
Abstract
Activation of endothelial cells (ECs) by proinflammatory stimuli triggers expression of cellular adhesion molecules including intercellular adhesion molecule 1 (ICAM-1) on the cell surface. Such molecules mediate the transendothelial migration of inflammatory cells, which is an early key step of atherogenesis. We have previously demonstrated that plasmin activates human inflammatory cells via the annexin A2 heterotetramer (A2t). Here we show that human umbilical vein endothelial cells (HUVECs) and human microvascular endothelial cells express high amounts of A2t, as shown by Western blotting, fluorescence microscopy and flow cytometry. Activation of HUVEC by plasmin led to cleavage of the annexin A2 subunit of the receptor complex, followed by the activation of Akt/nuclear factor (NF)-kB signaling, and phosphorylation of MAP kinases p38 and ERK1/2. Further, plasmin stimulates the NF-kB/p38-dependent expression of ICAM-1 by HUVEC. The plasmin-induced activation of cells was abolished when annexin A2 was down-regulated by small-interfering RNA. In vivo, we show co-localization of the ECs marker CD31 with the plasmin receptor A2t and ICAM-1 in human atherosclerotic plaques of human femoral arteries, which also exhibit activated NF-κB signaling as revealed by immunofluorescence staining for phosphorylated Iκbα. In addition, plasma of patients with advanced atherosclerosis exhibited enhanced plasmin activity and up-regulated levels of plasmin-α2-antiplasmin. These data point to a previously unrecognized functional role of plasmin in EC biology, which could be of particular relevance in the development of atherosclerosis.
Introduction
Recent studies on atherosclerosis had been focused on inflammation, providing new insight into the mechanisms of the disease. 1 Inflammatory cytokines such as tumor necrosis factor (TNF)-α are involved in vascular inflammation and stimulate the up-regulation of adhesion molecules such as intercellular adhesion molecule (ICAM)-1, ICAM-2, vascular cell adhesion molecule (VCAM)-1, E-selectin and P-selectin on the surface of endothelial cells (ECs), which then interact with specific ligands to recruit inflammatory cells such as circulating monocytes and T-cells to the endothelial wall, a process that is central to all stages of atherosclerosis.2,3 ICAM-1 has been implicated in the progression of autoimmune diseases and is critical for the firm arrest and transmigration of leukocytes out of blood vessels into tissues. It is becoming clear that immune cells not only dominate early lesions but also directly contribute to the development of atherosclerosis.4,5 Although excessive inflammatory and immune responses driven by proinflammatory cytokines are held responsible for cardiovascular events associated with atherosclerosis, the initiation of cytokine induction is still poorly understood, and the initial triggers for the expression of adhesion molecules remain obscure.6,7
Proteases, specifically serine proteases, play important roles in atherosclerosis. Contact activation takes place during any type of chronic inflammation, including atherosclerosis, and is invariably associated with the generation of plasmin.8,9 Latent matrix metalloproteinase (MMPs) expressed in atherosclerotic plaques by inflammatory cells (macrophages and foam cells) that can be activated by plasmin. 10 In the context of atherosclerosis plasmin is often linked to the degradation of the extracellular matrix, to the activation of latent metalloproteinases, and to proteo-lytic degradation of the vessel wall. 11 However, at the molecular level, apart from its fibrinolytic function, 12 plasmin is a potent cell activator. In platelets, plasmin induces aggregation, and in ECs, it promotes the biosynthesis of platelet-activating factor and chemotaxis. 9 Moreover, in human monocytes/macrophages, plasmin triggers the release of cytokines and proinflammatory lipid mediators and induces chemotaxis.6,13–16 In a mouse model of transplant artherosclerosis, carotid blood vessels from wild-type mice transplanted into Plg- / - recipient mice exhibited reduced proliferation and migration of smooth muscle cells into the intima, as well as reduced plaque area, inflammation and necrosis. 17 In humans, increased severity of the atherosclerosis is positively correlated with the expression of the specific plasminogen activator uPA. 18 Therefore, plasmin might play multiple roles in the development of atherosclerosis.
We have previously shown that plasmin activates human macrophages via cleavage of the annexin A2 tetramer, composed of annexin A2 and S100A2 (A2t). 6 Annexin A2 is a calcium-regulated, phospholipid-binding protein on ECs, macrophages, tumor cells and keratinocytes.19,20 Annexin A2 is as a key extracellular binding partner for pathogens and host proteins alike, and it also can be shed or secreted. 21
In the present study, we tested whether plasmin could activate human umbilical vein endothelial cells (HUVEC) and trigger increased expression of ICAM-1 via its proteolytic activity. Further, we investigated whether the components necessary for plasmin-mediated activation of ECs are present and co-localize in plaque material derived from human atherosclerotic femoral artery vessels. We also measured whether plasmin activity is enhanced and whether the contents of plasmin-α2-antiplasmin are up-regulated in plasma of patients with severe atherosclerosis compared with healthy controls.
Materials and methods
Cell isolation and culture
Primary HUVEC and human dermal microvascular cells (HMEC) were purchased from PromoCell (Heidelberg, Germany) and cultured to confluency at 37°C and 5% CO2 in EBM-2 basal medium (PromoCell) supplemented with Endothelial Cell Growth Medium 2 Supplement Pack. Macrophages were differentiated from monocytes isolated from buffy coats by Percoll gradient centrifugation as described previously. 6
Reagents
Purified human plasmin was purchased from Athens Research & Technology (#2008–04L, Athens, GA, USA) and was free of lipopolysaccharide as analyzed by the Pyrogent LAL assay (Lonza, Basel, Switzerland). Plasmin activity is given in Committee on Thrombolytic Agents (CTA) units/mL.13,22 The catalytic inhibitor of plasmin, D-Val-Phe-Lys chloromethyl ketone (VPLCK), the p38 MAPK inhibitor SB203580, and the MEK inhibitor U0126 were purchased from Calbiochem (San Diego, CA, USA). Chemically pure acetyl-11-keto-β-boswellic acid (AKβBA) was used as an inhibitor of the NF-κb.23,24 Lipopolysaccharide (LPS) was from Sigma, TNF-α was purchased from Peprotech (London, UK).
