
Abstract
Lung cancer is a malignancy characterized by high global incidence and mortality rates, with approximately 85% of cases classified as non-small cell lung cancer (NSCLC). Advances in molecular biology and targeted therapeutic agents have ushered NSCLC into the era of precision medicine. The echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) fusion gene represents a pivotal target for personalized treatment in NSCLC. Although ALK inhibitors exhibit significant efficacy against EML4-ALK-positive tumors, addressing drug resistance remains a major challenge. Identifying novel therapeutic targets and implementing combination therapies are essential for optimizing subsequent treatment strategies and improving overall prognosis. In the management of patients with EML4-ALK gene fusion, continuous monitoring of tumor progression and timely adjustment of treatment regimens based on disease status are critical to achieving long-term clinical benefits. This review comprehensively summarizes recent advances in the clinicopathological features, targeted drug development, resistance mechanisms, and potential therapeutic targets associated with the EML4-ALK fusion gene.
Keywords
1. Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide and represents the most commonly diagnosed cancer in China. 1 It is broadly classified into small cell lung cancer and non-small cell lung cancer (NSCLC), with NSCLC accounting for approximately 85% of all cases and constituting the predominant histological subtype. 2 Key driver oncogenes in NSCLC include the epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), and Rat sarcoma (RAS) genes. 3 The ALK gene can form fusion proteins with various partners, driving aberrant tumor cell proliferation and playing a critical role in NSCLC pathogenesis. Patients with ALK fusion-positive lung cancer generally demonstrate superior survival outcomes and a wider array of therapeutic options compared to those with other driver mutations, leading to the colloquial designation of ALK fusions as the “diamond mutation”. To date, more than 90 distinct ALK fusion partners have been identified in lung cancer, with the echinoderm microtubule-associated protein-like 4 (EML4) gene being the most prevalent, accounting for over 85% of ALK-positive NSCLC cases. 4 The resultant EML4-ALK fusion protein exhibits potent oncogenic activity, promoting cell proliferation, inhibiting apoptosis, and enhancing metastatic potential, thereby playing a pivotal role in oncogenic signaling pathways.
Common clinical methodologies for detecting EML4-ALK fusions encompass reverse transcription-polymerase chain reaction (RT-PCR), immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), and next-generation sequencing (NGS). Protein kinases constitute a major class of therapeutic targets in lung cancer. The development and clinical approval of tyrosine kinase inhibitors (TKIs) have established kinase-targeted therapy as a prominent and dynamic area of research. Several ALK-TKIs, including crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib, have been developed for treating advanced NSCLC patients harboring ALK fusions, heralding a new era in lung cancer targeted therapy. Current management strategies emphasize timely molecular profiling upon diagnosis to guide initial targeted therapy, supplemented by ongoing dynamic monitoring to inform treatment adjustments in response to disease evolution, thereby aligning with the principles of precision medicine. However, clinical responses to these targeted agents vary, and not all patients achieve desired outcomes, due in part to intrinsic or acquired drug resistance.
This review provides a comprehensive and updated overview of the clinicopathological features, targeted drug development, and resistance mechanisms associated with the EML4-ALK fusion gene. Distinct from prior reviews, this article: (1) systematically integrates the heterogeneity of EML4-ALK variants with clinical outcomes and variant-specific resistance patterns; (2) proposes a variant-stratified framework for personalized treatment strategies; and (3) incorporates the latest advances in liquid biopsy and fourth-generation ALK-TKIs, offering both conceptual novelty and practical clinical insights.
2. Literature Search Strategy
We conducted a systematic literature search in PubMed, Web of Science, and CNKI (China National Knowledge Infrastructure) databases for articles published between January 2007 (when EML4-ALK was first identified) and December 2024. The search terms included combinations of the following keywords: “EML4-ALK”, “anaplastic lymphoma kinase”, “non-small cell lung cancer”, “ALK inhibitor”, “crizotinib”, “alectinib”, “brigatinib”, “lorlatinib”, “drug resistance”, “resistance mechanism”, “variant”, “liquid biopsy”, “combination therapy”, and “targeted therapy”. We also manually screened the reference lists of retrieved articles and relevant reviews to identify additional eligible studies.
Inclusion criteria were: (1) original research articles, clinical trials, observational studies, or systematic reviews/meta-analyses focusing on EML4-ALK-positive NSCLC; (2) studies reporting clinicopathological characteristics, ALK-TKI efficacy, resistance mechanisms, novel therapeutic strategies, or detection methodologies; (3) articles published in English or Chinese. Exclusion criteria were: (1) case reports with fewer than three patients (unless reporting a novel resistance mutation or exceptional response); (2) conference abstracts without full-text availability; (3) non-peer-reviewed articles or preprints; (4) studies focusing solely on other ALK fusion partners (non-EML4) or other malignancies without relevance to NSCLC.
A total of 168 articles were initially identified, of which 89 were included after title/abstract screening and full-text assessment based on relevance and quality. Priority was given to phase III randomized controlled trials, large real-world cohort studies, and high-impact mechanistic studies published in peer-reviewed journals.
3. EML4-ALK Fusion Gene
The genomic landscape of lung cancer harbors numerous fusion events, with the EML4–ALK fusion being especially prominent. First identified in anaplastic large cell lymphoma (ALCL) in 1994, the ALK gene resides on the short arm of chromosome 2 and encodes a receptor tyrosine kinase (RTK). 5 Predominantly expressed in neuronal tissues, ALK activates multiple signaling cascades—including the mitogen-activated protein kinase (MAPK), Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathways—that collectively drive the pathogenesis of cancers such as non-small cell lung cancer (NSCLC), ALCL, inflammatory myofibroblastic tumor, and neuroblastoma. ALK fusions occur in over 3% of NSCLC patients but in only approximately 0.2% of non-NSCLC cases. EML proteins bind microtubules and regulate key processes such as cell division and signaling. EML4, a member of this family, promotes chromosome segregation during mitosis and contains four domains: the tandem atypical propeller in EML proteins (TAPE), the hydrophobic motif in EML proteins (HELP), the trimerization domain (TD), and an unstructured basic region. 6 The TD domain is essential for ALK autophosphorylation and activation.
The EML4–ALK fusion protein was first identified in 2007 in a surgical specimen from an elderly male patient with lung adenocarcinoma.
