematurely and cause missing data. Patients were censored if they died, lost to follow-up, or withdrawn from the study.
Results
Patient Population
A total of 1005 patients enrolled across 74 sites between April 2015 and September 2016 with the final 3-year follow-up completed in October 2019. A total of 966 patients (1237 lesions) were included in the per-protocol population (excludes patients with major protocol deviations) for final analysis. At 36 months, 629 patients completed the final visit and 337 discontinued due to the following reasons: consent withdrawal 3.3%, lost to follow-up 8.4%, deaths 17.6%, investigator decision 4.6%, sponsor decision 0.03%, and other 0.7% (Figure 1).
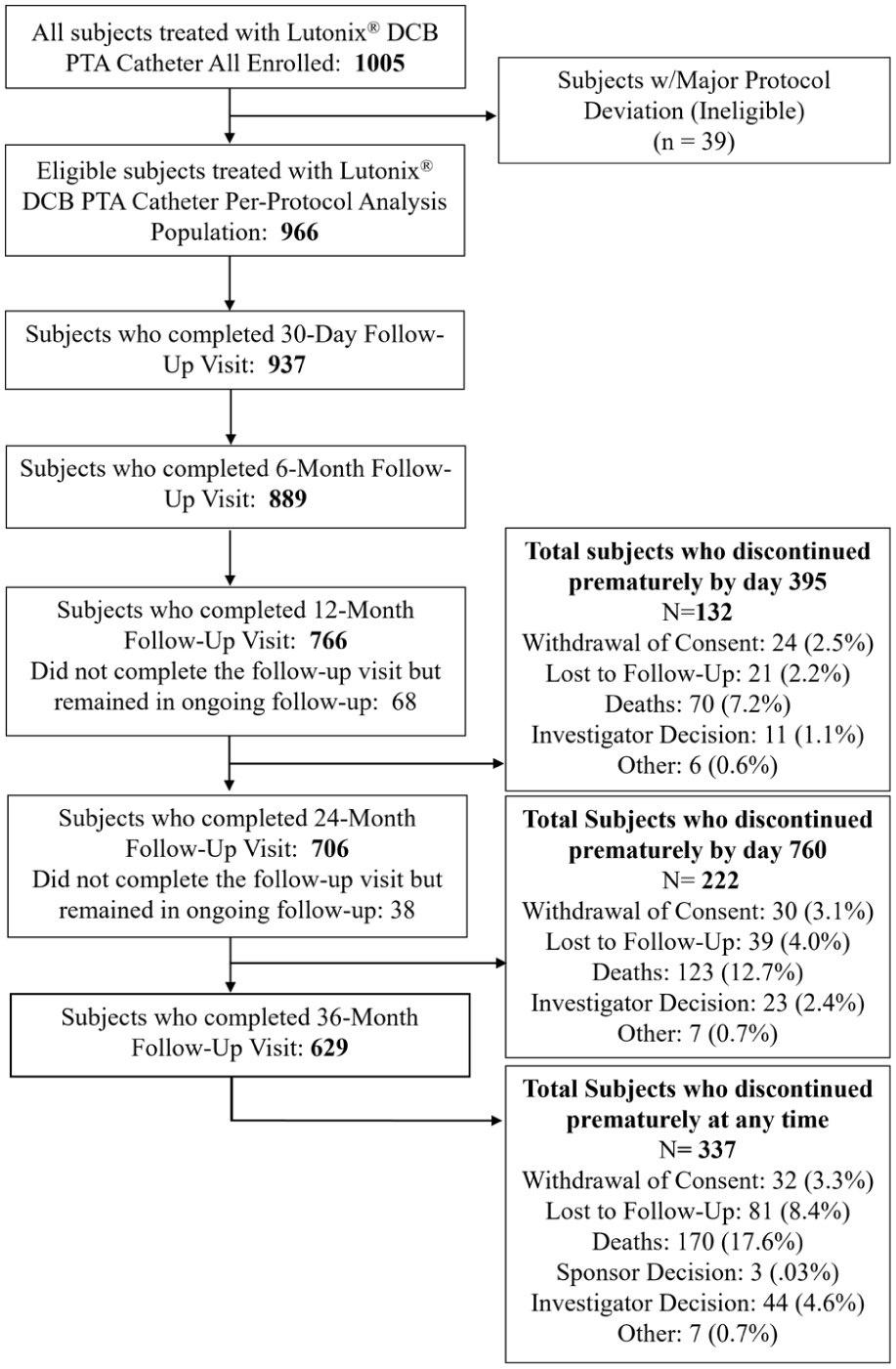
CONSORT flow diagram.