The following antibodies were used for immunoblotting or immunofluorescence: endothelial nitric oxide synthase (eNOS; Santa Cruz, Santa Cruz, CA, USA), annexin A2 (Abgent, San Diego, CA, USA), S100A10 (BD Biosciences, Bedford, MA, USA), ICAM-1 and ERK1/2 (Abcam, Cambridge, MA, USA), actin (Chemicon International, Temecula, CA, USA), p-Akt-(Ser473) (Upstate, Charlottesville, VA, USA), human CD31 (PECAM1, Novus Biologicals, Littleton, CO, USA). P-Iκbα, p-p38, p-JNK, JNK and p-ERK1/2 were from Cell Signaling (Beverly, MA, USA). Goat anti-mouse Alexa 488 and anti-rabbit Alexa 555 were purchased from Invitrogen (Grand Island, NY, USA). The following antibodies were used for flow cytometry: antiannexin A2 antibody (FITC) (#ABIN670363, antibodies-online.com), S100A10 (RDI, Concord, MA, USA), anti-CD31 (PECAM1, eBioscience, San Diego, CA, USA), PE anti-human ICAM-1 (Biolegend, San Diego, CA, USA), control mouse and rabbit IgG and PE-conjugated F(ab’)2 were from Dianova (Hamburg, Germany), von Willebrand factor (vWF) was from Dako (Hamburg, Germany) and IKK from Santa Cruz. FITC-labeled goat anti-mouse and anti-rabbit IgG (H+L) were purchased from Advanced Targeting Systems (San Diego, CA, USA).
Analysis of protein expression
Protein expression was analyzed by Western immunoblotting, fluorescence microscopy, and flow cytometry . For the cleavage of annexin A2, confluent monolayers of HUVEC were kept in fetal calf serum (FCS)-free EBM-2 medium for an additional 12 h prior to stimulation. The cells were then either left unstimulated or stimulated for 30 min with 0.43 CTA U/mL plasmin or equivalent amounts of catalytically inactivated plasmin (VPLCK-plasmin).14,22 For the analysis of phosphorylation of ERK1/2, p38, Akt and Iκbα, confluent monolayers of HUVEC were kept in FCS-free EBM-2 medium for an additional 12 h prior to stimulation with plasmin. In some experiments, the cells were pretreated for 30 min with 1 μmol/L of either SB203580, 25 U0216 or 10 μmol/L AKβBA 6 before plasmin stimulation. After the indicated treatment time, the cells were lysed with RIPA lysis buffer (50 mmol/L Tris-HCl [pH 7.4], 150 mmol/L NaCl, 0.1% sodium dodecyl sulfate, 0.25 mmol/L EDTA, 1% Triton, 1% deoxycholic acid) containing mammalian protease and phosphatase inhibitor cocktails (Calbiochem) and analyzed by Western immunoblotting. Expression of cell surface markers or proteins was analyzed by flow cytometry.
Reverse transcription and polymerase chain reaction analysis
Total RNA from 1 × 106 HUVEC stimulated with 0.43 CTA U/mL plasmin or equivalent amounts of catalytically inactivated plasmin (VPLCK-plasmin)
22
was isolated by Trizol reagent (Invitrogen) according to the manufacturer's instructions. One μg total RNA was reverse transcribed into single-stranded cDNA by incubation with Moloney murine leukemia virus reverse transcriptase (MMLV Reverse Transcriptase, Promega) and analyzed by semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR). ICAM-1 (319 bp) was detected with the forward primer 5
siRNA transfection
The small-interfering RNA (siRNA) targeting annexin A2 (sense strand: 5’ GUUACAGCCCUUAUGACAU 3’, anti-sense strand: 5’ AUGUCAUAAGGGCUGUAAC 3’) and control siRNA of identical nucleotide composition as siRNA, but in a random sequence were synthesized by GenePharma (Shanghai, China). For transient transfection, HUVEC were plated onto six-well plates and grown to >80% confluence. siRNA (100 nmol/L) was then delivered to cells for 72 h using Oligofectamine (Invitrogen) according to the manufacturer's instruction. After transfection, cells were harvested and tested for annexin A2 expression by Western blot analysis.
Chromozym plasmin activity assay
Chromozym PL (Roche, Basel, Switzerland) was used to measure the activity of plasmin. LP0668 heparin anticoagulation tubes were used to collect blood from patients with lower limb atherosclerosis (4 men and 4 women, ranging from 53 to 67 years of age [mean: 59.6 ± 5.2], body mass index [kg/ m2] range from 22.8 to 29.2 [mean: 26.6 ± 2.3], all with diabetes, n = 8) before surgery and healthy controls (4 men and 4 women, n = 8). The healthy control group was matched for age and gender with disease group. Equivalent amounts of plasma protein (5 μg) were diluted according to the manufacturer's protocol. Triplicates of each sample were dispensed into 96-well plates and incubated at 37°C. Absorbance changes were read at 405 nm. Plasmin concentrations in plasma were quantified with a plasmin-α2-antiplasmin enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN, USA).
Immunohistochemical analysis
Serial sections (5 μm) were obtained from human atherosclerotic femoral artery vessel specimens from six patients (2 men and 4 women) with hypertension, diabetes and arteriosclerosis obliterans, ranging from 66 to 83 years of age (mean: 71 ± 8). The patients undergoing vascular surgery for atherosclerotic complications were recruited from the outpatient clinic of Ruijin Hospital (Shanghai, People's Republic of China). The sections used for hematoxylin and eosin (H&E) and immunofluorescence staining of each group were from the same atherosclerotic vessel, which was splitted into two parts, one was paraffin-embedded (for H&E staining) and the other was optimal cutting temperature (OCT)-embedded (for immunofluor-escence staining). Paraffin-embedded 5 μm sections (from tissues fixed in 4% paraformaldehyde solution for 24 h, dehydrated and embedded in paraffin) were stained with H&E. Immunostaining of cryosections fixed in ice-cold acetone was performed using a previously described protocol. 26 To demonstrate the colocalization of ICAM-1, Annexin A2, S100A10 and phosphorylated Iκbawith an EC marker CD31 in human atherosclerotic sections, double staining was performed with the following antibodies: anti-human ICAM-1 (clone HM1, Abcam), anti-human annexin A2 (Abgent), anti-human S100A10 (BD Biosciences), anti-human phospho-Iκba (Cell Signaling) and anti-human CD31 (Novus Biologicals). Control samples were stained with either mouse or rabbit IgG (Santa Cruz). Primary antibodies were visualized with appropriate secondary antibodies conjugated to either Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen). DAPI (Sigma-Aldrich) was used to stain nuclei. All antibodies were diluted in antibody diluent (Dako, Hamburg, Germany) according to the manufacturer's protocols. Immunostaining was analyzed with a fluorescence microscope (Axio Scope A1, Zeiss). All study subjects gave their informed consent for participation. The study was approved by the Research Ethics Board of Renji Hospital, Shanghai JiaoTong University School of Medicine.