7
This fusion drives ligand-independent dimerization and constitutive activation of ALK, thereby stimulating multiple downstream signaling cascades, including the Ras/ERK, JAK/STAT, and PI3K/AKT pathways (Figure 1). As illustrated in the figure, Ras/ERK signaling primarily governs cell proliferation, whereas the JAK/STAT and PI3K/AKT pathways modulate cell survival and apoptosis. The coordinated activation of these pathways promotes aberrant proliferation, differentiation, and resistance to apoptosis, ultimately contributing to NSCLC pathogenesis. Whereas Ras/ERK signaling primarily governs proliferation, mTOR and STAT3 pathways modulate cell survival and apoptosis.
8
Distinct EML4–ALK fusion variants arise from variable genomic breakpoints. The ALK breakpoint most frequently lies within exon 20, occasionally within exon 19, whereas EML4 breakpoints exhibit greater variability.
9
This molecular heterogeneity yields a diverse array of fusion proteins; more than 17 variants (e.g., V1, V2, V3, V4, V5a) have been documented, with V1 (∼37%) and V3 (∼42%) predominating.
10
These structural differences are thought to account for variations in protein stability, inhibitor sensitivity, and the emergence of ALK resistance mutations. Depending on whether the EML4 TAPE domain is retained, variants are classified as long (V1, V2, V4) or short (V3, V5) (Figure 2). As shown in the figure, long variants contain the TAPE domain, which is associated with greater protein stability and more favorable clinical outcomes, whereas short variants lack this domain and exhibit higher intrinsic resistance to ALK-TKIs. Signaling pathways associated with the EML4-ALK fusion gene. This schematic illustrates the major downstream signaling pathways activated by the EML4-ALK fusion protein, including Ras/ERK (proliferation), JAK/STAT (survival), and PI3K/AKT (apoptosis resistance). These pathways collectively drive NSCLC pathogenesis Five common variants of EML4-ALK fusion gene. Long variants (V1, V2, V4) retain the TAPE domain and are associated with better prognosis, whereas short variants (V3, V5) lack this domain and exhibit higher intrinsic resistance. Abbreviations: MAM: adhesion-related domain; G-rich: glycine-rich region; TM: transmembrane domain; TK: tyrosine kinase domain; TD: trimerization domain; EML4-ALK V1: variant 1 (E13; A20); V2: variant 2 (E20; A20); V3: variant 3 (E6; A20); V4: variant 4 (E15del60; del71A20); V5a: variant 5a (E2; A20)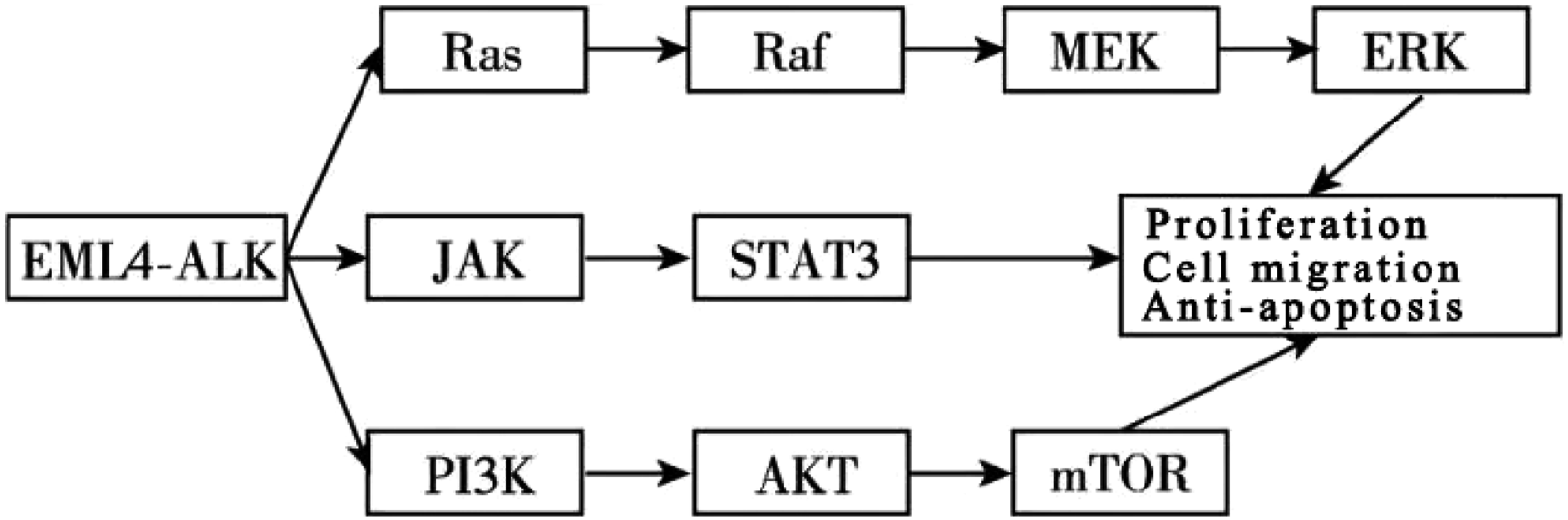

Owing to disparities in protein stability, long and short variants display distinct sensitivities to ALK inhibitors. Patients with long variants generally have better clinical outcomes and longer median progression-free survival (PFS) than those with short variants. 11
The distinct biological properties of EML4-ALK variants translate into clinically meaningful prognostic differences. Patients harboring long variants (V1, V2, V4) generally exhibit more favorable outcomes, with median progression-free survival (PFS) approximately 30%–50% longer than those with short variants (V3, V5) when treated with first- or second-generation ALK-TKIs. 11 Notably, the V3 variant is associated with a higher risk of early disease progression and shorter overall survival, particularly in patients receiving crizotinib or alectinib as first-line therapy. These findings support the integration of variant typing into baseline risk assessment, enabling the identification of high-risk patients who may benefit from more intensive upfront strategies, such as third-generation ALK-TKIs or combination regimens.
4. Clinicopathological Characteristics of EML4-ALK in NSCLC
EML4-ALK-positive NSCLC exhibits distinct demographic and clinical features. A nationwide real-world study (23,689 advanced NSCLC patients) reported an ALK positivity rate of 6.7%, with significantly higher frequencies in females, never-smokers or former light smokers, and patients with adenocarcinoma.12,13 Although ALK fusions can occur in other histological subtypes, they are most prevalent in lung adenocarcinoma, particularly invasive, mucin-rich, or pure solid nodule subtypes.14-16 Notably, ALK fusion prevalence is stage-dependent, reaching 19% in stage IV NSCLC compared with only 2%–7% in early-stage disease. 17 Recognition of these clinicopathological characteristics facilitates timely molecular testing, guides targeted therapy, and aids in predicting disease progression.
