
Abstract
Fibrosis, a non-physiological wound healing in multiple organs, is associated with end-stage pathological symptoms of a wide variety of vascular injury and inflammation related diseases. In response to chemical, immunological and physical insults, the body’s defense system and matrix synthetic machinery respond to healing the wound and maintain tissue homeostasis. However, uncontrolled wound healing leads to scarring or fibrosis, a pathological condition characterized by excessive synthesis and accumulation of extracellular matrix proteins, loss of tissue homeostasis and organ failure. Understanding the actual cause of pathological wound healing and identification of igniter(s) of fibrogenesis would be helpful to design novel therapeutic approaches to control pathological wound healing and to prevent fibrosis related morbidity and mortality. In this article, we review the significance of a few key cytokines (TGF-β, IFN-γ, IL-10) transcriptional activators (Sp1, Egr-1, Smad3), repressors (Smad7, Fli-1, PPAR-γ, p53, Klotho) and epigenetic modulators (acetyltransferase, methyltransferases, deacetylases, microRNAs) involved in major matrix protein collagen synthesis under pathological stage of wound healing, and the potentiality of these regulators as therapeutic targets for fibrosis treatment. The significance of endothelial to mesenchymal transition (EndMT) and senescence, two newly emerged fields in fibrosis research, has also been discussed.
Keywords
Introduction
Wound healing is an extremely critical and complex biological process by which organisms respond to injury and repair injured tissues in every organ of the body. Tissues can be injured by a wide variety of physical, chemical, immunological and pathological stresses including skin cut, chemical and viral-induced lung injury, alcohol-induced liver injury, high-glucose-induced liver and kidney injury, high blood pressure-induced heart and kidney injury.1,2 In response to injury or stress, a constellation of cellular and molecular events takes place in the wound area, collectively called the wound healing biological process. The wound healing process can be subdivided into three major phases: initial vascular injury associated inflammatory phase characterized by invasion of inflammatory cells in the injured site followed by the second proliferative phase characterized by wound closure, synthesis and deposition of granulation tissue and neovascularization which are essential for physiological wound healing; and third matrix remodeling phase when damaged and granulation tissues are replaced with newly synthesized fibrous matrix protein collagens. 3
Due to vascular injury in the wound area, mononuclear cells and macrophages infiltrate and synthesize different cytokines required for cell migration, activation, differentiation and synthesis of matrix proteins in the wound area. Local and immigrated fibroblasts become activated or undergo differentiation to myofibroblasts and produce required extracellular matrix (ECM) proteins including collagen, fibronectin, elastin and proteoglycans to replace the damage matrix and maintain the tissue homeostasis, a manifestation of scarless physiological wound healing. Physiological wound healing is achieved by an accurate balance between the rate of synthesis of ECM proteins and degradation of matrix proteins by matrix degrading proteolytic enzymes. The rate of synthesis of ECM proteins and degrading enzymes are tightly controlled at the level of transcription. 2 In controlling the physiological level of myofibroblast-synthesized matrix proteins accumulation in the wound area, serine protease urokinase type and tissue type plasminogen activator (uPA/tPA), plasmin and plasmin-dependent matrix metalloproteinase (MMP) play pivotal roles during wound healing. 2 The proteolytic activity of uPA/tPA/Plasmin/plasmin-dependent MMPs are blocked by plasminogen activator inhibitor-1 (PAI-1) and tissue inhibitor of metalloproteinase (TIMP1-4), and thus prevent further degradation of newly synthesized ECM proteins to accelerate wound healing. However, prolonged stress-induced inflammation and sustained cytokine activation of myofibroblasts leads to excessive collagen synthesis and deposition in the wound area and loss of tissue homeostasis, a pathological manifestation of wound healing with scar tissue or fibrosis.1,2,4
Almost ∼45% of total disease-related deaths are associated with abnormal fibroblast activation, fibrosis and organ failure. 5 There is no effective therapy for the treatment of fibrosis, an end stage pathological manifestation (a difficult-to-treat disease state) of a wide variety of diseases including systemic sclerosis (SSc), myocardial infarction (MI), glomerulosclerosis, tubulointerstitial fibrosis, myocardial infarction, chronic hypertension, idiopathic pulmonary fibrosis (IPF), and liver cirrhosis.2,6–11 In order to understand the basis of fibrogenesis, numerous in vitro and in vivo cellular and molecular studies have been undertaken in the last two decades. Using in vitro cell cultures (unstimulated controls, cytokine-induced in vitro model of fibrotic cells as well as cells isolated from fibrotic tissues), investigators dissected out the profibrotic and antifibrotic signaling pathways. Understanding the specific role of different transcriptional activators, coactivators, repressors, corepressors and epigenetic regulators involved in profibrotic and antifibrotic pathways help us to delineate the molecular basis of excessive collagen and other ECM protein synthesis under pathological situations of wound healing or fibrosis.12–15 Eventually, this knowledge on cytokine signaling, transcriptional and epigenetic regulation of matrix protein synthesis will be extremely helpful to design novel therapeutic strategies for treatment of patients with fibrosis. In this review, we discuss the role of a few key well-studied cytokines, transcriptional regulators and epigenetic regulators in myofibroblast differentiation, collagen synthesis (major ECM protein) and fibrogenesis in different organs and their significance in light of potential fibrosis therapy. Additionally, the newly emerged concepts on the link of fibrogenesis with two important biological processes, endothelial to mesenchymal transition (EndMT) and cellular senescence are also briefly discussed in light of therapeutic potentiality.
Key cytokines and growth factors involved in fibrogenesis
Transforming growth factor-beta and fibrosis
Transforming growth factor-beta (TGF-β) belongs to Th1 cytokines and is synthesized by a wide variety of cells including macrophages, mononuclear cells and fibroblasts. Closely related (structurally) TGF-β1, TGF-β2 and TGF-β3 form the TGF-β subfamily and their synthesis is cell type- and context-dependent with unique as well as similar functions. 16 TGF-β is a multifunctional cytokine because of its involvement in a number of biological processes including cellular proliferation, differentiation, migration, wound healing, and immunity.17–20 Therefore, deregulation in TGF-β signaling is associated with a variety of pathological conditions including cancer, inflammation, defective immune system, hypertrophy, and organ fibrosis.21,22 TGF-β is a major player in initiation and progression of fibrogenesis. In response to vascular injury, infiltrated mononuclear cells produce TGF-β and other growth factors in the wound area. As a chemo-attractant, TGF-β attracts neutrophils to the wound site and thus acts as an inflammatory cytokine in the initial stage of wound healing. TGF-β also induces migration of fibroblasts from the vicinity of wounds, and fibroblast to myofibroblasts differentiation. TGF-β-activated fibroblasts or differentiated myofibroblasts are the major cell-type that synthesizes collagen and other ECM proteins to heal the damaged tissues.1,4,22 However, sustained activation of myofibroblasts, due to chronic inflammation and TGF-β signaling, leads to the development of fibrosis and eventually organ failure.23,24
The unequivocal evidence of TGF-β-essentiality in pathological hypertrophy and fibrogenesis comes from a classical study by Schultz et al. 25 who demonstrated that while a suppressor dose of AngiotensinII is able to induce cardiac hypertrophy and cardiac fibrosis in wildtype mice, AngiotensinII fails to induce hypertrophy and fibrosis in TGF-β1 null mice in a immunocompromised background indicating the pivotal role of TGF-β in fibrogenesis. As TGF-β plays a pivotal role in fibrogenesis, it was originally thought that antibody neutralization of elevated TGF-β would be an effective therapeutic approach to control fibrogenesis. However, systemic TGF-β depletion will not be an ideal approach to halt fibrogenesis because TGF-β plays a crucial role in a number of important biological processes including cellular growth control and immunity. The outcome of a few clinical trials to treat SSc patients with TGF-β neutralizing antibody has been summarized recently. 14 As disruption of TGF-β signaling at the receptor level will be detrimental, it is important to identify downstream modulators which are involved in hyperactivation of TGF-β signaling and excessive collagen synthesis; controlling the activity of such modulators of profibrotic TGF-β signaling will be an ideal approach to controlling fibrosis without blocking other important TGF-β-regulated biological processes.