Statistical analysis
Values shown represent the mean ± SD where applicable. Statistical significance was calculated with the Mann-Whitney U test. Differences were considered significant for P < 0.05.
Results
ECs express the plasmin receptor annexin A2 heterotetramer
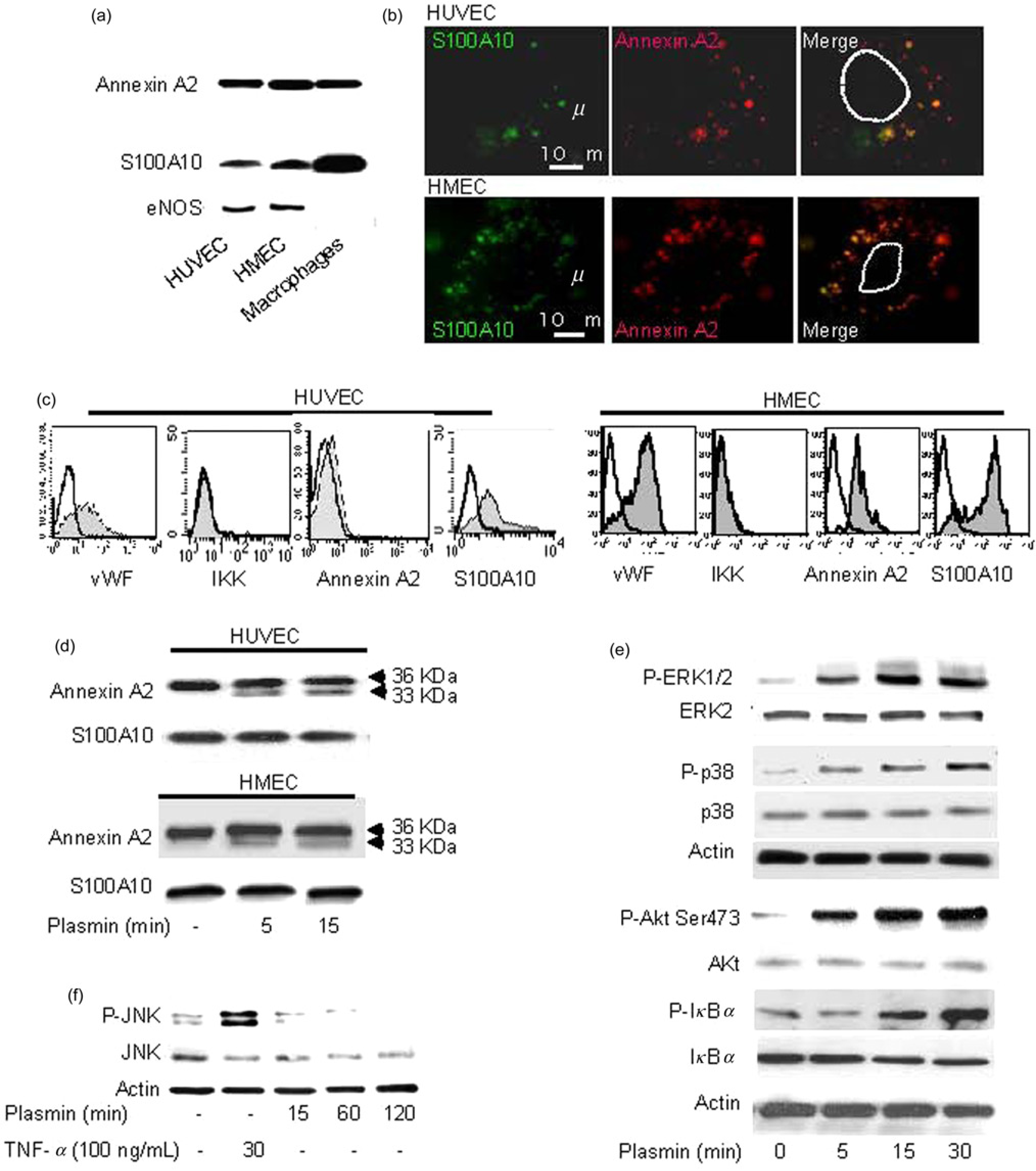
We have previously shown that plasmin is a potent activator of human monocytes and macrophages, where it binds to and cleaves the receptor annexin A2 heterotetramer (A2t), composed of annexin A2 and S100A2.6,22 It has been previously shown that ECs express the A2t on their surface. 27 We, therefore, compared the expression of annexin A2 and S100A2 in HUVEC, HMEC and human macrophages The data show that macrophages express more S100A10 than ECs, whereas the expression levels of annexin A2 were similar in all cell types (Figure 1a). Because, the majority of annexin A2 is expressed by ECs intracellularly, 28 we further performed microscopic and flow cytometric analyses of A2t in non-permeabilized ECs. The data demonstrate that annexin A2 and S100A10 are expressed on the surface of both types of ECs to similar extend where they co-localized forming A2t complex (Figures 1b and c). We, therefore, used in following experiments HUVEC to analyze the effects of plasmin on ECs.