5. Advances in EML4-ALK Targeted Therapies
ALK-TKIs for the Treatment of NSCLC
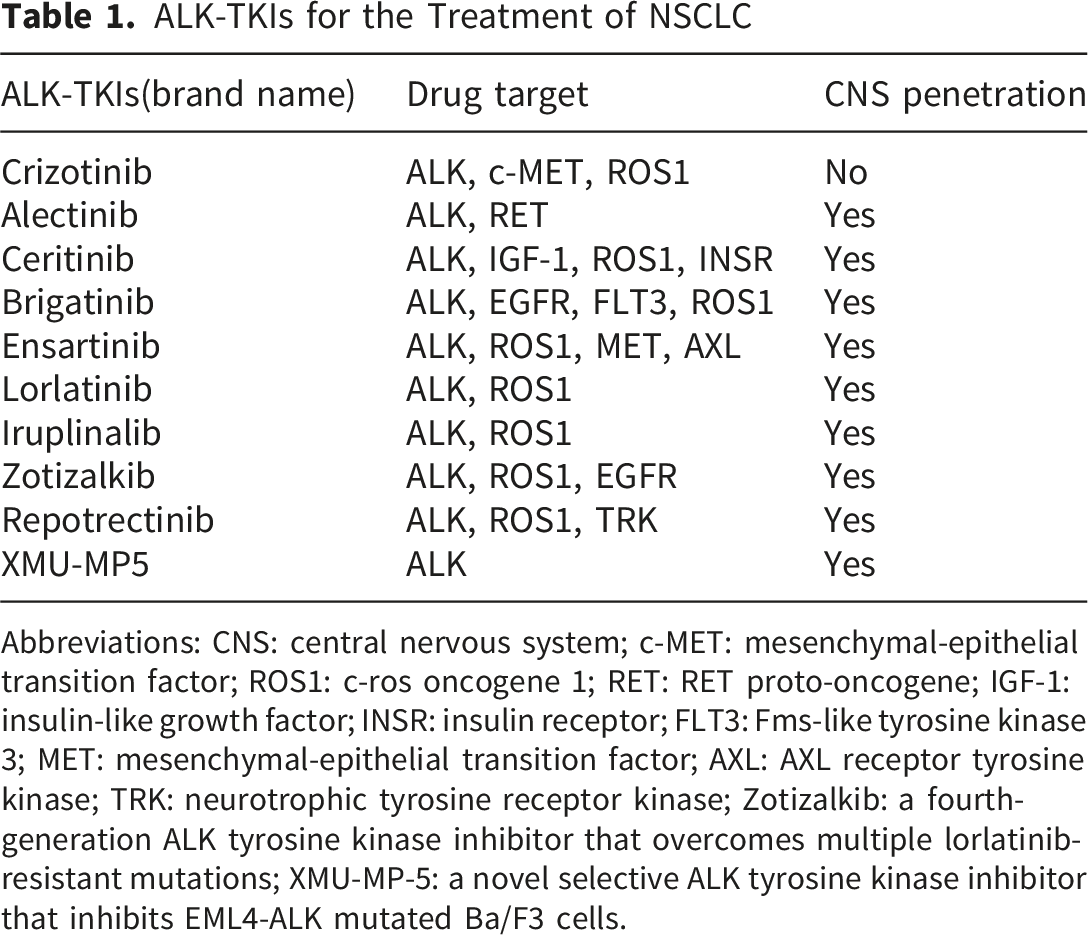
Abbreviations: CNS: central nervous system; c-MET: mesenchymal-epithelial transition factor; ROS1: c-ros oncogene 1; RET: RET proto-oncogene; IGF-1: insulin-like growth factor; INSR: insulin receptor; FLT3: Fms-like tyrosine kinase 3; MET: mesenchymal-epithelial transition factor; AXL: AXL receptor tyrosine kinase; TRK: neurotrophic tyrosine receptor kinase; Zotizalkib: a fourth-generation ALK tyrosine kinase inhibitor that overcomes multiple lorlatinib-resistant mutations; XMU-MP-5: a novel selective ALK tyrosine kinase inhibitor that inhibits EML4-ALK mutated Ba/F3 cells.
First-generation crizotinib provides a median PFS of ∼10 months but has limited blood-brain barrier penetration. Brain metastases occur in ∼30% of ALK-positive NSCLC patients at diagnosis and up to 58% within three years. 20 Second-generation TKIs (alectinib, ceritinib, brigatinib) offer enhanced CNS penetration and are preferred over crizotinib as initial therapy. Third-generation lorlatinib demonstrates superior potency and excellent CNS activity; a phase III trial (CROWN) showed prolonged 3-year PFS and reduced mortality risk versus crizotinib. 21 For brain metastases, alectinib, lorlatinib, and brigatinib are recommended, with crizotinib as a secondary option.