In pre-Smad era, several studies demonstrated the important roles of transcription factors including AP1 and Sp1 in the regulation of TGF-β-induced collagen synthesis in vitro and its significance in fibrosis.12,26,27 Over 15 years ago, discovery of mammalian Smad (C. elegans homolog of Sma and Drosophila homolog Mad) as the only known direct substrate of TGF-β receptor serine threonine kinase was a breakthrough in the field of TGF-β signaling research related to a wide variety of physiological and pathological cellular processes. Active TGF-β transduces its signal from cell surface to nucleus via canonical Smad-dependent pathway as well as non-canonical pathways including MAP kinase and PI3K/Akt/PKB kinase pathway. Upon binding to TGF-β constitutively active TGF-β receptor II transphosphorylates TGF-β receptor I and forms active TGF-β receptor I and II complex on the cell surface and phosphorylates the serine residues at SSXS motif of cytoplasmic Smad2 and Smad3. The phosphorylated active Smad2/Smad3 heterodimerize with Co-Smad Smad4 and translocate to the nucleus where Smads interact with Smad binding element (SBE) and also recruit p300 to the transcriptional complex of target gene. On the other hand, inhibitory Smad7 is also activated by Smad-dependent TGF-β signaling in a time-dependent manner and cytoplasmic Smad7 interacts with TGF-βR and blocks further Smad2/3 phosphorylation, thus controlling the magnitude of TGF-β-mediated signaling in a negative feedback loop.28–33 Overexpressed Smad2 and Smad3 stimulate collagen gene transcription in human dermal fibroblasts and potentiate TGF-β-induced collagen gene transcription in vitro. In contrast overexpressed Smad7 abrogates TGF-β-induced collagen gene transcription indicating TGF-β-induced collagen gene transcription is Smad-dependent and Smad7 is a potent negative regulator of TGF-β-induced profibrogenic responses.34,35 For Smad-induced collagen gene transcription, the physical and functional interaction of Smad complex with acetyltransferase p300 is essential.36–40 Interaction of Smad complex with Sp1 is also implicated in maximal stimulation TGF-β-induced collagen gene transcription.
41
TGF-β-induced non-Smad pathways including ERK1/2 MAPK, p38 MAPK and PI3K/Akt/PKB pathways also activate collagen synthesis independently or in coordination with TGF-β-activated Smads36,42 (Figure 1).
Schematic diagram showing TGF-β-signaling pathway and its link with major profibrogenic signaling network. TGF-β controls the activity and expression of major activators (red), repressors (blue) and epigenetic regulators (green) of profibrogenic signaling. This network is required for physiological wound healing. Deregulation in this network leads to fibrogenesis. (A color version of this figure is available in the online journal)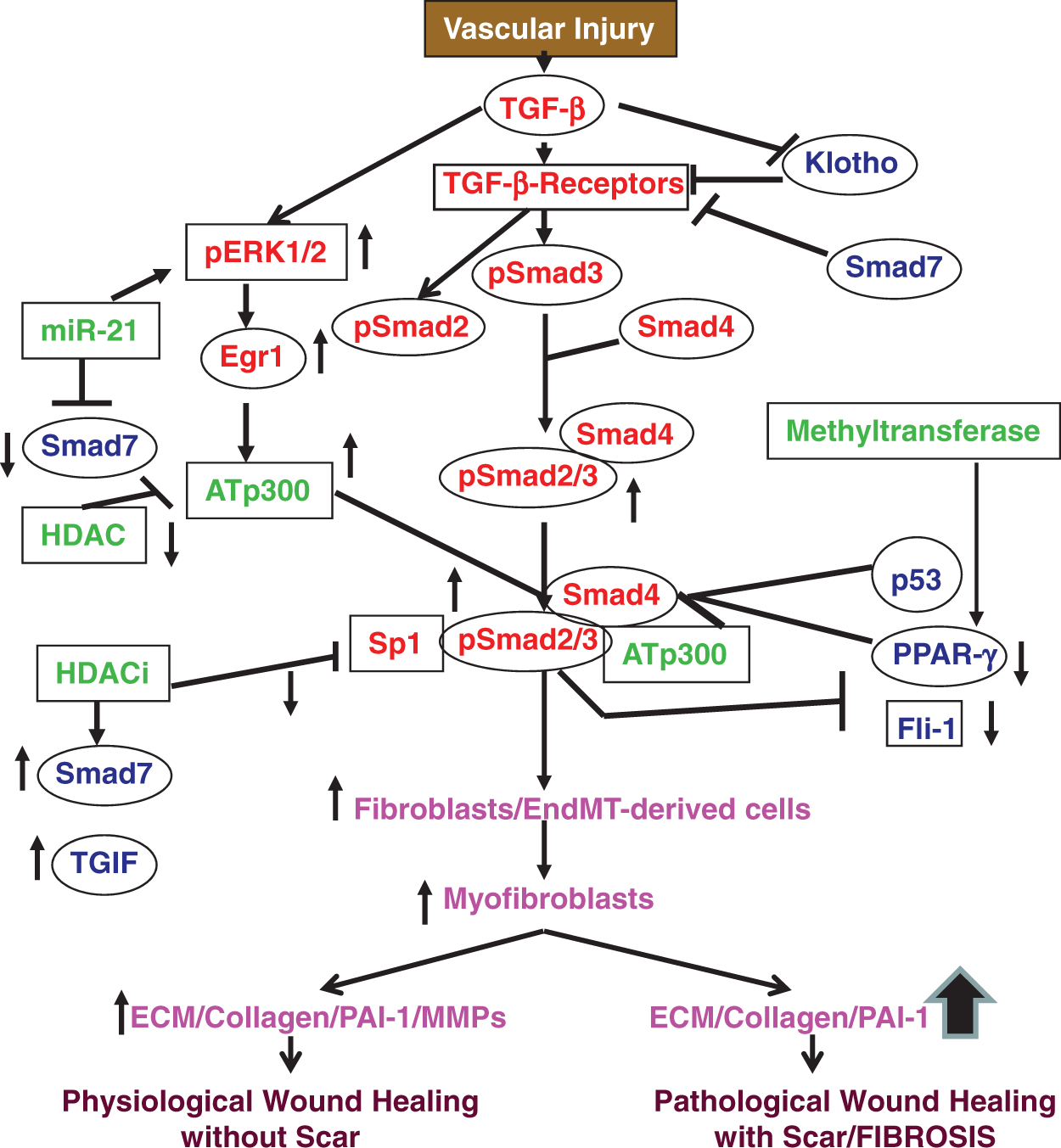
The significance of Smads in fibrogenesis is also evidenced by several in vivo studies using animal models of organ fibrosis. For example, while bleomycin treatment leads to skin and lung fibrosis in wildtype mice, Smad3 knockout mice are partially and completely protected from bleomycin-induced skin and lung fibrosis respectively.43,44 Interestingly, the Smad3 deficiency-associated protection of skin and lung from bleomycin-induced fibrosis is independent of inflammation because in both cases Smad3 deficiency fails to prevent bleomycin-induced early inflammation. Therefore, anti-inflammatory drugs are not suitable for treatment of already initiated fibrogenesis. The results from these in vivo studies suggest that Smad3 is required for fibroblast activation, myofibroblast differentiation and increased collagen synthesis in response to profibrotic responses.43,44 Most importantly, TGF-β production is not affected by Smad3 deficiency indicating that targeting Smad3 is a viable approach to controlling fibrosis without blocking global activity of TGF-β. Furthermore, adenovirus-mediated administration of a mutant form of Smad4 significantly inhibits scar formation in rat indicating suppression of increased Smad signaling is effective in prevention of scar formation during wound healing. 45 The essential role of Smads in fibrogenesis has also been evidenced in the UUO-induced mouse model of renal fibrosis where deletion of Smad4 is associated with decreased renal fibrosis. Like lung and skin, disruption of Smad4 is also associated with renal inflammation. 46 Similarly, Smad3 deficient mice are resistant to develop liver fibrosis in response to dimethylnitrosamine indicating that Smad3 deficiency protects mice from induced liver fibrogenesis. 47 Most importantly, skin fibroblasts derived from scleroderma patients show constitutively active nuclear localized Smad3.48,49 Additionally, spontaneous cardiac fibrosis in aged PAI-1 deficient mice is associated with constitutively active pSmad2 and pSmad3 in myocardial tissue and elevated collagen accumulation 23 suggesting activated Smad axis is involved in controlling fibrogenesis in human patients with skin fibrosis and in mouse models of skin, heart, lung and liver fibrosis. Collectively, these results implicate Smads in the pathogenesis of fibrosis. Selective suppression of Smad3 or Smad4, but not complete depletion, may be a viable therapeutic approach to control fibrosis. However, caution should be taken before such therapy because complete inactivation of Smad3/4 is associated with a wide variety of human cancers. 50
Interleukin-10 and fibrosis
IL-10, an 18 kDa protein synthesized by monocytes and lymphocytes (Th2 cells), is an anti-inflammatory and immunosuppressive cytokine. IL-10 transduces its signal via activation of JAK-STAT3, p38 MAPK and PI3-kinase pathways. 51 Importantly, IL-10 is a known antifibrotic cytokine and exogenous IL-10 significantly lowers fibrogenesis in different tissues including liver,52–55 lung,56,57 kidney, 58 and heart. 59 Deficiency of cellular S-adenosylmethionine (S-AdoMet), a metabolically active form of methionine that methylates DNA, RNA, and proteins, in liver is a salient feature of toxin-induced liver injury and decreased level of IL-10 has been reported in alcoholic cirrhosis. As exogenous S-AdoMet augments IL-10 synthesis, it has been inferred that anti-inflammatory and hepatoprotective activity of S-AdoMet may be partly due to increased IL-10 production. 52 Therefore, induction of IL-10 using specific inducer like S-AdoMet would be a viable approach to control fibrosis in liver. Furthermore, administration of IL-10 blocks CCl4-induced expression of profibrotic TGF-β, MMP-2, and TIMP and thus ameliorates CCl4-induced rat liver fibrosis. 54 The antifibrotic role of IL-10 has been further confirmed by the observations that hepatocyte-specific rat IL-10 expressing transgenic mice develop less liver fibrosis in response to CCl4 and thioacetamide treatment. As the number of CD8 + T cell decrease significantly in IL-10 expressing transgenic mice compared to wildtype mice, authors concluded that IL-10-mediated antifibrotic effect may be due to decreased CD8 + T cells. 53 In a recent study, Suh YG et al. 60 demonstrate that bone marrow cells infusion ameliorates CCl-4- induced mouse liver fibrosis via IL-10 because bone marrow cells derived from IL-10 knockout mice fails to ameliorate liver fibrosis. Furthermore, administration of inactivated Orf virus (ORFV, a member of Pox virus family and an immunomodulator) into pig-serum-induced or CCl4-induced liver fibrosis rat models abrogates liver fibrosis and also reverses established liver fibrosis via induction of IL-10 and IFN-γ 55 clearly indicating that IL-10 may be a potential therapeutic target for fibrosis treatment.