Plasmin triggers activation of the
Plasmin activates HUVEC
Stimulation of HUVEC or HMEC with plasmin led to the cleavage of annexin A2 yielding a proteolytic fragment of 33 kDa as detected in whole cell lysates. Under the same conditions, S100A10 remained unaffected (Figure 1d). We have previously shown that plasmin induces truncation of the annexin A2 N-terminus and dissociation of the A2t in monocytes. 22 These data suggest that plasmin might use A2t as a plasmin receptor and that it might activate ECs in a similar way as monocytes, macrophages and dendritic cells.6,22,29
Plasmin is an important protease playing role in many cellular processes including those inducing activation of antigen-presenting cells. In this study, we observed that both ERK1/2 and p38 MAPK were phosphorylated as early as five minutes after plasmin stimulation (Figure 1e). During the whole observation time, we did not detect any activation of stress-activated protein kinase/Jun N-terminal kinase (SAPK/JNK), the third family member of the MAP kinase family, although JNK activation was observed after stimulation of ECs with TNF-α (Figure 1f). The nuclear transcription factor NF-κb is a key player in the development and progression of chronic inflammatory diseases including atherosclerosis. 30 Indeed, plasmin induced the activation of Akt kinase, an upstream activator of NF-κb signaling, and phosphorylation of IκBa in HUVEC as shown by immunoblotting (Figure 1e). These data indicate that in HUVEC, plasmin triggers several signaling pathways, in particular, activation of p38, ERK1/2 MAPK and Akt/NF-κB.
Plasmin induces expression of ICAM-1 on HUVEC
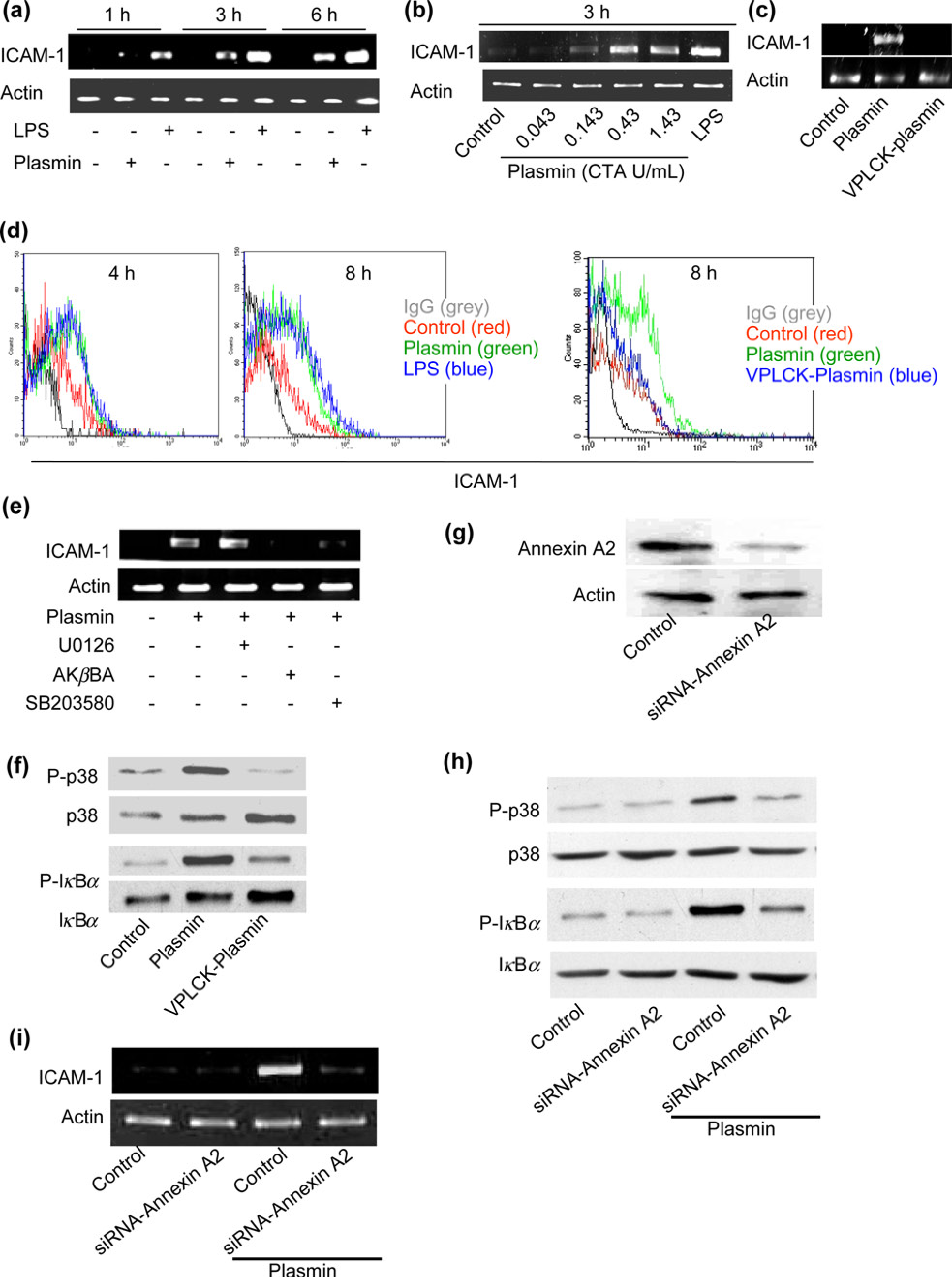
ICAM-1 is present in atherosclerotic lesions and is central to all stages of atherosclerosis. 4 We analyzed expression of ICAM-1 in plasmin-stimulated HUVEC by RT-PCR. We show that plasmin induced a marked time- and concentration-dependent increase of ICAM-1 mRNA levels comparable with those induced by the standard activator LPS (1 μg/mL), which served as a positive control (Figures 2a and b). Because the proteolytic activity of plasmin seems to be indispensable for the activation of signaling pathways in monocytes, macrophages and dendritic cells.6,15,22 We examined its role in ICAM-1 induction. Catalytically inactivated plasmin failed to stimulate the expression of ICAM-1 mRNA (Figure 2c). Thus, activation of this adhesion molecule by plasmin in HUVEC requires proteolytically active plasmin.