Additional novel ALK inhibitors are in development (Table 1). Iruplinalkib has been approved in China. 22 Fourth-generation inhibitors (zotizalkib, repotrectinib) overcome multiple lorlatinib-resistant mutations. 23 XMU-MP-5 is a preclinical selective ALK-TKI active against resistant mutations. 24
Clinical Profiles and Spectrum of Resistance Mutations for ALK-TKIs
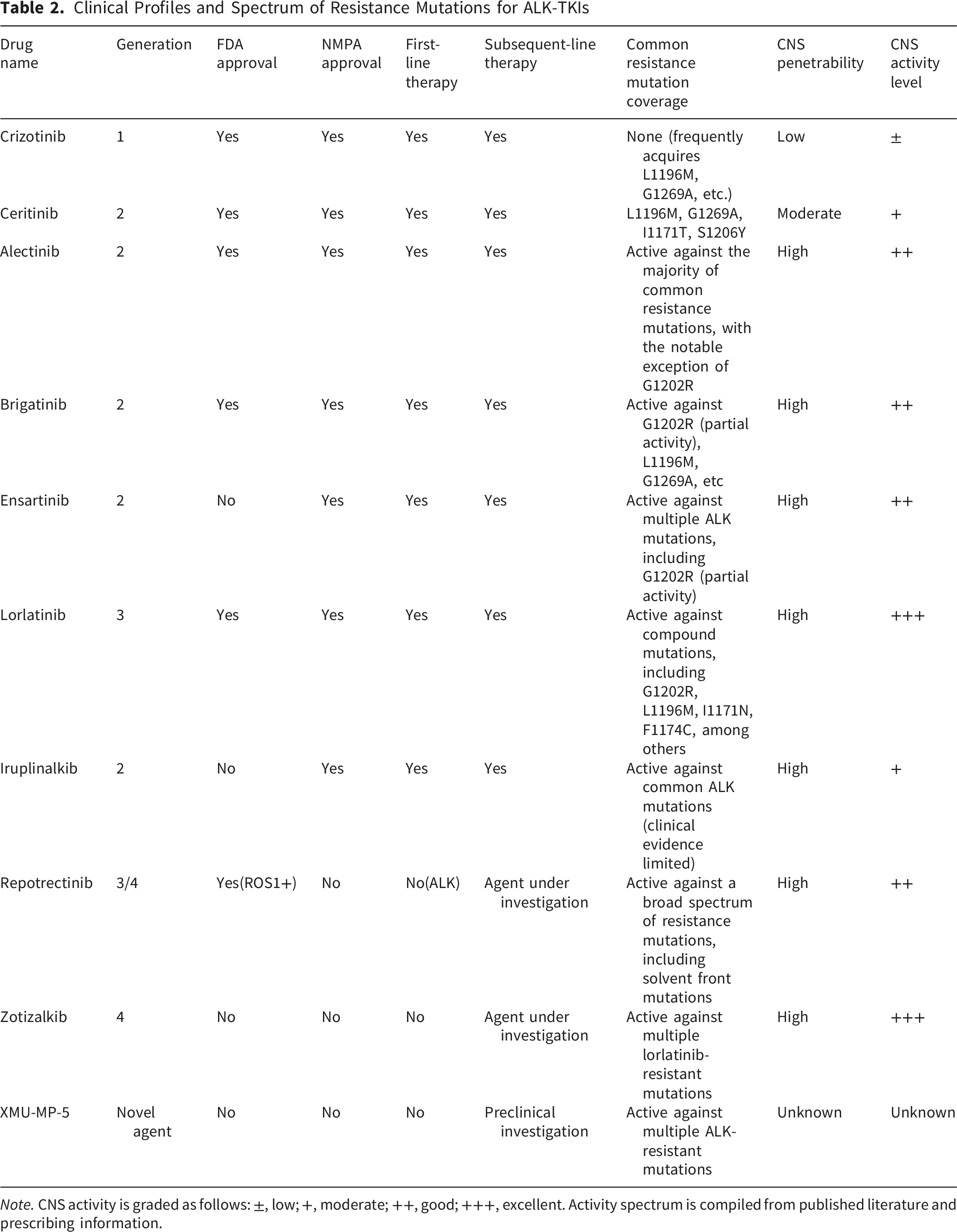
Note. CNS activity is graded as follows: ±, low; +, moderate; ++, good; +++, excellent. Activity spectrum is compiled from published literature and prescribing information.
Validation Status and Translational Limitations of Emerging Resistance Mechanisms in EML4-ALK-Positive NSCLC

6. EML4-ALK Drug Resistance
6.1. Clarification of Intrinsic vs. Acquired Resistance Definitions
Intrinsic (Primary) Resistance refers to the absence of initial response to ALK-TKI therapy or disease progression within the first 3–6 months of treatment despite adequate drug exposure. 28 This phenotype is driven by pre-existing molecular alterations that compromise ALK-TKI efficacy from treatment initiation. Acquired (Secondary) Resistance develops in patients who initially respond to ALK-TKIs but subsequently progress after a period of disease control (typically >6 months), mediated by therapy-induced clonal selection or de novo mutations. 29
6.2. Mechanisms of Intrinsic Resistance and Early Progression
6.2.1. Heterogeneity of EML4-ALK Fusion Variants (the Core Intrinsic Factor)
The short variants (V3, V5) of EML4-ALK have inherent structural and functional characteristics that lead to low ALK-TKI sensitivity: the lack of the TAPE domain leads to higher stability of the fusion protein and stronger constitutive activation of downstream Ras/ERK and mTOR pathways; meanwhile, the V3 variant can form a complex with NEK9/NEK7 to activate kinesin Eg5, leading to congenital enhancement of tumor cell migration and proliferation.26,30 These inherent properties make patients with V3 variant positive NSCLC prone to disease progression within 3–6 months after first/second-generation ALK-TKI treatment, which is the most important intrinsic resistance mechanism of EML4-ALK-positive NSCLC. Recent studies9,31 demonstrate that V3-positive tumors exhibit significantly shorter median progression-free survival (PFS) with crizotinib or alectinib (6.0–8.5 months vs. 12–15 months for V1/V2 variants). This variant-specific biology represents a form of intrinsic resistance that manifests as early clinical progression.
6.2.2. Congenital Activation of Bypass Signaling Pathways (No Drug-Induced Adaptive Change)
The congenital amplification/activation of oncogenic pathways independent of ALK is a key cause of intrinsic resistance, among which MET amplification is the most common and well-characterized one. 25 Tumor cells with congenital MET amplification can activate the PI3K/AKT pathway through HGF-MET signaling independent of ALK, so that the downstream oncogenic signaling remains active even after ALK is inhibited by TKIs, resulting in early treatment failure. 32 In addition, the congenital overexpression of EGFR/HER3 and the hyperactivation of SRC kinase also belong to the intrinsic bypass activation that leads to early progression.
6.2.3. Intrinsic Abnormalities of the Tumor Microenvironment
Cancer-associated fibroblasts (CAFs) in the tumor microenvironment of EML4-ALK-positive NSCLC have inherent paracrine regulation effects: CAFs can secrete HGF to activate MET signaling of tumor cells, and simultaneously induce the congenital upregulation of integrin β1 in lung cancer cells, forming a CAF-tumor cell crosstalk loop that is independent of ALK signaling. 27 This inherent microenvironmental abnormality reduces the sensitivity of tumor cells to ALK-TKIs from the beginning of treatment, leading to early progression.