Matrix metalloproteinase-8 (MMP-8) cleaves IL-10 and thus the level of active IL-10 in a particular cellular environment depends on the level of active MMP-8. Interestingly, MMP-8 knockout mice are partially resistant to develop lung fibrosis in response to bleomycin compared to wildtype mice, and the level of full length active IL-10 is higher in MMP-8 knockout lung compared to wildtype indicating MMP-8-deficiency-associated reduced lung fibrosis may be due to increased level of active IL-10. 56 This notion has been further supported by in vitro experiments where unlike wildtype lung fibroblasts, bleomycin fails to stimulate collagen synthesis in MMP-8 knockout lung fibroblasts with elevated IL-10 and phospho-STAT3. Furthermore, depletion of IL-10 using IL-10 antibody in MMP-8 knockout cells increases bleomycin-induced collagen synthesis strongly indicating that MMP-8 deficiency-associated decreased profibrotic responses are mediated via antifibrotic IL-10. 56 Furthermore, administration of methyl-palmitate, a fatty acid ester, ameliorates bleomycin-induced excessive collagen deposition and decreases NF-κB-activation in a mouse model of lung fibrosis possibly via induction of IL-10 level indicating the therapeutic potentiality of IL-10 for fibrosis treatment. 57 Antifibrotic activity of IL-10 has also been evidenced in renal fibrosis. Intravenous injection of recombinant human IL-10 in rats with anti-thymocyte antibody-induced glomerulosclerosis ameliorates intraglomerular inflammation and fibrosis. 61 Additionally, spleen-derived IL-10 is known to prevent obesity-induced inflammation and fibrosis in kidney. Splenectomy aggravates obesity-induced inflammation and fibrosis in the glomerulus, and IL-10 treatment decreases high-fat diet mediated obesity-induced inflammation and fibrosis in glomerulus indicating a potent antifibrotic nature of IL-10. 58 Similarly, IL-10 knockout mice are more susceptible to induced myocardial fibrosis and left ventricular dysfunction compared to wildtype mice in response to isoproterenol (ISO) and transverse aortic constriction (TAC). 59 Most importantly, systemic administration of recombinant mouse IL-10 reduces ISO-induced and TAC-induced myocardial fibrosis and improves left ventricular function. IL-10-mediated suppression of ISO-induced myocardial fibrosis may be due to suppression of NF-κB-induced inflammatory pathway and IL-10-mediated suppression of NF-κB is STAT3-dependent. 59 Taken together, it is reasonable to conclude that IL-10 protects different organs from severity of fibrosis. Therefore, selective induction of IL-10 or recombinant IL-10 may be an attractive therapeutic approach for the treatment of fibrosis. However, determination of dose and the duration of IL-10 therapy for treatment of fibrosis are crucial because long term therapy by high dose IL-10 may increase inflammation and fibrogenesis as has been hinted upon by Sun et al. 62 Additionally, the serum level of IL-10 is significantly higher in SSc patients and positively correlated with disease severity.63,64
Interferon-gamma and fibrosis
Interferon gamma (IFN-γ), mostly synthesized by T-cells and NK cells, belongs to class II Interferon and is involved in regulation of early immune responses. IFN-γ transduces its signal via activation of JAK-STAT pathway where JAK phosphorylates Stat-1α· Activated Stats dimerize and translocate to the nucleus and activate or repress IFN-γ responsive genes. 65 IFN-γ is a potent inhibitor of collagen gene transcription and blocks TGF-β-induced collagen synthesis in vitro37,66–68 implicating the significance of IFN-γ as an antifibrotic cytokine. Several molecular mechanisms have been reported for antagonistic action of IFN-γ on TGF-β-induced profibrotic responses. For example, IFN-γ antagonizes TGF-β-induced collagen synthesis via activation of JAK-Stat-1α pathway where activated Stat-1α competes with TGF-β-activated pSmad2/3 for limiting amount of p300, an essential factor of induced collagen synthesis. Activated Stat-1α sequesters p300 from activated Smad complex and thus abrogates TGF-β-induced collagen synthesis. 37 Later Dooley et al. 69 demonstrated that IFN-γ activates Y-box binding protein-1 (YB-1) which inhibits collagen synthesis either by direct interaction with collagen gene promoter or by sequestrating p300 from active Smad complex and via activation of inhibitory Smad, Smad7. 69 However, IFN-γ is able to inhibit collagen gene transcription in Smad7-depleted dermal fibroblasts indicating Smad7 is not involved in IFN-γ-induced suppression of basal as well as TGF-β-induced collagen synthesis in dermal fibroblasts. 37 Furthermore, Interleukin-18 (IL-18)-mediated inhibition of basal as well as TGF-β-induced collagen gene transcription is via induction of IFN-γ and suppression of TGF-β in normal and SSc dermal fibroblasts. Furthermore, IL-18-mediated inhibition of collagen synthesis is IFN-γ-dependent and through induction of Ets-1 and ERK1/2 MAPK pathway. 70 Involvement of IFN-γ-induced ERK1/2 MAPK in IFN-γ-mediated suppression of collagen synthesis is consistent with the previous observation that IFN-γ-induced ERK1/2 mediated phosphorylation of C/EBPβ suppresses collagen synthesis in dermal fibroblasts. 71 Similarly, IFN-γ also antagonizes TGF-β-induced α-SMA, fibronectin and PAI-1 expression in renal tubular epithelial cells via activation of ERK1/2 MAPK and Stat3 signal transduction pathways. 72 In addition, IFN-γ is also involved in HGF-mediated suppression of collagen synthesis in SSc fibroblasts indicating IFN-γ is a potent inhibitor of profibrotic responses. 73