Plasmin induces
To investigate whether increased mRNA levels are correlated with increased protein levels, we performed flow cytometric analysis and found that the expression of HUVEC surface ICAM-1 after four hours of stimulation was up-regulated and that this was even more prominent after eight hours of stimulation and also required proteolytically active plasmin (Figure 2d). The ICAM-1 expression induced by 0.43 CTA U/mL plasmin was similar to that induced by 1 μg/mL LPS at both time points (Figure 2d), suggesting that plasmin is able to markedly induce ICAM-1 protein expression on the surface of HUVEC. To define, by which signaling pathways plasmin induced expression of ICAM-1, we pretreated HUVEC for 30 min with inhibitors to p38 MAPK (SB203580, 1 μmol/L), the MAPK/ERK kinase pathway (U0126, 10 μmol/L) and an inhibitor of NF-κB (AKβBA, 10 μmol/L) before stimulation with plasmin for additional three hours. RT-PCR revealed that pretreatment of HUVEC with SB203580 and AKβBA significantly inhibited the plasmin-induced ICAM-1 expression. In contrast, inhibition of ERK1/2 by the MEK inhibitor U0126 did not affect the plasmin-mediated induction of ICAM-1 (Figure 2e). The data also demonstrated that treatment with VPLCK-Plasmin did not activate the p38 and NF-KB signaling pathways compared with plasmin stimulation in HUVEC (Figure 2f). To demonstrate the link between the cleavage of annexin A2 and the activation of the plasmin-induced signaling pathways in HUVEC, we employed annexin A2 siRNA transfections to effectively reduce endogenous annexin A2 protein expression in HUVEC (Figure 2g). This significantly inhibited plasmin-induced activation of p38 and NF-KB signaling pathways (Figure 2h) and ICAM-1 expression (Figure 2i) in HUVEC. These data imply the pivotal role of annexin A2 in the plasmin-mediated induction of ICAM-1 in human ECs.
ECs in human atherosclerotic femoral artery lesions express high levels of ICAM-1 and plasmin receptor
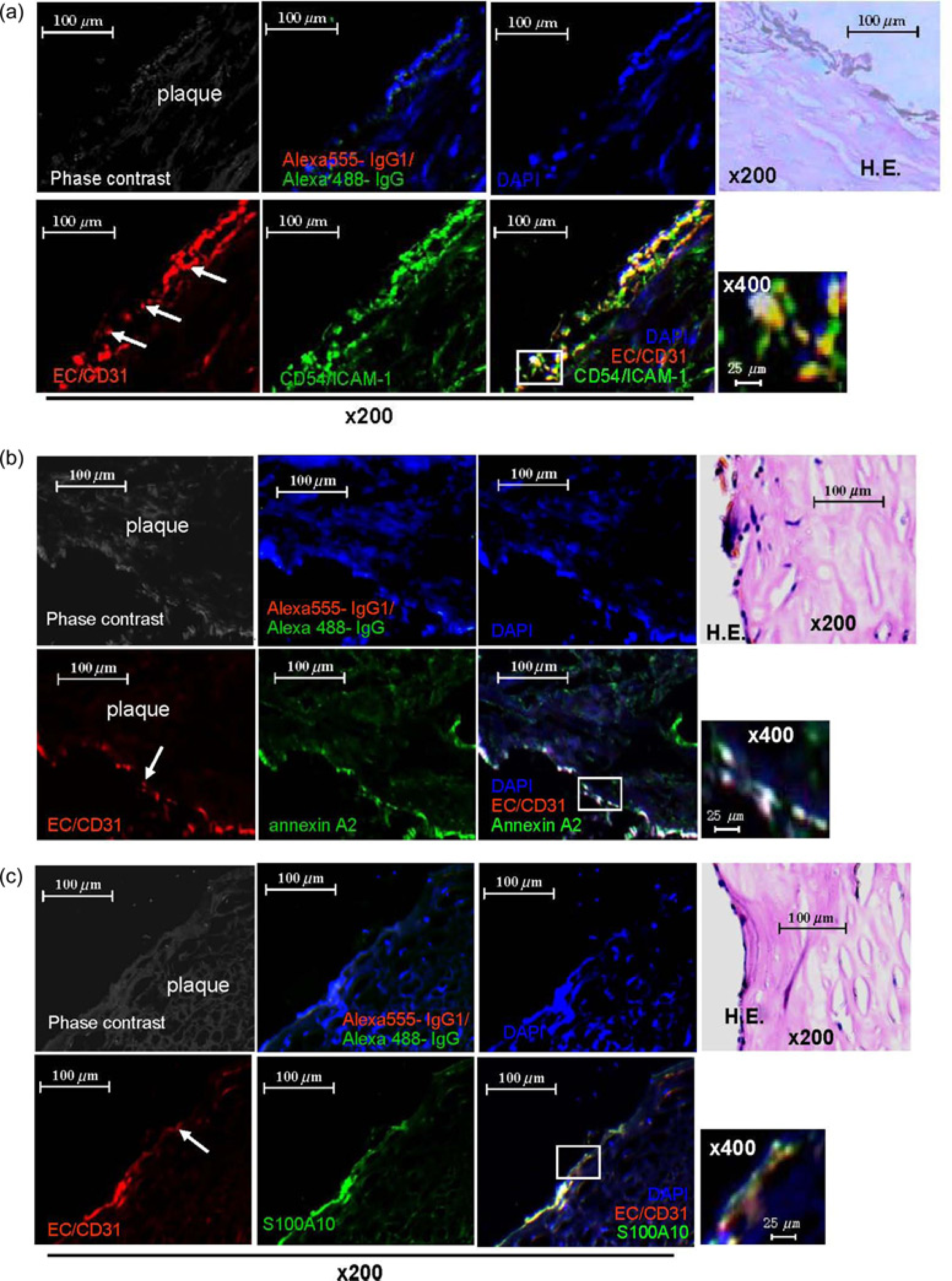
Multicolor fluorescence microscopy of sections from human atherosclerotic femoral artery plaques stained for the human ECs marker CD31 as well as ICAM-1 revealed co-localization of CD31 and ICAM-1 indicating that in atherosclerotic plaques, ECs express ICAM-1 (Figure 3a). Similarly, CD31+-ECs co-localized in atheromatous plaques with the plasmin receptor complex consisting of annexin A2 (Figure 3b) and S100A10 (Figure 3c). These results indicate that ECs within atherosclerotic plaques express plasmin receptor and ICAM-1 on their surface. We have previously shown that plasmin is ubiquitously present in human atherosclerotic lesions 29 implicating that plasmin might directly activate ECs via A2t resulting in high expression of ICAM-1.