6.2.4. Intratumoral Heterogeneity in the Diagnostic Phase
The inherent spatial and clonal heterogeneity of tumors may lead to the failure of routine detection (FISH/RT-PCR) to capture the minor subclones with ALK-TKI resistance (e.g., subclones with MET amplification or V3 variant). 33 These resistant subclones are the dominant clones at the initial stage of treatment, resulting in rapid early progression, which is a diagnostic-related intrinsic resistance factor. 9
6.3. Acquired Resistance of EML4-ALK
Genomic instability is a fundamental hallmark of cancer. It drives tumor mutagenesis, enabling evasion of therapeutic interventions and immune-mediated destruction, ultimately leading to acquired drug resistance. Resistance mechanisms pose a significant challenge in targeted therapies and constitute an inevitable limitation of TKI treatment. Approximately one-third of patients receiving ALK-TKIs develop resistance through secondary mutations, resulting in disease progression. In a representative case, a 56-year-old female with EML4-ALK-positive adenocarcinoma experienced disease progression after two months of first-line alectinib therapy. Next-generation sequencing of pleural effusion identified two novel mutations alongside the original alteration, including E803Q in exon 14—a new secondary mutation conferring resistance to most second-generation ALK-TKIs. 34
Resistance mechanisms in EML4-ALK-driven cancers are primarily categorized as on-target or off-target. On-target resistance arises from ALK fusion amplification or secondary mutations that enhance kinase activity or reduce inhibitor binding affinity, thereby diminishing ALK-TKI efficacy. Off-target resistance occurs through activation of bypass signaling pathways—such as EGFR, KIT, SRC, and MET—which reactivate downstream effectors to promote tumor cell proliferation and survival. 35 The EML4-ALK fusion oncoprotein primarily drives carcinogenesis and resistance through sustained Ras/ERK signaling activation. Optogenetics and live-cell imaging studies reveal that EML4-ALK-containing cytoplasmic protein condensates modulate oncogenic signaling by sequestering RTK adaptor proteins like GRB2 and SOS1. Subsequent release of these sequestered adaptors can resensitize RTK signaling, reactivate ERK pathways, and ultimately promote cancer cell survival through acquired tolerance mechanisms. 36
Variations in protein stability and kinase activity among EML4-ALK variants contribute to differential TKI sensitivity and treatment duration. Patients with the V1 variant demonstrate superior response rates and treatment efficacy with crizotinib compared to those with non-V1 variants, 5 potentially due to the earlier emergence of resistance mutations in non-V1 subtypes. Acquired ALK resistance mutations also occur more frequently in patients with the EML4-ALK V3 variant. 37 These variant-specific resistance patterns complicate the achievement of optimal therapeutic outcomes in EML4-ALK-positive patients receiving targeted therapies.
Current clinical practice often employs sequential ALK-TKI regimens to overcome resistance; however, this approach risks generating compound mutations—new EML4-ALK variants harboring multiple ALK resistance mutations that present additional therapeutic challenges. Analysis of 12 EML4-ALK-positive patients identified 10 resistance-associated variants, including three mutants (L1196P, C1237Y, and L1196M/G1202R) demonstrating pan-inhibitor resistance, typically emerging after multiple lines of TKI therapy. 38 These findings highlight limitations in sequential ALK-TKI strategies.
While ALK-positive NSCLC patients may maintain tumor control for approximately six months after TKI discontinuation, clinical guidelines do not recommend therapy interruption while awaiting efficacy loss. More effective approaches include timely transition to alternative agents or combination therapies that incorporate novel drugs with ALK-TKIs.
The spectrum of acquired resistance mutations differs markedly between EML4-ALK variants. V3-positive tumors are more prone to developing solvent front mutations (e.g., G1202R) and compound mutations following sequential TKI therapy, which confer pan-ALK inhibitor resistance.37,38 This observation carries important clinical implications: for V3 patients, sequential monotherapy strategies may be less effective, and alternative approaches such as upfront lorlatinib or combination regimens should be prioritized. Furthermore, routine monitoring of circulating tumor DNA (ctDNA) for emerging resistance mutations is particularly critical in V3 patients, enabling timely detection of resistance and early intervention before clinical progression.
7. Novel Therapeutic Strategies for EML4-ALK-Positive NSCLC
7.1 Combination Therapies
Despite the sequential development of ALK-targeted agents, most patients eventually develop drug resistance. Beyond the pursuit of new TKIs, complementary strategies are required to overcome resistance to ALK inhibitors. When resistance to ALK-TKI monotherapy emerges, combination regimens utilizing multiple anticancer agents may yield superior outcomes for patients with EML4-ALK-positive NSCLC.
7.1.1. ALK-TKIs Combined With SRC Inhibitors
SRC is a protein frequently hyperactivated in cancers. Combined ALK and SRC inhibition induces cell death in EML4-ALK-positive NSCLC cells through dual targeting of the phosphorylated proteome and suppression of mTOR pathway activity. 39 Targeting SRC signaling is therefore important for managing NSCLC with acquired resistance to ALK-TKIs. Consequently, combining ALK and SRC inhibitors may represent a more effective first-line strategy for EML4-ALK-positive patients.
7.1.2 Dual Targeting of MET and Integrin β1
Cancer-associated fibroblasts (CAFs) modulate tumor cell sensitivity to ALK-TKIs via multiple concurrent signaling pathways. Hepatocyte growth factor (HGF)-MET signaling drives CAF-mediated paracrine resistance, while CAF co-culture activates integrin β1 in lung cancer cells. Simultaneous inhibition of MET and integrin β1 can suppress this deleterious crosstalk between CAFs and tumor cells, thereby overcoming CAF-mediated resistance in EML4-ALK-driven cancers and potentially improving treatment outcomes for resistant NSCLC. 40 However, the clinical relevance of this mechanism remains to be established. Current evidence is limited to co-culture systems and xenograft studies, with no prospective clinical trials evaluating combined MET/integrin β1 inhibition in ALK-positive NSCLC patients. Translational limitations include the heterogeneity of CAF subtypes in human tumors, the lack of non-invasive biomarkers to monitor CAF activation, and potential toxicity from long-term integrin blockade. Therefore, while this strategy holds promise, its clinical application awaits further validation in early-phase trials.
7.1.3. ALK-TKIs Combined With JAK1/2 Inhibitors
Liquid-liquid phase separation (LLPS) is a process by which proteins and nucleic acids form dynamic, membrane-less organelles via molecular interactions within cells. The EML4-ALK oncoprotein can undergo LLPS to form membrane-less cytoplasmic granules, leading to aberrant activation of downstream signaling and contributing to therapy resistance. 41 Notably, a subset of human EML4-ALK lung adenocarcinomas exhibits biomarkers of squamous cell carcinoma (SCC) and a propensity for squamous transformation. These tumors often demonstrate resistance to ALK-TKIs—a phenomenon promoted by EML4-ALK phase separation via JAK-STAT pathway activation, which drives squamous transdifferentiation and drug resistance. 42 This evidence underscores the remarkable plasticity of EML4-ALK-mutant NSCLC in facilitating adenosquamous transition. Combining JAK1/2 inhibitors with ALK-TKIs may thus effectively counter this form of resistant disease.Nevertheless, it is critical to recognize that the concept of LLPS-driven resistance remains largely preclinical. To date, no validated method exists to detect LLPS in routine patient tissue or liquid biopsies, and direct evidence linking LLPS to clinical outcomes in ALK-positive NSCLC is lacking. Furthermore, pharmacologically targeting phase separation without disrupting normal cellular functions poses a substantial challenge. Consequently, while the JAK-STAT pathway is druggable with approved inhibitors, the clinical utility of combining JAK1/2 inhibitors with ALK-TKIs requires prospective validation in biomarker-enriched patient cohorts.