The antifibrotic activity of IFN-γ has also been well documented by various in vivo studies using animal models of organ fibrosis. Bleomycin instillation in mice significantly increases the level of profibrotic TGF-β and matrix protein collagen in lungs. However, cotreatment with IFN-γ reduces bleomycin-induced TGF-β and collagen synthesis indicating a potential therapeutic value of IFN-γ for treatment of lung fibrosis. 74 The therapeutic efficacy of IFN-γ has also been tested in silica-induced experimental rat model of lung fibrosis. IFN-γ treatment significantly reduces the silica-induced elevated levels of TGF-β, IL-4 and collagen synthesis suggesting IFN-γ ameliorates lung fibrosis via suppression of profibrotic cytokines. 75 Furthermore, IL-12 mediated amelioration of bleomycin-induced lung fibrosis in mice is also due to induction of IFN-γ as evidenced by the observations that i) IL-12-induces the level of IFN-γ, and ii) administration of IFN-γ antibody blunts the antifibrotic activity of IL-12 in this murine model of lung fibrosis. 76 Additionally, the antifibrotic effect of IFN-γ has been pre-clinically tested in two animal models of liver fibrosis (150 rats with CCl4-induced liver fibrosis and 196 rats with dimethylnitrosamine-induced liver fibrosis) and also in 47 patients with Hepatitis B virus-induced liver fibrosis. 77 IFN-γ effectively ameliorates liver fibrosis in animal models and patients with liver fibrosis. According to this clinical study, a higher dose of IFN-γ is more effective for fibrosis treatment. The effects of low and high doses of IFN-γ have also been tested in post-lamenectomy peridural fibrosis, a common pathological state in post lumber surgery. Under this setting, low dose of IFN-γ is more effective and safe for treatment of peridural fibrosis in rats. In contrast, high dose of IFN-γ increases the number of inflammatory cells compared to controls and low dose IFN-γ group. 78 Taken together the results of these in vitro and in vivo studies, it is apparent that IFN-γ has potentiality as a drug for fibrosis therapy. However, determination of an efficient dose of IFN-γ using a large population of animals with induced organ fibrosis is needed for testing efficacy and toxicity of IFN-γ before clinical trials in humans because effect of low versus high dose of IFN-γ for treatment of fibrosis is controversial. It is important to mention that at present, the outcomes of IFN-γ-based clinical trials are controversial. While the results of some clinical trials suggest that IFN-γ therapy has a modest beneficial effect on survival and amelioration of tissue fibrosis in patients with dermal fibrosis, chronic hepatitis-C virus-induced liver fibrosis and idiopathic pulmonary fibrosis (IPF), the outcomes of several other IFN-γ-based clinical trials for fibrosis therapy are negative (Reviewed in Ref. 79).
Key transcription factors involved in fibrogenesis: potentiality as therapeutic targets
Eukaryotic gene expression is largely controlled at the level of transcription and post transcriptional levels. Large numbers of transcription factors are involved in the regulation of expression of a specific gene in a cell type-, organ specific-, physiological condition- and developmental stage-specific manner. Involvement of these factors in different signal transduction pathways determines the level of expression of a specific gene.80,81 Therefore, expression level of a transcription factor and its activity (modulated by phosphorylation, acetylation, and methylation) are critical for the physiological level of expression of a specific target gene. Abnormality in transcription factors is associated with a number of human diseases. Like many other pathological conditions, fibrosis is also associated with abnormality in activity of transcription factors and coactivarors. Excessive synthesis and abnormal accumulation of Type I collagen, the major ECM protein, in different tissues is the hallmark of organ fibrosis. The expression of Type I collagen genes (COL1A1 and COL1A2) are tightly and coordinately regulated at the level of transcription by more than a dozen transcription factors and coactivators in a context dependent manner.12,15 Here, we update the status of a few key transcriptional activators, repressors, coactivators and epigenetic regulators of collagen gene expression which are altered in fibrotic tissues and have potentiality as therapeutic targets for fibrosis treatment.
Activators of matrix protein synthesis with therapeutic potentiality
Sp1 and fibrosis
The Specificity protein-1 (Sp1), a 95–105 kDa protein, belongs to the zinc finger family transcription factor and is involved in selection of a wide variety of RNA Polymerase II gene promoters with GC-box and stimulation of transcription.82–84 While Sp1 is involved in stimulation of Type I collagen gene expression in fibroblasts,26,85–87 the expression levels of collagen and several other extracellular matrix protein genes are significantly reduced in Sp-1-depleted fibroblasts. Furthermore, decoy Sp1 oligonucleotide inhibits collagen gene transcription in vivo in mouse skin indicating Sp1 is an ideal target to reduce collagen and other ECM synthesis and may be useful for fibrosis therapy. 88 It is known that proteosome inhibitors reduce Type I collagen synthesis and stimulate MMP-1 synthesis. For example, proteosome inhibitor Bortezomib or Velcade, an FDA approved drug for multiple myeloma treatment, suppresses basal and TGF-β-induced collagen gene transcription and activates MMP-1 expression. While suppression of collagen gene transcription by Bortezomib is due to blocking Sp1 binding to collagen gene promoters without affecting Smad-signaling, activation of MMP-1 transcription is due to Bortezomib-induced binding of AP-1 to MMP-1 promoter, suggesting proteosome inhibitor imparts its dual antifibrotic action via suppression of Sp1-dependent matrix protein synthesis and induction of AP-1-dependent matrix protein degrading enzyme MMP-1 synthesis. 89 Most importantly, Sp1 binding activity to collagen I gene promoter is positively correlated with the increased level of collagen mRNA expression in SSc fibroblasts 90 further implicating Sp1 in fibrogenesis.
The role of Sp1 in induced extracellular matrix protein synthesis in kidney and the significance of Sp1-depletion in attenuation of kidney fibrosis have been documented by several in vitro and in vivo studies. Chae et al. 91 reported that ring Sp1 decoy oligonucleotide inhibits serum-induced proliferation of kidney fibroblasts and synthesis of TGF-β and fibronectin in vitro. Most importantly, administration of ring Sp1 oligonucleotide blunts UUO-induced ECM synthesis and renal fibrosis. As ring-Sp1 decoy oligonucleotides are more resistant to nuclease-mediated degradation compared to phosphorothioted double stranded Sp-1 decoy, using ring Sp1 decoy oligonucleotide may be a fruitful approach to control fibrosis. 91 Elevated level of Sp1 expression in glomerulus and proximal tubule is reported in different forms of glomerulonephritis patients compared to healthy controls 92 indicating a probable link of elevated Sp1 activity and human renal fibrosis. Using chimeric decoy oligonucleotide containing NF-kB and Sp1-binding sites, Kim et al. 93 demonstrated that chimeric decoy oligonucleotide significantly ameliorated UUO-induced inflammation and tubulointerstitial fibrosis in mice suggesting suppression of Sp1 activity may reduce profibrogenic processes and renal fibrosis. 93 Furthermore, AngII-induced increased inflammation and worse renal fibrosis in Smad7 knockout mice is also associated with significant upregulation of Sp1. 94 Together, these results suggest Sp1 is a potent positive regulator of fibrogenesis and targeting Sp1 will be an ideal approach to control renal fibrosis.