Baseline Characteristics
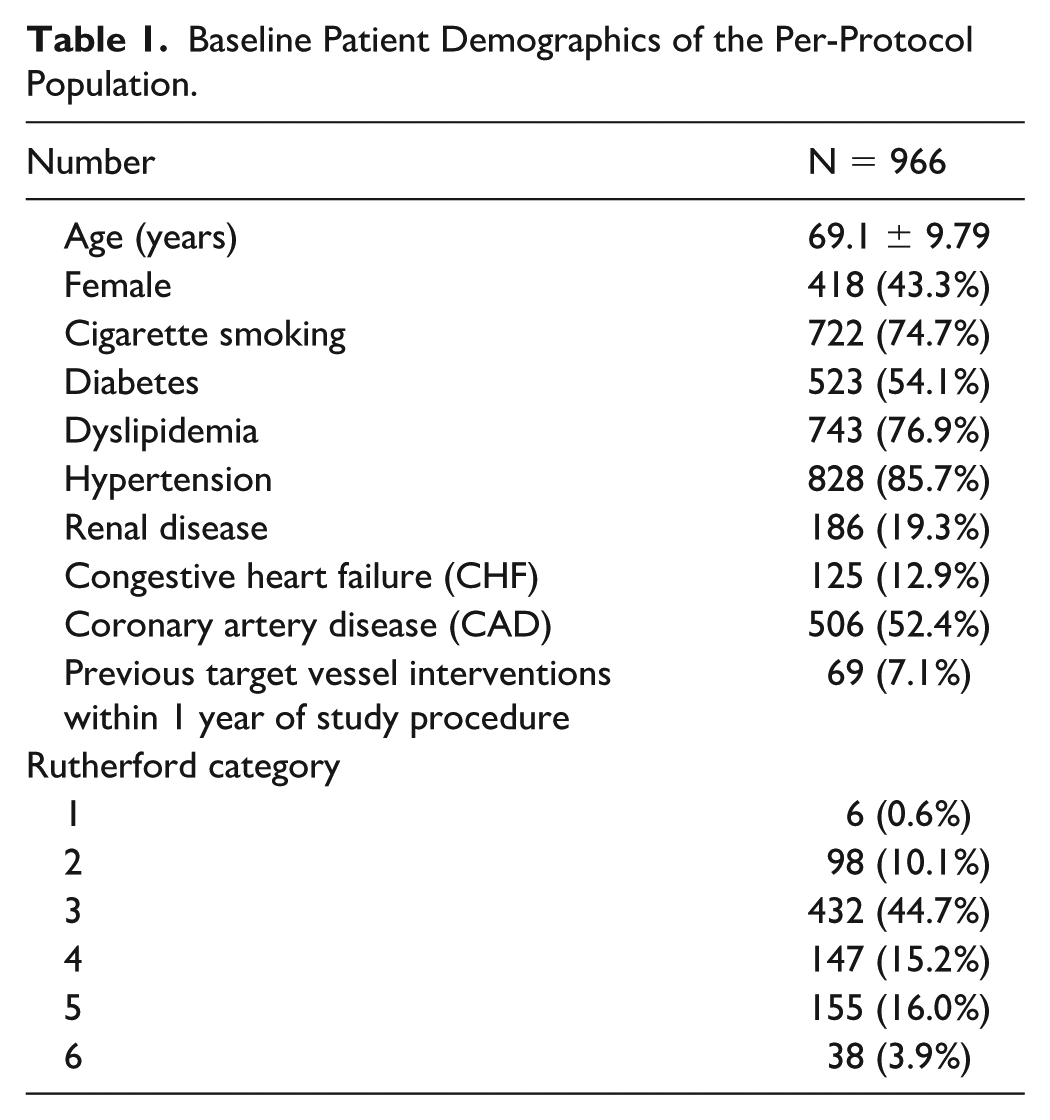
Baseline patient demographics and lesion characteristics are provided in Tables 1 and 2. The mean age of the study population was 69.1 years. The majority of patients were male (56.7%), smoked or previously smoked (74.7%), had diabetes (54.1%), and had at least one type of cardiovascular disease (71.2%). Rutherford category 3 was most prevalent (44.7%) and critical limb-threatening ischemia (CLTI) patients (Rutherford 4-6) represented 35.1% of the per-protocol patients. The majority of the target lesions were de novo (93.3%), located in the SFA (85.2%), with mild-to-severe calcification (88%). The mean target lesion length was 75.0 ± 43.93 mm, and the baseline stenosis was 86.7% ± 12.5% per visual estimate by investigators.