7.1.4 ALK-TKIs Combined With Immunotherapy
Immunotherapy represents a leading frontier in oncology and has demonstrated significant antitumor activity in NSCLC. Combining targeted therapy with immunotherapy may reduce early disease recurrence. 43 Although immune checkpoint blockade (ICB) has been used for years in advanced NSCLC, its efficacy in EML4-ALK-driven NSCLC remains under active investigation. Numerous novel immunotherapy-based approaches are emerging, including oncolytic virus immunotherapy, autologous tumor-infiltrating lymphocyte (TIL) therapy, and combinations of existing immunotherapies with mRNA vaccines. 44 Incorporating ALK-TKIs with immunotherapeutic agents represents another promising strategy to overcome resistance in EML4-ALK-positive NSCLC. Accumulating evidence suggests that for patients developing resistance to ALK-TKI monotherapy, combination regimens targeting EML4-ALK will likely constitute a crucial long-term strategy for achieving optimal clinical outcomes.
7.2. Exploring Novel Therapeutic Targets
7.2.1. Heat Shock Protein 90 (Hsp90)
Heat shock protein 90 (Hsp90) is an ATP-dependent molecular chaperone that contributes to protein homeostasis and represents a critical facilitator of malignant transformation and progression. The EML4-ALK fusion protein strongly depends on Hsp90, as its TAPE domain requires Hsp90 for structural stability. Hsp90 inhibitors disrupt TAPE domain homeostasis, promote degradation of the EML4-ALK fusion protein, and thereby induce tumor cell death. 45 Consequently, Hsp90 is considered a promising therapeutic target for inducing cancer cell death. Hsp90 inhibitors exhibit potent anticancer activity, particularly against EML4-ALK variants containing the TAPE domain.
7.2.2. EGFR Family
The EGFR (also known as Human Epidermal Growth Factor Receptor, HER) family comprises four receptor tyrosine kinases: ErbB1/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4. Notably, HER3 possesses low intrinsic tyrosine kinase activity. HER3 mutations or overexpression inhibit apoptosis, accelerate proliferation, and promote immune evasion in cancer cells. HER3 overexpression may serve as a potential marker for poor prognosis in ALK-positive NSCLC patients, especially those with EML4-ALK V1 or V2 variants. 46 Both ErbB receptors and AKT (a component of the PI3K/AKT pathway) exhibit compensatory signaling upregulation in EML4-ALK-positive NSCLC cell lines following ALK-TKI treatment. Concurrent inhibition of ALK along with ErbB receptors or AKT disrupts the Ras/MAPK and PI3K/AKT oncogenic pathways and enhances apoptosis in EML4-ALK-positive NSCLC cells. 47 Thus, AKT and ErbB receptors represent potential targets for preventing adaptive resistance to ALK-TKIs in NSCLC.
7.2.3. Kinesin Eg5
The EML4-ALK V3 variant assembles into a complex with NIMA-related kinase 9 (NEK9) and NEK7 on interphase microtubules, leading to NEK7 activation and phosphorylation of its downstream targets. This process promotes increased cell length and enhanced migratory capacity, 48 underlying the heightened resistance of the V3 variant to ALK-TKIs. Kinesin Eg5 is a downstream target of the EML4-ALK V3-NEK9-NEK7 complex and its associated signaling pathways. Phosphorylation of kinesin Eg5 drives the acquisition of an elongated, mesenchymal-like morphology. This morphological shift facilitates cancer cell metastasis and constitutes a recognized hallmark of cancer. 49 Therefore, targeting kinesin Eg5 holds potential therapeutic value for EML4-ALK V3-driven NSCLC.
7.3. Exploring Novel Detection Technologies
Advances in identifying novel targets and detecting EML4-ALK fusion mutations necessitate the development of new diagnostic methods to guide precision therapy. While FISH, RT-PCR, and IHC represent established techniques for ALK fusion detection, liquid biopsy offers a less invasive alternative capable of capturing tumor heterogeneity. Primary and metastatic tumors release cells into the peripheral circulation, forming circulating tumor cells (CTCs); the detection of CTCs constitutes one form of liquid biopsy. 50 Analyzing peripheral blood components such as CTCs, cell-free DNA (cfDNA), and circulating tumor DNA (ctDNA) enables the detection of genomic alterations conferring ALK-TKI resistance. This approach facilitates dynamic disease monitoring, guides treatment decisions, and predicts potential resistance mechanisms. 51 Consequently, liquid biopsy provides novel diagnostic and monitoring strategies for EML4-ALK-positive NSCLC patients.
Emerging liquid biopsy technologies, including CTC analysis, serve as a critical bridge connecting lung cancer patients with precision medicine. The development of resistance to ALK inhibitors can accelerate the progression of leptomeningeal metastasis (LM), a refractory complication occurring in approximately 10.3% of ALK-positive NSCLC cases. Analyzing cerebrospinal fluid (CSF) cfDNA via next-generation sequencing (NGS) can elucidate LM resistance mechanisms and monitor responses to intrathecal therapies such as pemetrexed, 52 thereby expanding treatment options for EML4-ALK-positive patients with LM. Furthermore, a retrospective study developed a potential reference measurement procedure utilizing reverse transcription digital PCR (RT-dPCR) for the quantitative detection of EML4-ALK V1 and V3 fusion variants alongside a reference gene (ALK-ref) in NSCLC. This method demonstrated high specificity and sensitivity, enabling the detection of rare targets with low copy numbers and thereby improving the accuracy and comprehensiveness of fusion gene testing. 53
7.4. Translational Limitations of Emerging Resistance Concepts
The preceding sections have discussed two emerging mechanisms of resistance in EML4-ALK-positive NSCLC: CAF-mediated TME signaling and LLPS-driven oncogenic adaptation. While scientifically compelling, it is important to explicitly acknowledge their current stage of validation and translational barriers (Table 3). Both concepts are supported primarily by preclinical evidence from cell lines, murine models, and limited patient-derived samples. Direct clinical proof—such as prospective correlations with treatment outcomes in independent cohorts or successful interventional trials—is absent. Key limitations include: (1) absence of standardized, clinically deployable assays for detecting CAF activation states or LLPS in patient specimens; (2) lack of specific inhibitors targeting LLPS without off-target effects; (3) heterogeneity of CAF subtypes and tumor microenvironment across patients; and (4) unknown safety profiles of long-term integrin or JAK inhibition when combined with ALK-TKIs. Therefore, these mechanisms should be viewed as promising scientific hypotheses that require further translational research, including the development of companion diagnostics and phase I/II clinical trials, before they can inform routine clinical decision-making.