The profibrotic role of Sp1 in liver fibrosis has also been tested in vivo using animal models. Injection of ring-Sp1 decoy oligonucleotide through tail vein in CCl4-induced liver fibrosis model decreases the levels of TGF-β and extracellular matrix proteins and ameliorates liver cirrhosis indicating the profibrotic role of Sp1 in liver fibrosis. 95 Furthermore, transfected decoy Sp1 oligonucleotide blunts Sp1 binding to profibrotic cytokine, α-SMA and collagen gene promoters and prevents their transcription and profibrotic responses. 96 The leptin-induced stimulation of collagen gene transcription is also due to Sp1 activation by phosphorylation, and its increased binding to collagen gene promoter in rat HSC further indicating the profibrogenic role of Sp1. 97
Sp1 is also involved in chemokine CCL-18-stimulated collagen synthesis in human lung fibroblasts. CCL-18 chemokine induces Sp1 phosphorylation, DNA binding activity and target gene expression in human lung fibroblasts without altering TGF-β production and its activation. Interestingly, CCL-18-stimulated collagen synthesis is not affected by neutralizing TGF-β antibody or ALK5 inhibitor SB431542, a potent inhibitor of TGF-β receptor I kinase. However, overexpression of dominant mutant Sp1 blunts CCL-18-induced collagen synthesis 98 strongly indicating Sp1 plays a significant role in CCL-18-induced collagen synthesis. Like in kidney and liver, ring Sp1 decoy oligonucleotide blocks bleomycin-induced TGF-β, MMP13 and fibronectin synthesis in lungs further indicating that Sp1 is profibrotic factor and an important target to control lung fibrosis. 99 Sp1 also contributes in myocardin related transcription factor A (MRTF-A)-induced collagen gene transcription in lung fibroblasts. 100 Collectively, the results of all these in vitro and in vivo studies firmly suggest that Sp1 is an important profibrotic factor and targeting Sp1 may be a suitable approach to control fibrosis in multiple organs.
Early growth response-1 and fibrosis
Early growth response-1 (Egr-1) gene codes for a zinc-finger transcription factor of 80-82 kDa protein and belongs to a family of five members (Egr1-4, and NGF1-B). Egr-1 controls target gene expression via direct interaction with specific DNA sequences as well as interaction with transcriptional regulators on transcriptional complex. Egr-1 directly interacts with acetyltransferase p300/CBP and augments transcription of target genes, 101 and it is involved in a number of biological processes including cellular growth, proliferation, differentiation and matrix remodelling. 101 Importantly, Egr-1 stimulates expression of acetyltransferase p300, an essential profibrotic regulator. Egr-1 binding to the p300 promoter is essential for this stimulation in skin fibroblasts indicating Egr-1 and p300 are interdependent for driving important biological processes. 102 In response to vascular injury, Egr-1 may activate matrix remodeling genes and profibrotic genes such as TGF-β and PDGF and contribute to pathogenesis of vascular and matrix remodeling. 103 In a lung ischemia/reperfusion (I/R) mouse model, I/R-induced elevated Egr-1 level is associated with pathological tissue remodeling or fibrosis. 103 Furthermore, the cardioprotective effect of calcium channel blockers verapamil, diltiazem and nifedipine on ischemia-reperfusion (I/R)-induced cardiac tissue injury in rats may be due to calcium channel blocker-mediated downregulation of I/R-induced profibrotic Egr-1 mRNA and protein. 104 Similarly, the cardioprotective effect of N-butyl haloperidol iodide on I/R-induced myocardial damage is also due to inhibition of Egr-1 expression. 105
In the UUO model of kidney fibrosis, while the elevated level of Egr-1 protein is associated with increased myofibroblasts differentiation, DNA enzyme mediated depletion of Egr-1 blunts myofibroblasts differentiation and increased collagen synthesis in obstructed kidneys indicating Egr-1 is a promising target for renal fibrosis therapy. 106 Using inducible TGF-β transgenic mice in the presence and absence of Egr1, Lee et al. 107 demonstrate that Egr1 is required for TGF-β-induced lung epithelial cell apoptosis and lung fibrosis. Additionally, this study also demonstrates that TGF-β-induced apoptosis is required for lung fibrosis in this murine model of lung fibrosis. 107 Interaction of Egr-1 with the collagen gene promoter binding site is essential for stimulation of collagen gene expression in dermal fibroblasts. TGF-β fails to stimulate collagen synthesis in Egr-1 knockout mouse embryonic fibroblasts 108 indicating an essential role of Egr-1 in TGF-β-induced collagen synthesis. Furthermore, while Egr-1 knockout mice are resistant to bleomycin-induced skin and lung fibrosis, fibroblast-specific Egr-1 overexpressing mice show more collagen deposition in the wound bed compared to wildtype controls in response to incisional wound 109 clearly indicating Egr-1 is a potent profibrotic transcription factor and contributor in skin fibrogenesis. Importantly, Egr-1 level is elevated in SSc fibrotic skin. Therefore, targeting Egr-1 in fibrotic tissues may be a potent therapeutic approach to controlling fibrosis.
Together these findings suggest that Egr-1 is a key profibrotic factor for matrix remodeling in skin, lung and kidney. In contrast to these findings, Pritchard and Nagy 110 reported that Egr-1 deficiency is associated with augmentation of HSC activation in a CCl4-induced model of liver fibrosis compared to controls. The levels of collagen, α-SMA and fibrosis are significantly higher in CCl4-exposed Egr-1 knockout liver compared to CCl4-exposed wildtype liver indicating Egr-1 is hepatoprotective rather than a promoter of liver fibrogenesis. Further studies from the same investigators show that Egr-1-mediated hepatoprotection in response to CCl4-mediated liver injury is due at least in part via induction of TNF-α that is required for timely cell cycle progression of hepatocytes exposed to CCl4. In contrast, the level of TNF-α is decreased in Egr-1 knockout mice and thus worsens liver injury after exposure to CCl4. 111 Similarly, Egr-1 expression is significantly elevated in liver of wildtype mice fed with ANIT (alpha-naphthylisothiocyanate) diet, a bile duct epithelial cell and liver toxicant. Interestingly, the extent of tissue fibrosis in Egr-1 knockout mice is higher compared to wildtype mice fed with ANIT diet, indicating that elevated Egr-1 is not a contributor to liver fibrosis in wildtype mice in response to ANIT diet. Egr-1 deficiency is also associated with increased expression of β6-integrin that is involved in TGF-β activation. Therefore, Egr-1 deficiency is not protective from liver fibrogenesis, rather it worsens liver fibrosis and thus Egr-1 is an antifibrotic transcription factor in this context. 112 Based on these observations, it is reasonable to conclude that Egr-1 plays a significant role in fibrogenesis in most of the organs except liver (under the described experimental settings) as a potent profibrotic transcription factor, and thus selective suppression of Egr-1 in those fibrotic tissues may be a viable approach to control fibrosis. However, systemic downregulation of Egr-1 may not be an ideal approach as it may affect the liver’s protective mechanism from fibrogenesis in response to toxicants and other profibrotic signals because Egr-1 is an important negative regulator of hepatic fibrosis.