Baseline Patient Demographics of the Per-Protocol Population.
Baseline Lesion Characteristics of the Per-Protocol Population.

Of 1235 lesions.
Lesions of longer length were found in subjects that had chronic total occlusion (CTO) (98.1 mm) or required stenting (89.7 mm) during the index procedure, and lesions of shorter length were found in subjects that required only pre-dilation (69.8 mm) or cutting/scoring balloons (62.1 mm) as adjunctive treatment prior to DCB.
Target lesions were distributed equally between the left and right limbs of subjects. Access site was through the femoral artery in the majority (97.1%) of the subjects, and a CTO catheter or re-entry device was necessary for target lesion crossing in 12.2% of the subjects. The majority of the subjects (97.4%) presented with no thrombus prior to the index procedure. Nontarget lesions were treated in 40.1% of the subjects, with 50.4% of these nontarget lesions located below the knee.
Procedural Characteristics
pre-dilation was performed in 80.0% of all patients and post-dilation or adjunctive procedures were performed on the target lesion in 69.0% of the patients. Mean transit time, balloon inflation pressure, and total duration of inflation were 35.0 seconds, 8.0 atm, and 153 seconds, respectively. Successful Lutonix DCB placement was recorded for 99.6% of study devices. Acute device and procedural success were 93.0% (95% CI: 91.2%, 94.5%) (Table 3).
Overview of Procedural Characteristics (Per-Protocol Population).
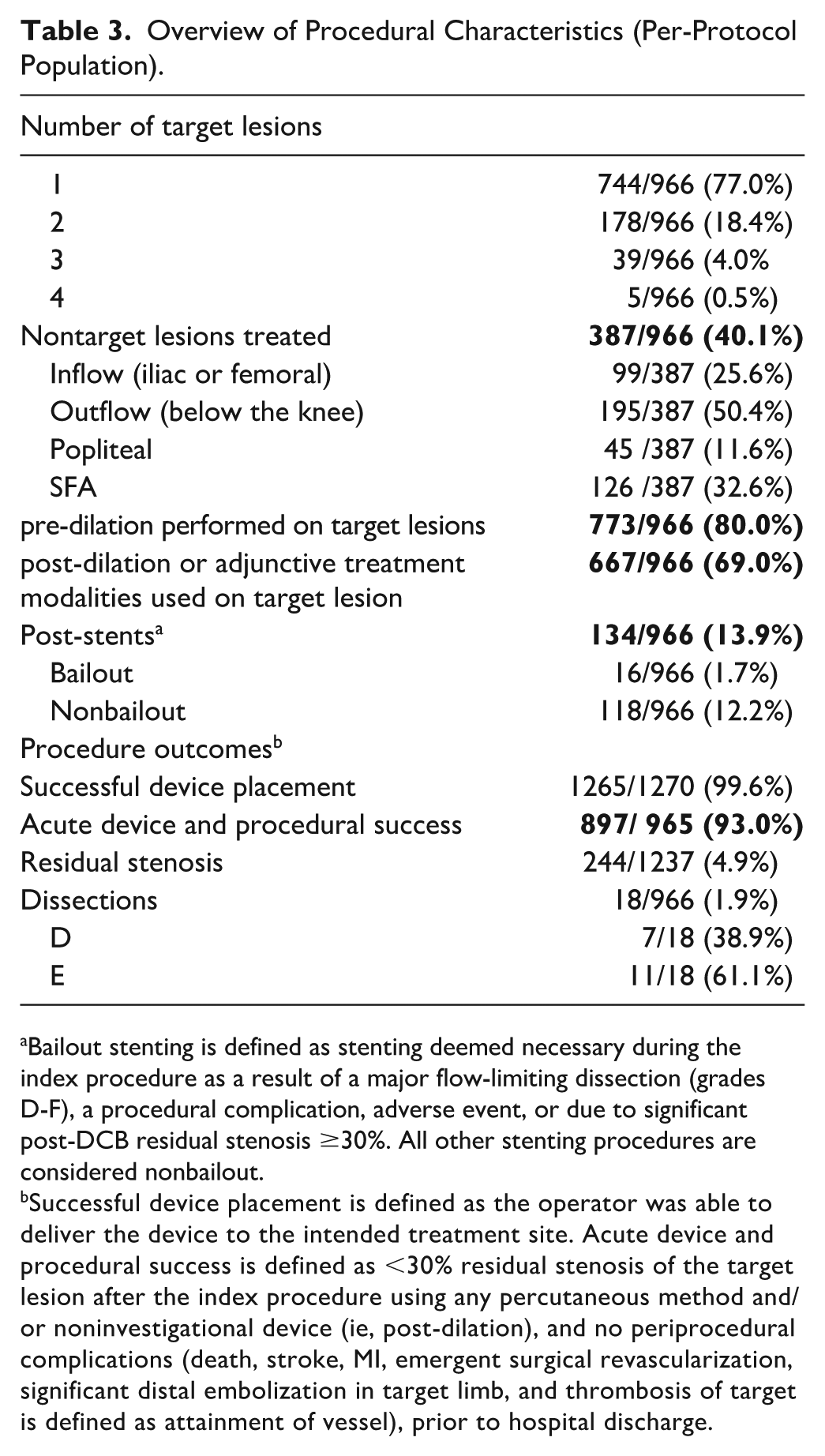
Bailout stenting is defined as stenting deemed necessary during the index procedure as a result of a major flow-limiting dissection (grades D-F), a procedural complication, adverse event, or due to significant post-DCB residual stenosis ≥30%. All other stenting procedures are considered nonbailout.