Co-localization of
Activated NF-κB is present in ECs derived from human atherosclerotic femoral artery lesions
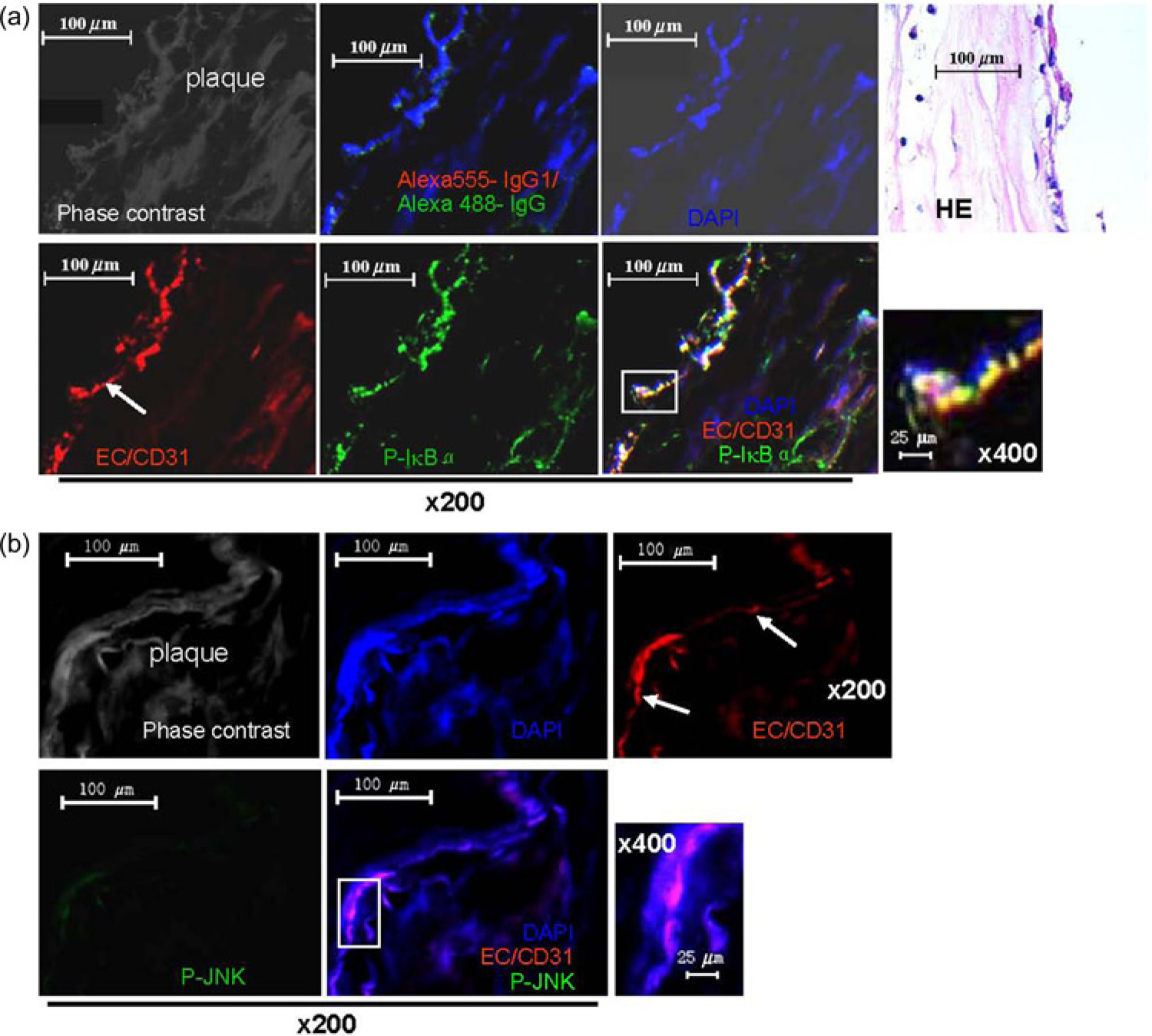
The expression of ICAM-1 is dependent on the activation of NF-KB signaling. 31 Therefore, we examined whether NF-KB is activated in vivo in ECs in atherosclerotic patients using double immunofluorescence microscopy. Notably, immu-nofluorescence staining for IkBα phosphorylated on Ser32 or phospho-JNK revealed that quite a few cells in the human atherosclerotic femoral artery plaques express p-IκBα, and that there is co-localization of the EC-specific marker CD31 with p-IKBα in atheromatous plaques (Figure 4a), but nearly no phospho-JNK expression (Figure 4b), which is consistent with the in vitro results (Figure 1e and f). Together, these data demonstrate that ECs in atherosclerotic lesions exhibit NF-KB activation in terms of phosphorylation of IκBα and that they represent the main source of the cellular adhesion molecule ICAM-1 in atherosclerosis.