7.5. Protein Degraders
Protein degraders, represented by proteolysis-targeting chimeras (PROTACs) and molecular glues, are a novel generation of targeted therapeutic strategies for EML4-ALK-positive NSCLC, which specifically degrade the EML4-ALK fusion protein through the ubiquitin-proteasome system instead of only inhibiting its kinase activity, thus showing potential advantages in overcoming drug resistance caused by ALK target mutations and amplification. 54 At present, the research on ALK protein degraders is in the stage of parallel preclinical optimization and early clinical development, and no candidate drugs have entered Phase III clinical trials yet. 55
In preclinical studies, ALK-PROTACs such as CPD-1224 and ALX-0141 (NVL-520) have shown stronger anti-tumor activity than traditional ALK-TKIs in EML4-ALK-positive NSCLC cell lines and xenograft models containing drug-resistant mutations such as G1202R and L1196M, 56 which can significantly degrade the fusion protein and inhibit the downstream Ras/ERK and PI3K/AKT signaling pathways, and still have inhibitory effects on TKI-resistant compound mutation models. Phase I clinical trials such as NCT04863325 have preliminarily verified the clinical effectiveness of NVL-520 (ALX-0141), with an objective response rate of about 23% and a disease control rate of 60% in patients with drug resistance after multiple lines of ALK-TKI treatment. 57 In terms of safety, the existing preclinical and Phase I clinical data show that the off-target toxicity of ALK protein degraders is significantly lower than that of traditional multi-target ALK-TKIs, 56 and the main toxicities are Grade 1-2 gastrointestinal reactions and hematological toxicity, without reports of severe liver and kidney toxicity. However, the long-term medication safety data are still lacking, and there is no special evaluation on central nervous system (CNS) toxicity. Some preclinical studies have shown that PROTACs may cause mild damage to blood-brain barrier endothelial cells due to their large molecular weight, 48 and their potential CNS toxicity needs to be further verified.
CNS penetration and intracranial anti-tumor activity are the main bottlenecks in the development of ALK protein degraders. 48 Most PROTACs are difficult to penetrate the blood-brain barrier due to their large molecular weight (usually >1000 Da) and low lipophilicity. The intracranial drug concentration in preclinical models is only 5%∼10% of that in peripheral blood, and the inhibitory effect on EML4-ALK-positive brain metastasis models is significantly weaker than that of third-generation TKIs such as lorlatinib.
7.6. Drug Repurposing
Drug repurposing refers to the application of drugs approved for other diseases in the treatment of EML4-ALK-positive NSCLC, which has the core advantages of short research and development cycle, low cost and sufficient safety data, and can be quickly transformed into clinical treatment schemes. 49 The research on drug repurposing in EML4-ALK-positive NSCLC is mainly focused on three types of drugs: anti-angiogenic agents, immunomodulators and epigenetic regulators, among which anti-angiogenic agents have the most sufficient clinical evidence when combined with ALK-TKIs.
Anti-angiogenic agents represented by anlotinib and bevacizumab have been widely used in the off-label combination with ALK-TKIs in clinical practice for the treatment of EML4-ALK-positive NSCLC patients with brain metastases. 43 Real-world studies have shown that anlotinib combined with alectinib can significantly improve the intracranial objective response rate of patients with brain metastases, with the rate reaching 45%, which is significantly higher than that of alectinib monotherapy. Its mechanism is to inhibit tumor angiogenesis and destroy the tumor microenvironment of the blood-brain barrier, thereby increasing the intracranial concentration of ALK-TKIs. Immunomodulators such as hydroxychloroquine, an anti-malarial drug, have been confirmed in Phase II clinical trials (NCT03296817) that its combination with crizotinib can improve the disease control rate (72%) and prolong the median progression-free survival in patients with progressive disease after first-line crizotinib treatment by inhibiting the autophagy pathway. 44 Epigenetic regulators such as vorinostat, a HDAC inhibitor, have shown the effect of reversing the intrinsic resistance of EML4-ALK V3 variant in preclinical studies, 50 but there is no clinical data to verify its efficacy yet.
In terms of safety, since the repurposed drugs are all approved drugs, the single-drug safety data are clear, and the toxicity after combination with ALK-TKIs is mainly superimposed toxicity without new severe toxicity. 49 The main toxicities of anlotinib combined with alectinib are hypertension and proteinuria, which are typical toxicities of anti-angiogenic drugs, and the main toxicity of hydroxychloroquine combined with crizotinib is retinopathy, which can be effectively controlled by regular dose adjustment and relevant examinations.43,44 The CNS activity of repurposed drugs varies significantly with the drug type 49 : anti-angiogenic drugs have no direct ALK inhibitory activity, but can indirectly enhance the CNS efficacy of the combination regimen by improving the intracranial concentration of ALK-TKIs; hydroxychloroquine and vorinostat have moderate CNS penetration and can reach effective therapeutic concentrations in the cranium, but their single drugs have no obvious inhibitory effect on intracranial lesions and only play an auxiliary role in combination therapy.
At present, the research and development of drug repurposing in EML4-ALK-positive NSCLC is mainly focused on the combination of approved drugs and ALK-TKIs, and most of them are in the stage of Phase II clinical trials or real-world studies. 49 The research focus in the future is to carry out large-sample Phase III randomized controlled trials to verify the efficacy and safety of the combination regimen, and explore the individualized selection of repurposed drugs based on the EML4-ALK variant subtype, such as giving priority to the combination of anti-angiogenic drugs for patients with V3 variant with high risk of early progression and brain metastasis.30,49
7.7. Early Treatment Strategies: Beyond Sequential Monotherapy
Proposed Early Treatment Strategies for EML4-ALK-Positive NSCLC: Risk-Stratified Recommendations

Note. Evidence level definitions: Phase III = randomized controlled trial; real-world = prospective or retrospective cohort studies; preclinical = cell lines/animal models only.