Successful device placement is defined as the operator was able to deliver the device to the intended treatment site. Acute device and procedural success is defined as <30% residual stenosis of the target lesion after the index procedure using any percutaneous method and/or noninvestigational device (ie, post-dilation), and no periprocedural complications (death, stroke, MI, emergent surgical revascularization, significant distal embolization in target limb, and thrombosis of target is defined as attainment of vessel), prior to hospital discharge.
Seventy-four subjects (7.7%) were reported to have device and/or procedure-related adverse events during the index procedure. Four subjects (0.4%) experienced a device failure, malfunction, or defect during the index procedure; however, no adverse events were associated with these occurrences.
Adjunctive procedures prior to DCB, were performed on 56.7% of the target lesions and the most prevalent procedure was atherectomy (49.6%), followed by cutting/scoring balloon (8.6%). Adjunctive procedures post-DCB were performed in 18.3% of subjects and only 15.3% of subjects had both adjunctive procedures. Mean residual stenosis following adjunctive treatment prior to DCB was 43.4%, which was reduced to 4.9% after the post-DCB adjunctive treatments.
Efficacy Outcomes
The primary efficacy endpoint, freedom from TLR at 12 months, by proportional analysis was 88.6% (95% exact CI: 86.3%, 90.6%). Reasons for reintervention were 10.6% clinically-driven (CD) and 5.4% the result of DUS findings. Kaplan-Meier estimate of primary patency (Core Lab) of target lesions at 12 months was 83.7% (81.1%, 86.0%), in which all subjects without a confirmed TLR or loss of patency were censored at day 395 or the date of study discontinuation, whichever occurred first.
Freedom from TLR of the per-protocol population by Kaplan-Meier estimates at 36 months was 74.4% (95% CI: 71.2%, 77.2%) (Figure 2 and Table 4). Kaplan-Meier estimate of freedom from TVR at 36 months was 69.5% (95% CI: 66.2%, 72.6%) (Table 4). At 36 months, there was a total of 422 reinterventions on the target limb, including repeat events. Of these reinterventions, 305 (72.3%) involved the target lesion. The majority of the reinterventions were CD (91.7%) and/or DUS-driven (36.0%).