Repressors of matrix protein synthesis with therapeutic potentiality
Anti-Smad7 and fibrosis
The inhibitory Smad7, a 46 kDa protein, is a negative regulator of Smad-dependent TGF-β signaling. Smad7 is regulated by TGF-β in a time-dependent manner and controls the magnitude of TGF-β signaling via direct interaction with TGF-β activated TβR and blocking further phosphorylation of R-Smads Smad2 and Smad3. While overexpressed Smad7 inhibits TGF-β-induced collagen gene transcription, 34 the level of both basal and TGF-β-induced collagen gene transcription are higher in Smad7-depleted skin fibroblasts 37 suggesting the role of Smad7 as a negative regulator of collagen synthesis. Thus under the normal wound healing process, Smad7 controls the magnitude of TGF-β-induced collagen synthesis and deregulation of Smad7 may impair physiological wound healing. Adenovirus expression of Smad7 blunts bleomycin-induced Type I collagen synthesis and lung fibrosis via suppression of bleomycin-induced Smad2/3 phosphorylation in vivo suggesting activation of Smad7 may be a viable approach to control injury-related lung fibrosis. 113 Similarly, adenovirus-mediated overexpression of Smad7 blunts bile duct ligation (BDL)-induced collagen synthesis and liver fibrosis in rats. 114 Furthermore, Asiatic acid blunts CCl4-induced HSC activation, myofibroblast differentiation, liver damage and liver fibrosis in rats and antiproliferative activity of Asiatic acid on CCl4-induced liver fibrosis is associated with increased expression of Smad7 protein in vivo. While Asiatic acid stimulates Smad7 and prevents TGF-β-induced Smad2/3 activation in HSC-T6 cells, in Smad7 deficient cells Asiatic acid fails to block activation clearly indicating that Asiatic acid exerts its antifibrotic effect via activation of antifibrotic Smad7. Thus activation of Smad7 may be an attractive approach to control injury related liver fibrosis. 115
The antifibrotic role of Smad7 in liver fibrosis has also been demonstrated in transgenic mice where hepatocyte-specific overexpression of Smad7 improves CCl4-induced liver fibrosis. 116 Along with Smad7, coexpression of uPA significantly blocks CCl4-induced myofibroblast differentiation and liver fibrosis more efficiently further indicating activation of Smad7 is a viable approach to control liver fibrosis. 117 The increased antifibrotic efficiency may be due to increased uPA and plasmin-dependent MMP activation and increased ECM degradation in the presence of uPA as well as direct suppression of collagen gene transcription by excess negative regulator Smad7. Additionally, Curcumin-induced elevated Smad7 has also been implicated in Curcumin-mediated amelioration of CCl4-induced liver fibrosis. 118 Importantly, decreased level of Smad7 has been reported in different fibrotic tissues. For example, while the level of phosphoSmad2/3 and PAI-1 are elevated in SSc skin and in explanted SSc fibroblasts, the level of Smad7 is significantly decreased indicating a negative correlation of Smad7 with fibrogenesis. Furthermore, adenovirus-mediated overexpression of Smad7 decreases elevated Smad signaling and PAI-1 expression in SSc fibroblasts clearly indicating the negative role of Smad7 in fibrogenesis. 119 The decreased levels of Smad7 in SSc skin may be due to elevated levels of profibrotic microRNA-21 in SSc skin because Smad7 is a direct target of microRNA-21. 120 The level of Smad7 is also decreased in a UUO-induced rat model of renal fibrosis compared to controls.121,122 Additionally, the reduction of Smad7 in fibrotic kidney may be due to increased degradation and ubiquitination activity of Smad7, which is increased in UUO kidney. 122 Using ultrasound-mediated inducible Smad7 gene transfer, two independent studies show that ultrasound-induced kidney specific overexpressed Smad7 completely blocks UUO-induced Smad2/3 activation, myofibroblast differentiation and tubulointerstitial fibrosis indicating that Smad7 delivery using this safe and effective procedure (ultrasound-mediated organ specific inducible system) may have new therapeutic potentiality for fibrosis treatment.121,123,124
Recently, Chung et al. 125 demonstrated a novel molecular mechanism of overexpressed Smad7-mediated amelioration of renal fibrosis. In the UUO model of renal fibrotic tissue, while the levels of antifibrotic microRNA miR-29b (target collagen directly) are significantly decreased, the expression of profibrotic microRNA miR-21 and miR-192 are increased. In contrast, in ultrasound-mediated Smad7 overexpressing kidney, the levels of miR-29b are increased and miR-21 and miR-192 are decreased suggesting overexpressed Smad7 may block renal fibrosis via activation of antifibrotic microRNA and repression of profibrotic microRNAs. 125 It is important to note that Smad7 mRNA is a direct target of profibrotic miR-21. 120 Additionally, in Smad7 knockout mice, UUO further downregulates miR-29b and stimulates the expression levels of miR-21 and miR-192 and thus promotes renal fibrosis. In vitro data reveals miR-29b, miR-21 and miR-192 are downstream of Smad7 activity.125,126 Antifibrotic activity of Smad7 has also been documented in the heart where postconditioning decreases myofibroblast differentiation, collagen synthesis and fibrosis in rat myocardium after ischemia/reperfusion and improves cardiac function. Interestingly, postconditioning-mediated reduction of myocardial fibrosis is also associated with increased levels of Smad7. 127 Similarly, Curcumin-mediated inhibition of myofibroblast differentiation, elevated collagen synthesis and fibrosis in rat infarcted myocardium, is associated with elevated levels of Smad7 and decreased activation of Smad2/3 suggesting Curcumin-mediated improvement of infarcted heart may be through anti-Smad7. 128 Collectively, these in vitro and in vivo results strongly suggest that Smad7 is a potential drug target for effective treatment of fibrosis in different organs.
Fli-1 and fibrosis
Friend leukemia integration-1 (Fli-1), a 52 kDa protein, belongs to the Ets- protein family and is a negative regulator of collagen gene transcription. While Ets family protein Ets-1 stimulates collagen gene expression, Fli-1 abrogates that stimulation in human dermal fibroblasts indicating Fli-1 is a negative regulator of collagen synthesis. 129 In mouse embryonic fibroblasts derived from Fli-1+/+, Fli-1+/− and Fli-1−/− mice, the level of Fli-1 is inversely related to the expression level of Type I collagen. Furthermore, the level of Fli-1 is significantly lower in SSc fibrotic skin and explanted SSc fibroblasts compared to healthy controls clearly indicating a reduced level of Fli-1 may be responsible for elevated collagen synthesis and accumulation in SSc fibrotic skin. 130 Furthermore, the level of collagen mRNA and protein are significantly upregulated in skin derived from mice with mutant Fli-1 compared to wildtype controls further confirming Fli-1 is a negative regulator of profibrogenic responses and Fli-1 is required to maintain the dermal tissue homeostasis. 131 As connective tissue growth factor (CTGF), a downstream factor of TGF-β, plays a significant role in fibrogenesis132,133 and CTGF is also regulated by Ets-1 and Fli-1 in a positive and negative manner respectively, it is reasonable to interpret that TGF-β-induced suppression of Fli-1 causes induction of CTGF and may contribute to fibrogenesis. 134 The transcriptional activity of Fli-1 is not only regulated by protein level but also modulated by its posttranslational modifications including acetylation and phosphorylation. In response to TGF-β, cellular PCAF acetyltransferase acetylates Fli-1 at 380 lysine residue (380 K), decreases its stability and reduces interaction with collagen gene promoter, and thus stimulates collagen gene expression. 131 Collectively, these reports from Dr. Trojanowska’s laboratory certainly establish that Fli-1 is a repressor of collagen synthesis and epigenetic modification of Fli-1 controls its activity. Selective pharmacological inhibition of pCAF-mediated Fli-1 acetylation using small molecule (drugable) inhibitors may stabilize Fli-1 which may in turn block increased collagen synthesis in fibrotic tissues.
Fli-1 is also cardioprotective because Fli-1 knockdown mice express more collagen in heart and develop more fibrosis compared to wildtype mice in response to a cardiotonic steroid marinobufagenin (MBG)-induced uremic cardiomyopathy model with progressive cardiac fibrosis. Like skin fibroblasts, Fli-1 inhibits collagen synthesis in cardiac fibroblasts and overexpressed Fli-1 blocks MBG-induced collagen synthesis. MBG-induced PKC-δ-mediated phosphorylation of Fli-1 at threonine 312 decreases its nuclear accumulation and thus in the absence of repressor Fli-1, MBG imparts its stimulatory influence on collagen synthesis. 135 MBG-mediated suppression of Fli-1 activity contributing to MBG-induced hypertension and cardiac fibrosis has been further evidenced by observations that patients with preeclampsia display increased levels of plasma MBG and decreased levels of Fli-1. Additionally, administration of monoclonal antibodies against MBG in partially nepherectomized rats, increases the level of Fli-1 and decreases cardiac and vascular fibrosis.136,137 Taken together, it is quite reasonable to interpret that induction of Fli-1 activity or stabilizing the cellular Fli-1 by post-translational modifications may be an ideal approach to decrease collagen synthesis and ameliorate fibrogenesis. It is also evident from these in vitro and in vivo studies that pharmacological inhibition of MBG may also be a potential therapeutic approach to control fibrosis via activation of antifibrotic Fli-1.