Plasmin activity is enhanced in Atherosclerosis
We next investigated whether circulating plasmin could directly activate ECs in vivo. As production of plasmin can be examined by measuring the circulating plasmin-α2-antiplasmin, we analyzed its concentrations in plasma by ELISA. Concentrations of plasmin-α2-antiplasmin were significantly elevated in patients with atherosclerosis compared with healthy controls (1170.74 ± 464.24 versus 91.51 ± 44.26 ng/mL, P< 0.0001, n
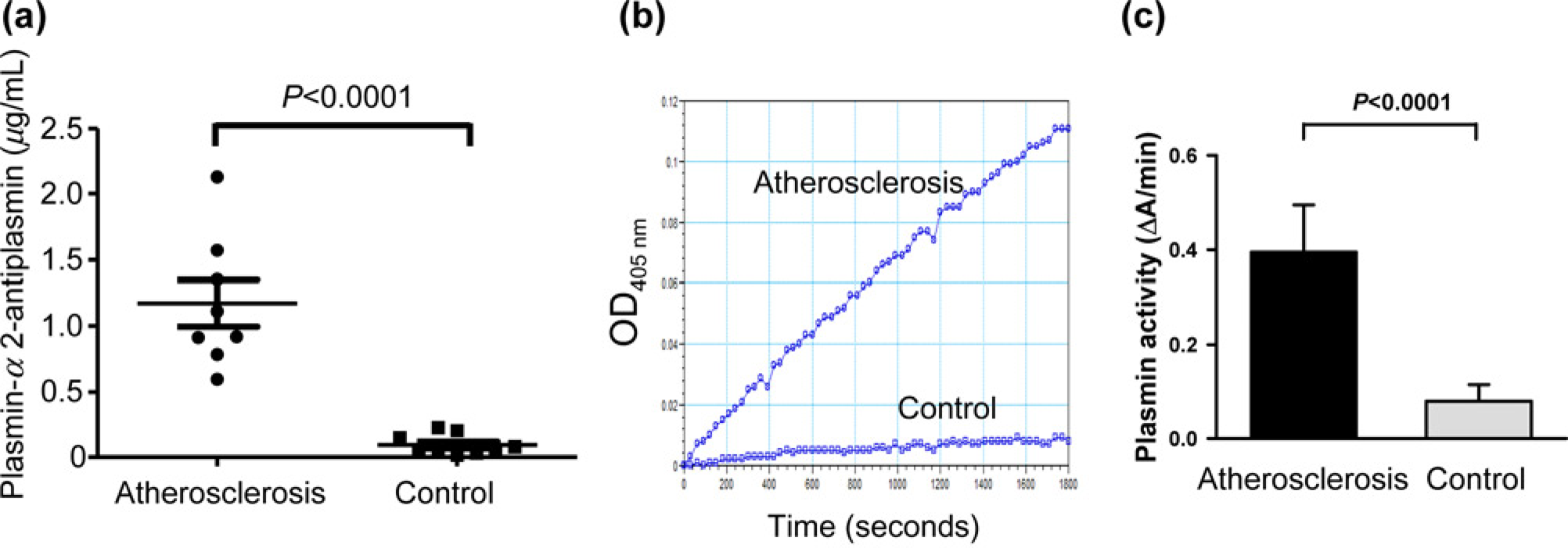
Enhanced plasmin activity in blood of patients with severe atherosclerosis compared with healthy controls. (a) Total plasmin-α2-antiplasmin levels in the plasma derived from eight patients with atherosclerosis and eight control subjects was measured by
Discussion
The development of atherosclerotic plaques is a multistep inflammatory process.1,32,33 The first step involves an altered endothelial function with a change in normal homeostatic responses, which is directly responsible for the recruitment of circulating leukocytes and their uptake into inflamed tissue. The transendothelial migration of leukocytes is a key event mediated by cell adhesion molecules, such as ICAM-1, on the vascular endothelium. 34 In addition to its well-known role in leukocyte emigration, ICAM-1 has now been shown to have other critical functions including transmission of intracellular signals 35 leading to rearrangement of the actin cytoskeleton aiding leukocyte diapedesis and initiating the activation of proinflammatory cascades that can perpetuate an inflammatory response.36,37 Thus, CAM-1 acts as an adhesion molecule and a signal transducer in ECs regulating endothelial permeability and efficient transendothelial migration of leukocytes. Hence, ICAM-1 expression has been associated with the progression of autoimmune diseases and especially with the process of atherosclerosis.4,5 Elucidation of the signaling that is indispensible for ICAM-1 expression is required to design suitable therapies targeting ICAM-1 expression, as well as ICAM-1-mediated inflammatory responses.
Contact activation takes place in any type of chronic inflammation including atherosclerosis, and is invariably associated with the generation of plasmin.8,9 Plasmin is a potent cell activator that induces significant increases in the cell-surface tissue factor activity in HUVEC. 38 Plasmin is also a potent chemoattractant for ECs. Intact catalytic activity of plasmin and an interaction with the integrin αvβ3 are required for this process. 39 However, it was not known whether plasmin could directly trigger ECs to express the adhesion molecule ICAM-1 on the cell surface. Here we show for the first time that the serine protease plasmin up-regulates the cell adhesion molecule ICAM-1 on the surface of HUVEC. Plasmin cleaves annexin A2, that was indispensable for the plasmin-induced activation of NF-κB and MAP kinases, as well as expression of ICAM.
A similar function was reported for TNF-α, endotoxic LPS, and even high salt intake, which all induce EC activation, resulting in up-regulation of the endothelial ICAM-1 expression.3,40 At the molecular level, it has been previously reported that LPS triggers ICAM-1 expression in ECs through p38 signaling. 41 Salt intake has been shown to control the production of transforming growth factor (TGF)-β1-required activation of p38 in ECs. 40 The TNF-α-induced ICAM-1 expression has been shown to be dependent on the activation of the NF-κB signaling pathway in ECs. 2 The increased expression of ICAM-1 on J774.2 macrophages by endotoxin was shown also to involve the activation of NF-κB. 42 In agreement with these data, our study shows that plasmin induces ICAM-1 gene expression in HUVEC via NF-κB and p38 activation, but not via ERK1/2 activation.
Annexin A2 is a cell-surface plasmin receptor which is a calcium-regulated, phospholipid-binding protein on endothelial cells, macrophages, tumor cells and keratinocytes.19,20 Consistent with our previous studies in monocytes and macrophages,6,22 here we also demonstrate that plasmin activates HUVEC signaling via the cell surface membrane-linked receptor annexin A2t and induces proteolytic cleavage of annexin A2. Annexin A2 is targeted by plasmin through lysine in position 27, 22 which yields a 33-kDa fragment of annexin A2. Because the first 14 amino acids of the N-terminal domain of annexin A2 are required as binding domain for S100A10, 43 cleavage in position 27 inevitably leads to dissociation of the annexin A2t, Newly generated cleavage fragments can act alone or bind to functional molecules such as β2GPI (β2 glycoprotein I) to activate ECs. 44 For example, cleavage fragments could activate known or unknown transmembrane receptors such as the toll-like receptor 4, through which the annexin A2t tetramer was reported to activate human and murine macrophages. 21 The inhibition of annexin A2 expression by siRNA-annexin A2 27 affects the plasmin-induced signaling pathways and ICAM-1 expression in HUVEC. These results suggest that annexin A2 is involved in plasmin -stimulated HUVEC activation and stress the crucial role of the annexin A2 heterotetramer.