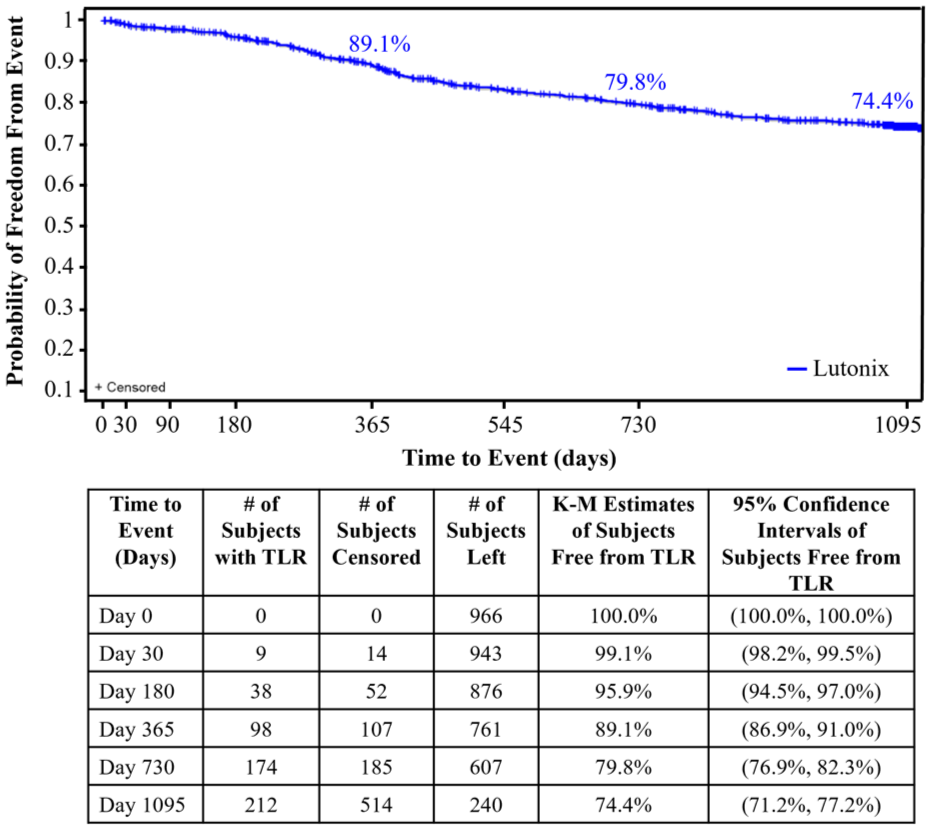
Freedom from TLR of the per-protocol population by Kaplan-Meier.
Secondary Endpoints Through to 36 Months.
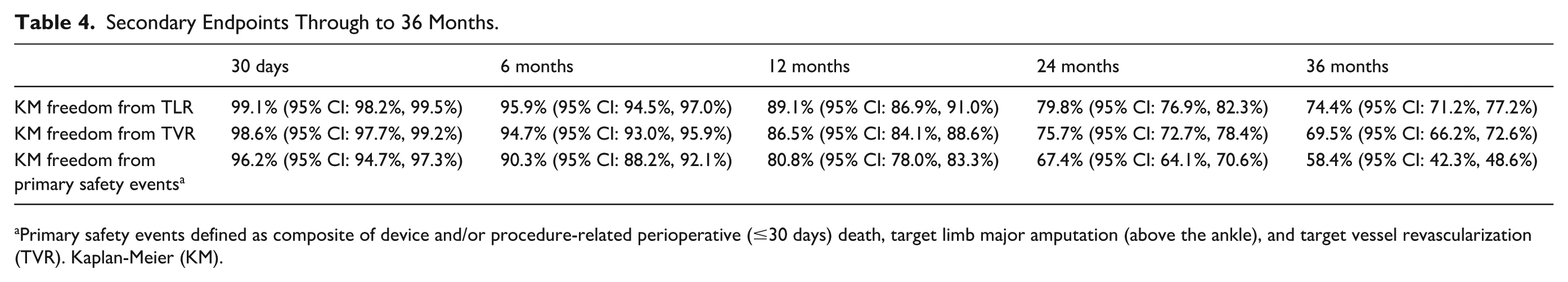
Primary safety events defined as composite of device and/or procedure-related perioperative (≤30 days) death, target limb major amputation (above the ankle), and target vessel revascularization (TVR). Kaplan-Meier (KM).
Safety Outcomes
Acute device and procedural success, defined as attainment of <30% residual stenosis of the target lesion after the index procedure using any percutaneous method and/or noninvestigational device, and no periprocedural complications prior to hospital discharge, was achieved at a rate of 93.0% (95% CI: 91.2%, 94.5%).