PPAR-gamma and fibrosis
Peroxisome proliferator-activated receptor-gamma (PPAR-γ) is a 58 kDa nuclear hormone receptor and is involved in a number of biological processes including lipid and glucose metabolism, adipocyte differentiation and insulin sensitivity.138–140 PPAR-γ is synthesized by a wide variety of cells and acts as both a transcriptional activator and repressor in a gene-specific and context-dependent manner.39,140–143 Over the last decade, numerous independent studies from different laboratories established that PPAR-γ is a negative regulator of profibrotic signal-induced collagen synthesis and blunts fibrogenesis in a wide variety of organs in experimental animal models of fibrosis. PPAR-γ is activated by pharmacological agents like thiazolidinedione (TZD) derivative rosiglitazone, troglitazone, pioglitazone and natural compound 15d-PGJ2.140,141 Ligand-activated PPAR-γ prevents TGF-β-induced myofibroblast differentiation and collagen synthesis in vitro in culture cells.142,144–146 While ligand-activated PPAR-γ blunts TGF-β-induced myofibroblast differentiation and collagen synthesis in dermal fibroblasts, 142 PPAR-γ deficiency in mouse embryonic fibroblasts is associated with elevated TβR-I level, constitutive activation of Smad signaling, constitutive Smad-p300 interaction and increased collagen synthesis and secretion. 143 This data has been supported by an in vivo observation that mice with fibroblast-specific deletion of PPAR-γ are more susceptible to develop bleomycin-induced skin fibrosis compared to wildtype mice. 147 Additionally, 15d-PGJ2 fails to suppress elevated collagen synthesis in PPAR-γ null fibroblasts suggesting PPAR-γ is a potent negative regulator of Smad-dependent TGF-β-induced profibrotic responses. 143 Ligand activated PPAR-γ prevents TGF-β-induced collagen synthesis via sequestration of essential coactivator p300 from TGF-β-induced pSmad complex on the collagen gene promoter. Inhibition is not due to altered Smad phosphorylation, heterodimerization, nuclear translocation or DNA interaction. Thus disruption of Smad-p300 complex by PPAR-γ ligand is one of the molecular mechanisms of PPAR-γ-induced abrogation of TGF-β-induced collagen synthesis in skin fibroblasts. 39 It is important to note that in SSc skin, the Smad-p300 axis is constitutively active, 49 an essential protein–protein interaction in TGF-β-induced collagen synthesis.37–39 Therefore, disruption of Smad-p300 complex using PPAR-γ ligands is an attractive therapeutic approach to control fibrosis in SSc.
The significance of PPAR-γ and its ligands in antifibrotic activity has also been documented in vivo. In a rat model of type 2 diabetes, administration of troglitazone, a PPAR-γ ligand, ameliorates diabetic glomerulosclerosis. 148 The antidiabetic drug pioglitazone, a PPAR-γ agonist, ameliorates CCl4-induced liver fibrosis. 149 Similarly, administration of the PPAR-γ ligand rosiglitazone, an antidiabetic drug, ameliorates bleomycin-induced dermal lipoatrophy, myofibroblasts differentiation and dermal fibrosis without affecting Smad activation. 150 Importantly, the level of PPAR-γ is significantly lower in SSc skin and lung tissue and also in explanted SSc skin fibroblasts. 151 The decreased level of PPAR-γ in SSc fibroblasts may be due to increased TGF-β signaling because normal fibroblasts exposed to TGF-β show decreased expression of PPAR-γ and TGF-β-mediated suppression of PPAR-γ is Smad-dependent. 151 In a recent study, Mutlu et al. 152 reported that the proteosomal inhibitor Bortezomib blunts bleomycin-induced skin and lung fibrosis in mice via suppression of Smad-dependent TGF-β-induced profibrotic signaling. Importantly, Bortezomib stimulates the expression of cellular PPAR-γ and blocking PPAR-γ activity diminishes Bortezomib-induced inhibition of profibrotic responses indicating PPAR-γ activation by the proteosomal inhibitor Bortezomib may be an ideal approach to attenuate skin and lung fibrosis. 152 In contrast to this study, Pujols et al. 153 reported that low doses of the proteosomal inhibitor MG262 abrogate TGF-β-induced collagen synthesis in nasal fibroblasts. However, inhibition of TGF-β-induced profibrotic responses is not due to upregulation of PPAR-γ rather due to suppression of NF-κB activation and inhibition of proinflammatory cytokines. This discrepancy may be due to cell-type differences or due to differences in proteosomal inhibitors. As protease inhibitors Bortezomib and MG262 can induce apoptosis, determination of the proper doses of protease inhibitor for fibrosis therapy is critical as has been hinted upon by Pujols et al. 153 The antifibrotic action of PPAR-γ in lung fibrosis has also been evidenced in db/db mice. The db/db obese mice with impaired leptin signaling, are resistant to develop bleomycin-induced lung fibrosis indicating leptin signaling is required for bleomycin-induced lung fibrosis. Importantly, leptin potentiates TGF-β-induced profibrotic responses via suppression of PPAR-γ. In db/db mice, the level of PPAR-γ is significantly higher compared to lean wildtype mice. In the absence of leptin activity in db/db mice, elevated PPAR-γ prevents bleomycin-induced lung fibrosis. 154 These results from different in vitro and in vivo studies provide substantial evidence in favor of the antifibrotic activity of PPAR-γ. Therefore, activation of PPAR-γ using the FDA approved antidiabetic drug pioglitazone, the proteosomal inhibitor Bortezomib, or new drugable small molecule agonists of PPAR-γ may be an attractive approach to controlling fibrogenesis in different organs.
p53 and fibrosis
p53 is a negative growth regulator of a wide variety of cells. p53 is also involved in senescence and apoptosis in a cell-type and context-dependent manner. 155 Cellular p53 activity is regulated by posttranslational modification including phosphorylation, acetylation and also through proteosomal degradation. 156 Along with its involvement in several biological processes, p53 also plays a critical role in matrix remodeling and tissue homeostasis by regulating ECM proteins including fibronectin and collagen synthesis and controlling cellular proteolytic activity of uPA/tPA/PAI-1/MMPs/TIMPs axis.38,157–160 While ectopically expressed p53 abrogates TGF-β-induced collagen synthesis in dermal fibroblasts, p53 null mouse embryonic fibroblasts produce significantly elevated Type I collagen compared to wildtype controls. The negative regulation of collagen by p53 is at the level of transcription indicating p53 is a negative transcriptional modulator of profibrotic responses. 38 Furthermore, pharmacologic stimulation of p53 blunts TGF-β-induced collagen synthesis. Abrogation of Smad-dependent TGF-β-induced collagen synthesis is not due to attenuation of TGF-β receptor level activation of Smads or nuclear translocation, rather abrogation is due to disruption of Smad-p300 interaction by overexpressed p53 in dermal fibroblasts. Additionally, overexpressed p300 reverses p53-mediated suppression of collagen gene transcription. 38 Therefore, pharmacologic activation of p53 may be an approach to control the magnitude of collagen synthesis in fibrotic tissues. 38 This has been supported by the observation that overexpressed p53 also abrogates Type I collagen synthesis in lung fibroblasts. 160 Consistent with these in vitro observations, Dagher et al. 161 demonstrate that inhibition of p53 activity in vivo, using pifithrin in a rat model of acute kidney injury, develops worse fibrosis compared to vehicle treated animals with acute kidney injury. The inhibition of p53 is also associated with increased epithelial to mesenchymal transition (EMT) and induced collagen synthesis. Similarly, p53 knockout mice show worse kidney injury compared to wildtype mice in response to ischemia as characterized by increased/prolonged inflammation due to reduced leukocyte apoptosis, increased fibronectin and collagen deposition and worse fibrosis. 162 Elevated collagen synthesis in vivo in p53 knockout mice is consistent with previous in vitro observations that p53 deficiency is associated with elevated levels of collagen synthesis.38,160 These results collectively establish the anti-inflammatory and antifibrotic roles of p53. Therefore, specific induction of p53 in affected kidney and other organs may reduce inflammation and associated organ fibrosis. The antifibrotic role of p53 has also been studied in lung using animal models of lung fibrosis. Administration of nicotinamide, an inhibitor of collagen synthesis in vitro, blunts bleomycin-induced collagen synthesis and fibrosis in lungs. The antifibrotic effect of nicotinamide may be due to decreased expression of profibrotic TGF-β, increased levels of p53 and increased apoptosis. 163 In contrast to these reports, Pujols et al. 153 show that in nasal fibroblasts, protease inhibitor MG262 inhibits TGF-β-induced collagen synthesis and that it is associated with elevated p53 levels. However, the p53 inhibitor pifithrin (1μM) fails to reverse MG262-mediated inhibition of collagen synthesis. Therefore, in nasal fibroblasts, elevated p53 may not be responsible for protease inhibitor-mediated inhibition of collagen under this experimental setting. However, the concentration used in this study (1μM) may not be sufficient to block the function of MG262-induced elevated levels of cellular p53.