(1) Upfront lorlatinib monotherapy. The phase III CROWN trial demonstrated superior PFS and intracranial control with lorlatinib versus crizotinib, and real-world data show that V3 patients receiving lorlatinib as first-line treatment achieve outcomes comparable to long-variant patients. This is the preferred option for high-risk patients. (2) ALK-TKI plus anti-angiogenic agent. Real-world studies (e.g., anlotinib plus alectinib) have shown improved intracranial objective response rates (45% vs. alectinib alone) in patients with brain metastases. This combination may be considered for patients with CNS involvement, particularly when lorlatinib is not available or tolerated. However, phase III confirmatory trials are lacking.
8. Clinical Framework for Resistance Monitoring and Management
Based on emerging clinical evidence and consensus guidelines,30,31,40 we propose a clinically actionable framework for resistance monitoring and management in EML4-ALK-positive NSCLC:
8.1. Timing and Modality of Re-biopsy
Resistance monitoring should be initiated at the first sign of disease progression, defined radiologically by RECIST v1.1 criteria or by emergence of new symptoms suspicious for CNS progression. 19 For patients with isolated oligoprogression, tissue re-biopsy of the progressing lesion is recommended to distinguish on-target from off-target resistance mechanisms. For patients with systemic progression or when tissue biopsy is technically challenging, liquid biopsy using plasma circulating tumor DNA (ctDNA) is the preferred approach, given its ability to capture intra-tumoral heterogeneity and detect emerging resistance mutations with high sensitivity. 40 In patients with suspected CNS progression, cerebrospinal fluid (CSF) ctDNA analysis offers a unique window into the intracranial resistance landscape. 41
8.2. Interpretation of Resistance Mechanisms and Treatment Sequencing
The detection of secondary ALK mutations (e.g., G1202R, L1196M, I1171N) should guide the selection of subsequent ALK-TKIs based on the spectrum of activity against specific mutations. For patients with solvent front mutations (e.g., G1202R), third-generation lorlatinib is the preferred option, whereas second-generation TKIs may retain activity against certain gatekeeper mutations (e.g., L1196M) in the absence of compound mutations.19,30 In patients with off-target resistance (e.g., MET amplification, EGFR activation, or phenotypic transformation), combination strategies targeting the bypass pathway should be considered, with re-biopsy results guiding the selection of specific combination partners.25,29
8.3. Dynamic Monitoring and Adaptive Treatment
Beyond the time of overt progression, emerging data support the use of serial ctDNA monitoring to detect molecular progression before radiographic progression. 40 Early detection of emerging resistance mutations enables timely treatment adaptation, potentially improving outcomes. For patients with V3 variant-positive tumors, intensified ctDNA surveillance is particularly warranted given their higher propensity for early resistance mutation emergence. 30
9. A Proposed Treatment Algorithm for EML4-ALK-Positive NSCLC
To translate complex molecular and clinical evidence into practical patient management, we establish a standardized treatment framework for EML4-ALK-positive NSCLC. This systematic framework incorporates variant stratification, baseline risk assessment, first-line treatment selection, resistance monitoring, and post-progression management.
9.1. Step 1: Diagnosis and Molecular Characterization
Upon diagnosis of advanced NSCLC, confirm ALK fusion positivity via IHC, FISH, or NGS. If EML4-ALK is identified, determine the specific variant type (V1, V2, V3, V4, V5a, etc.) using NGS or RT-PCR when feasible. Variant typing is crucial for risk stratification: long variants (V1, V2, V4) are associated with favorable prognosis, whereas short variants (V3, V5) confer higher risk of early progression and resistance. Concurrently, assess baseline brain metastases via contrast-enhanced MRI.
9.2. Step 2: First-Line Therapy Selection
For
For
9.3. Step 3: Routine Monitoring and Early Detection of Resistance
Perform radiological assessment (chest CT, brain MRI) every 8–12 weeks. Concurrently, consider serial plasma ctDNA monitoring every 3–6 months, especially for V3 variant patients who have a higher propensity for early emergence of resistance mutations. Detection of emerging ALK resistance mutations (e.g., G1202R, L1196M, I1171N) in ctDNA may precede radiographic progression and enable timely treatment adaptation.
9.4. Step 4: Management at Disease Progression
Upon confirmed disease progression (RECIST v1.1), perform re-biopsy of the progressing lesion (tissue) or liquid biopsy (plasma ctDNA) to identify the resistance mechanism. For isolated CNS progression, consider stereotactic radiosurgery or whole-brain radiotherapy while continuing the same ALK-TKI, or switch to an ALK-TKI with better CNS penetration (e.g., lorlatinib). For systemic progression, classify resistance as:
9.5. Step 5: Ongoing Dynamic Adaptation
Repeat resistance profiling at each progression event. For patients with V3 variant or compound mutations, upfront use of lorlatinib or combination regimens should be prioritized over sequential monotherapy to prevent accumulation of pan-resistant mutations.
10. Future Perspectives
The EML4-ALK fusion gene represents a significant oncogenic driver in non-small cell lung cancer (NSCLC). EML4-ALK-positive NSCLC is typically characterized by adenocarcinoma histology, occurrence in never- or light-smokers, and a higher incidence in females. Although current diagnostic techniques have inherent limitations, they sufficiently support routine clinical management, enabling most EML4-ALK-positive patients to receive beneficial targeted therapies.
Nevertheless, the emergence of drug resistance and tumor recurrence remains a major obstacle to achieving long-term disease control. Determining the optimal sequencing and administration of ALK-TKIs, alongside developing effective strategies to overcome resistance, constitutes a critical challenge in the field. ALK-TKI monotherapy has inherent limitations, underscoring the need for novel therapeutic approaches. These may include targeting alternative pathways or implementing rational combination regimens.
Advances in sequencing technologies and ongoing translational research will facilitate the selection of precise and efficient ALK testing methodologies. Ultimately, tailoring targeted therapies based on individual patient profiles and molecular characteristics—employing either sequential or combination strategies to circumvent resistance mechanisms—will be crucial for improving clinical outcomes in EML4-ALK-positive NSCLC.
By integrating variant-specific biological heterogeneity with emerging therapeutic strategies, this review provides a clinically actionable framework for risk stratification and treatment selection in EML4-ALK-positive NSCLC. The variant-stratified perspective offers a novel conceptual lens for understanding the heterogeneous responses to ALK-TKIs and may inform the design of future clinical trials and combination regimens.
Integrating variant-specific biology into clinical practice, we propose a preliminary framework for variant-stratified management of EML4-ALK-positive NSCLC:
Footnotes
Acknowledgements
We would like to thank all participants in this study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Scientific Research Foundation of the Science and Technology Department of Sichuan Province (Grants 2017SZ0066).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.