Freedom from primary safety events by Kaplan-Meier estimates (composite off all-cause perioperative death and from the following: index limb amputation, index limb reintervention, and index-limb-related death) through 30 days was 98.2% (95% CI: 97.2%, 98.9%) (Table 4). The all-cause mortality of the preprotocol population is shown in Table 5. All-cause death of the full cohort at 36 months was 17.2% (173/1005). The most common Clinical Events Committee (CEC)-adjudicated causes of death were cardiovascular and malignancy. Independent adjudication by the CEC determined that none of the deaths were related to the study device; 6 were possibly or potentially related to the study procedure.
Mortality, Amputation, and Major Amputation of the Preprotocol Population.
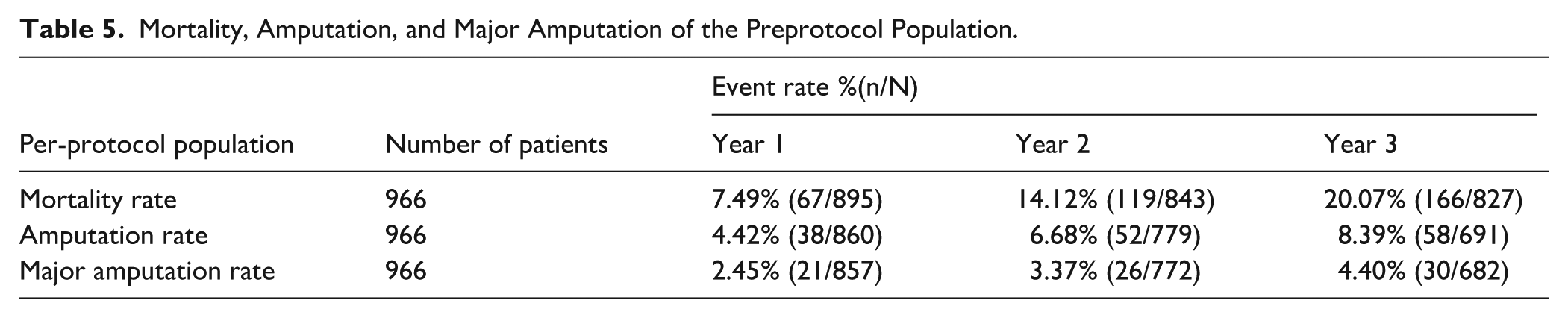
Major amputation rates at 12, 24, and 36 months are shown in Table 5. At 36 months, 682 subjects had evaluable data and 652 achieved freedom from major amputation for a rate of 95.6% (95% CI: 93.8%, 97.0%). All target limb amputations were deemed not related to the index device or procedure with the exception of one below-the-knee amputation.
Discussion
The data presented here examine the durability of safety and efficacy results of Lutonix DCB in the treatment of FP, in a complex real-world cohort, which includes a large female population (43.3%), a high prevalence of comorbidities including hypertension (85.7%) and dyslipidemia (76.9%), and a history of cardiovascular disease (71.2%). Compared with other DCB registries, the Lutonix SAFE Registry (n = 1005) had more calcified lesions (88%) than Lutonix SFA Global Registry (n = 691) (50.2%), 5 IN.PACT Global Registry (n = 1535) (68.7%), 3 and RANGER All Comers SFA Registry (n = 172) (26%), 12 and more patients with CLTI, 35.1%, 9%, 11.1%, and 12%, respectively. However, in the Lutonix SAFE Registry, the mean lesion length was shorter than in the other registries, 75, 101, 120, and 129 mm, respectively (Table 6).3,5,12
DCB Registries.