Large amounts of plasmin were observed in all layers of human atherosclerotic aorta sections, 29 including the intima which is an EC layer. 45 Our vivo data show that CD31+-ECs are co-localized with the high level expression of annexin A2t, which are known plasmin receptors 22 in human atherosclerotic plaques. Together with our vitro data, it is reasonable to expect occurrence of plasmin-mediated activation of EC in atherosclerotic lesions. Consistent with our vivo data show that CD31+-ECs are co-localized with ICAM-1 and the NF-KB signaling pathway is activated in ECs in human atherosclerotic plaques. Inhibition of NF-KB activation in HUVEC by AKβBA markedly decreased the plasmin-induced production of the adhesion molecule ICAM-1. As an inhibitor of NF-KB (AKβBA) 23 has been shown to affect the development of atherosclerosis in apoE-/- mice treated with LPS to mimic a systemic infection, 46 NF-KB as a target might present a promising strategy for the treatment of human atherosclerosis. We intend to conduct a clinical study to block NF-KB in vivo to find its function in atherogenesis.
Because of the increased plasmin generation in humans (detected as circulating PAP complexes), high levels of PAP were predictive for myocardial infarction in two large clinical studies47,48 and was associated with increased sub-clinical atherosclerosis in a third study, 49 we have performed a study, where we demonstrate that patients with lower limb atherosclerosis have elevated plasmin concentrations and increased plasmin activity compared with controls. These data are consistent with the findings of another group who shows increase of PAP levels from 2.20 nmol/L in the control group to 24.06 nmol/L in patients and a correlation of the increased PAP levels with cardiovascular events. 47 Plasmin as an inducer of ICAM might be partially responsible for the development of the disease.
In conclusion, our data demonstrate that the serine protease plasmin triggers expression of the adhesion molecule ICAM-1 on human ECs via the annexin A2t heterotetramer and several downstream signaling pathways (Figure 6). Increased generation of plasmin in human atherosclerotic lesions in close proximity to the annexin A2t-expressing ECs point to plasmin-mediated activation of ECs and provides a novel link between proteolysis and atherogenesis. Blockage of ICAM-1 expression via functional pathway inhibitors or receptor antagonists could be useful in treating chronic inflammatory diseases, including atherosclerosis. The plasmin receptor – annexin A2 – may harbor therapeutic potential for the treatment of human atherosclerosis.
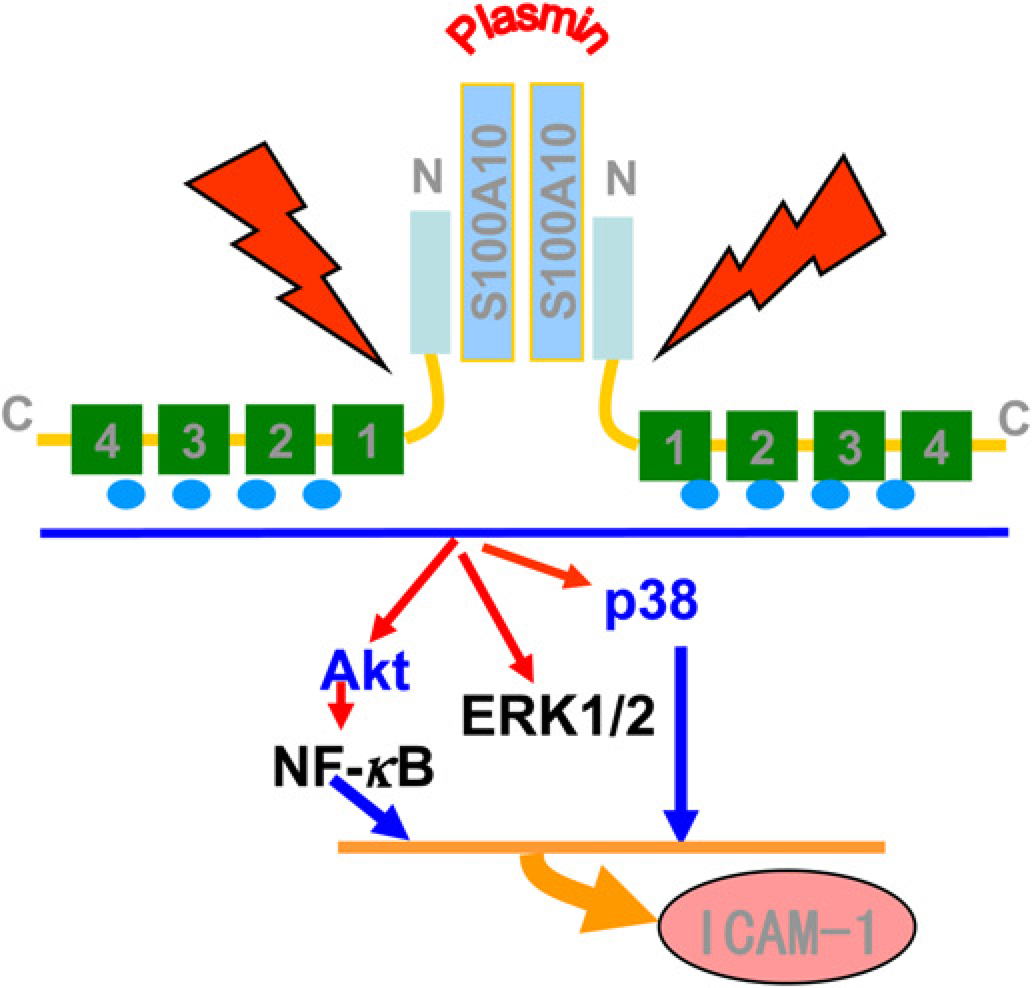
Scheme for the plasmin-induced activation of
Footnotes
Acknowledgements
This research was supported by a grant from the physician funding project of Shanghai Jiao Tong University Medical school (No: 09XJ21053), the Shanghai Scientific Research Innovation Program of Shanghai Education Commission (EC) (No. 11YZ59), the Shanghai Pujiang Program of the Shanghai Science and Technology Committee (No. 10PJ1407300), the German Research Foundation (DFG), the National Key Project for Basic Research (973) (No 2011CB503905), the New Scholars Foundation of Ministry of Education of China (No 20110073120103), and the National Natural Science Foundation of China (No. 81202298).