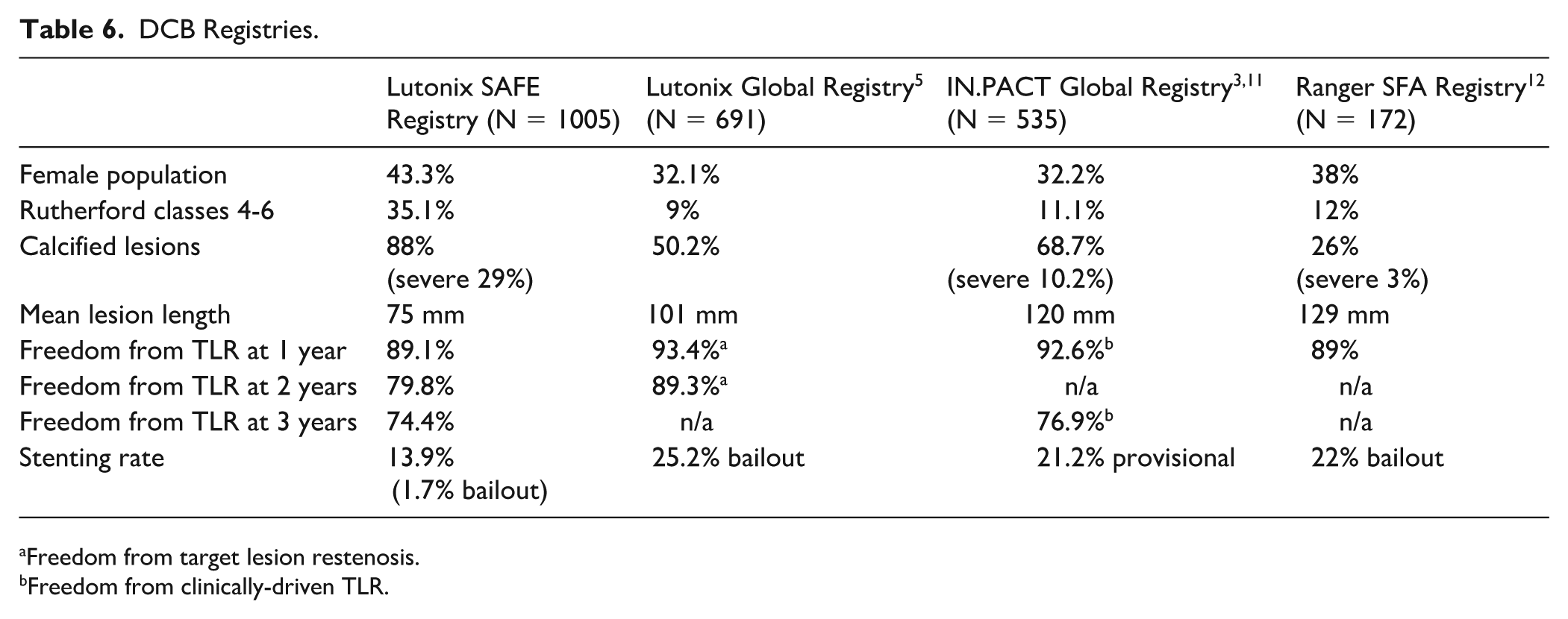
Freedom from target lesion restenosis.
Freedom from clinically-driven TLR.
The results presented here demonstrate comparable clinical outcomes to these other registries. At 1 year, freedom from TLR was 89.1% in the Lutonix SAFE Registry, freedom from target lesions restenosis was 93.4% in the Lutonix Global SFA Registry, 5 freedom from CD-TLR was 92.6% in the IN.PACT Global Registry 3 and freedom from TLR was 89% in the Ranger All Comers SFA Registry. 12 The Lutonix SAFE Registry confirmed prior data showing 1-year effectiveness of the Lutonix DCB in treating FP lesions.
At 24 months, freedom from TLR was 79.8% versus 89.3% when compared with Lutonix Global SFA Registry; 5 freedom from target lesion restenosis was 89.3% for the overall population, 88.2% for long lesions up to 500 mm, and 84.6% for ISR. Clinically-assessed primary patency was 75.6% at 24 months.
Here, at 3 years, the Lutonix freedom from TLR was 74.4%. Although not directly comparable, in the IN.PACT Global Registry, 11 freedom from CD-TLR through 36 months was 76.9%. In the IN.PACT, there was a higher rate of baseline total occlusion (35.5% vs 27%), but lower severe calcification (10.2% vs 29.0%), and lower CLTI (11.1% vs 35%). In contrast, the IN.PACT SFA Japan Trial 13 showed a freedom from CD-TLR of 84.4% at 36 months. In this cohort, however, only 7.4% of lesions were severely calcified, and most patients were claudicants versus CLTI.
The overall stenting rate in Lutonix SAFE Registry was 13.9%. Of these, only 16/134 (1.7%) were considered bailout. In this registry, patients with dissections prior to DCB were not excluded as in the LEVANT II study. 2 Also, despite the high prevalence of calcified disease the need for bailout stent was surprisingly low. This may have been accounted for by the relatively shorter lesions included in this registry and the change in the IFU that encouraged prolonged balloon inflation of at least 2 minutes. Also, this registry had a high rate of vessel preparation using atherectomy, which could account for the lower rate of bailout stenting. A higher rate of bailout stenting 25.2%, was seen in the Lutonix Global SFA Registry, 5 which had more long lesions (>140 mm) (35.7%) and ISR (33.7%). The provisional stenting rate was also higher in the IN.PACT Global Registry (21.2%) 11 and the bailout stenting rate in the Ranger All Comers SFA Registry (22%). 12
The safety of paclitaxel DCB and the Lutonix balloon is now well established, with the original concerns of DCB increased mortality 14 have largely subsided. Several independent studies concluded comparable long-term mortality rates of drug-eluting devices compared with non-drug-eluting devices.15-20 Specific to Lutonix, a meta-analysis of 1093 patients treated with Lutonix versus 250 patients treated with balloon angioplasty (PTA) in LEVANT 1 (The Lutonix Paclitaxel-Coated Balloon for the Prevention of Femoropopliteal Restenosis), LEVANT 2, and the LEVANT Japan Clinical Trial, reported a 5-year hazard ratio (HR) for mortality of 1.01 (95% CI: 0.68, 1.52) with no mortality differences between DCB angioplasty and PTA. 17
The mortality rate reported here (14.1% at 2 years and 20.07% at 3 years) is slightly higher than Lutonix SFA Global Registry, 5 5.9% at 2 years, and IN.PACT Global Registry 11 11.6% at 3 years. No equivalent data are available for the Ranger. This is most likely reflective of the larger number of CLTI patients included in the Lutonix SAFE Registry versus IN.PACT Global Registry. 11 The major amputation rate reported here (3.37% at 2 years and 4.7% at 3 years) was higher than those reported in other large registries, such as the Lutonix Global SFA Registry (1% at 2 years) 5 and the IN.PACT Global Registry (1% at 3 years). 11
Collectively, these prospective registry data of Lutonix SAFE DCB support long-term durability and efficacy with a low-bailout stenting rate, supporting DCB usage when a “leave nothing behind” approach is favored. A recent multicenter registry-based randomized trial showed the TVR rate was significantly lower in the drug-eluting devices (which included both stents and DCBs) compared with non-drug-eluting devices, after 1 year in CLI patients. However, this benefit was not sustained with longer follow-up. 19 Interpretation of this SWEDEPAD data and extrapolation to DCBs efficacy is challenging, as stents and balloons were grouped together and only TVR HRs are reported, in which there is no way of knowing if the reintervention occurred on the same lesion or elsewhere in the vessel. Furthermore, no data are reported on lesion length, CTO, calcification, and/or residual stenosis after initial treatment and bailout stenting rate, making comparisons challenging.
Limitations of the Study
The registry is limited by its single-arm design with no comparative control. Due to IFU changes throughout the duration of the study, efficacy results may have been lower, as not all procedures were performed based on “best practice,” considering appropriate balloon sizing to allow for full-wall apposition, pressure >7 atm, inflation time >120 seconds.
This registry did not capture follow-up Rutherford class or ankle-brachial index, therefore limiting the ability to quantify symptom relief or quality of life improvement.
Footnotes
Ethical Considerations
The trial was registered on the National Institutes of Health website (ClinicalTirals.gov; NCT02424383). The study was approved by the institutional review board (IRB) at each site, before commencement.
Consent to Participate
Informed consent was obtained from all subjects prior to enrolement in the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors disclosed the registry was supported by BD. BD provided financial support for the research, analysis and publication.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Nicolas Shammas receives research grants from BD, Philips, Shockwave, Abbott, and Angiodynamics; Consultant Angiodynamics; Speaker for Angiodynamics, Boehringer Ingelheim, Amgen, and Bayer. Dion Franga declared no conflicts of interest. Erin Moore is an investigator for BD (Covera Study), Speaker for WL Gore, Terumo, and Medtronic. The author(s) Edward Yoo, Dion Franga, and George L. Mueller declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. George Mueller declared no conflicts of interest. Thomas P. Davis, BD speaking and advisory, Stryker advisory and training programs, stock owner and advisor of Verge Medical, stock owner of Cardioflow, Abbott, Advisory board. Phillips speaker and Advisory Board. Edward Woo declared no conflicts of interest.
Data Availability Statement
The data sets generated during the SAFE study are available in the ClinicalTrial.Gov repository: https://clinicaltrials.gov/study/NCT02424383?term=Lutonix%20SAFE%20Registry&rank=1&tab=results.