In a recent study, Huang et al. 164 demonstrate that a small molecule inhibitor (TM5275) of PAI-1, a potent inhibitor of the uPA/tPA fibrinolytic system and contributor to fibrosis, prevents murine lung fibrosis induced by intranasal instillation of adenovirally expressed constitutively active TGF-β1. Treatment of this murine model of lung fibrosis with TM5275, increases uPA/tPA activity, myofibroblast apoptosis, induction of p53 and decreases synthesis of ECM proteins including collagen and fibronectin. Therefore, TM5275-mediated suppression of lung fibrosis is not only due to decreased PAI-1 activity and increased uPA/tPA activity but also due to elevated levels of antifibrotic p53. This study further suggests that an inhibitor of PAI-1, a designated contributor in fibrogenesis may be an ideal drug for fibrosis therapy via induction of p53 and uPA/tPA. 164 Taken together, it is reasonable to conclude that p53 is a potent endogenous anti-inflammatory and antifibrotic factor. While lack of p53 is associated with increased collagen, fibronectin and other ECM synthesis, activation of p53 blocks fibrogenesis. Pharmacological induction of cellular p53 either at the level of synthesis or by reducing its proteosomal degradation will be helpful in maintaining physiological matrix biosynthesis in injured organs and may be an alternate therapeutic approach to control organ fibrogenesis.
Klotho and fibrosis
Klotho is an anti-aging gene and a known suppressor of several aging phenotypes including osteoporosis, arteriosclerosis and short life span. Klotho deficiency is associated with chronic kidney diseases, vascular calcification, cardiac hypertrophy and an accelerated aging process.165–169 Klotho gene codes for a 116 kDa transmembrane protein that acts as a coreceptor for FGF23 signaling. Adenovirus-mediated overexpression of Klotho prevents endothelial dysfunction via induction of nitric oxide synthesis and reducing increased blood pressure in the Otsuka Long-Evans Tokushima Fatty (OLETF)-induced rat model of atherosclerotic disease. Importantly, overexpressed Klotho ameliorates perivascular fibrosis in this atherosclerotic rat model indicating an antifibrotic role of Klotho. 170 In spontaneously hypertensive rats, the level of Klotho is significantly decreased in kidneys. Administration of AngII receptor blockers Fosinoopril and Valsartan in hypertensive rats stimulates Klotho synthesis and decreases PAI-1 and MMP9 expression indicating Klotho may play a significant role to protecting the kidney from fibrogenesis. 171 A recent study demonstrates that Klotho imparts its antifibrotic action and ameliorates excessive ECM synthesis via direct interaction with TGF-β receptor II and prevents TGF-β-interaction with its receptors thus blocking TGF-β-induced profibrotic responses. 172
As seen in the hypertensive rat kidney, the level of expression of Klotho is decreased in chronic kidney disease induced by UUO. In addition, mice with a haplo-dose of Klotho are more susceptible to fibrogenesis and present with worse fibrosis in response to UUO as compared to UUO-wildtype mice. 173 Increased collagen deposition in haplo-Klotho kidneys may be due to increased TGF-β activation because Klotho prevents TGF-β signaling by direct interaction with TGF-β receptor II. Furthermore, recombinant Klotho downregulates the expression of α-SMA and the PAI-1 gene. Interestingly, TGF-β also inhibits Klotho expression in renal epithelial cells suggesting Klotho deficiency contributes to renal fibrosis in the UUO model. 173 Significantly elevated Wnt signaling has been implicated in fibrogenesis in a wide variety of fibrotic tissues including kidney, skin, heart, lung and liver.174–178 Interestingly, soluble Klotho acts as an antagonist of Wnt signaling 179 and while Klotho deficiency is associated with increased Wnt signaling; soluble Klotho binds to Wnt and blocks Wnt signaling 179 thus blunting Wnt-induced fibrogenesis. Increased Wnt signaling in UUO-wildtype mice is associated with renal fibrosis. In contrast, Wnt signaling is significantly decreased in UUO-Klotho transgenic mice that are associated with decreased tubulo-interstitial fibrosis. Furthermore, Klotho heterozygous mice show increased Wnt signaling and elevated fibrosis compared to wildtype mice in response to UUO suggesting the magnitude of UUO-induced kidney fibrosis depends on the level of Klotho protein. 174 Therefore, pharmacological activation of antifibrotic Klotho or administration of recombinant Klotho, an antagonist of TGF-β and Wnt signaling, in injured tissues may be an attractive therapeutic approach to maintain physiological wound healing and reduce the progress of already initiated fibrogenesis.
Epigenetics of fibrogenesis
Epigenetic factors including acetyltransferase, methyltransferase, deacetylases, and microRNAs play an important role in the regulation of eukaryotic gene expression. However, like deregulated transcription factors, deregulated epigenetic factors also play a pivotal role in the initiation and progression of a variety of human diseases.15,180–185 Like other eukaryotic genes, expression of collagen, the major ECM protein, is also influenced by epigenetic regulators including acetyltransferases, deacetylases, methyltransferases and microRNAs.
Acetyltransferase p300 and fibrosis
The nuclear phosphoprotein p300 is a ∼300 kDa protein with different structural and functional domains including cysteine-histidine rich CH1, CH2 and CH3 domains important for protein–protein interaction, a bromodomain and a histone acetyltransferase (HAT) domain. With intrinsic acetyltransferase activity, p300 acetylates specific lysine residues in histones and transcription factors and modulates the expression of genes whose products are involved in cellular growth, proliferation, apoptosis and essential for embryonic development.13,15,186 Acetyltransferase p300 plays an essential role in Smad-dependent TGF-β-induced collagen synthesis in human dermal fibroblasts and the expression of p300 is stimulated by TGF-β. 36 TGF-β-induced stimulation of acetyltransferase p300 is Smad-independent and ERK1/2-MAPK-dependent. The Egr-1 transcription factor, a potent profibrotic factor as described earlier, is essential for TGF-β-induced p300 expression in vitro and in vivo. 102 Importantly, TGF-β-induced p300 stimulation is associated with increased histone acetylation of the collagen gene promoter and elevated collagen gene transcription.39,102 While overexpressed p300 stimulates basal and TGF-β-induced collagen synthesis in a dose dependent manner, TGF-β fails to stimulate collagen synthesis in p300 depleted fibroblasts suggesting p300 sensitizes fibroblasts to TGF-β-induced profibrotic responses. 40 Furthermore, acetyltransferase activity of p300 is required for maximal stimulation of TGF-β-induced collagen synthesis indicating epigenetic regulation of collagen gene expression. 36 Acetyltransferase p300 also integrates profibrotic TGF-β signaling and antifibrotic IFN-γ signaling in skin fibroblasts. Disruption of TGF-β-induced Smad-p300 complex formation by IFN-γ-induced Stat-1α abrogates TGF-β-induced collagen synthesis. 37 Similarly, disruption of Smad-p300 complex formation by overexpressed p53 or ligand activated PPAR-γ completely abrogate TGF-β-induced collagen synthesis suggesting interaction of p300 with activated Smad and intrinsic acetyltransferase activity of p300 are essential for elevated collagen synthesis in response to profibrotic signaling.38,39,102
The significance of p300 in organ fibrogenesis has also been supported by several observations made in animal models of fibrosis and SSc patients. The level of p300 is significantly higher in fibrotic skin derived from a murine model of skin fibrosis, sclerodermatous-graft versus host disease (Scl-GVHD), in SSc skin and explanted SSc skin fibroblasts compared to healthy controls.40,102 The level of p300 is significantly increased in AngII-induced cardiac fibrosis in mice and increased p300 levels in AngII-induced fibrotic tissues are associated with increased acetylation of histone3 lysine 9 (H3K9) residue (H3K9 is acetylated by p300) (Ghosh AK et al., unpublished data). The finding that elevated p300 constitutively interacts with Smad2/3 in SSc skin fibroblasts 49 further establishes a firm link of acetyltransferase p300 with fibrosis.36–40,49,102 As acetyltransferase activity of p300 is essential for maximal stimulation of TGF-β-induced collagen synthesis, 36 selective normalization of increased acetyltransferase activity of p300 and histone acetylation in fibrotic tissues using p300-specific small molecule inhibitors may be a suitable therapeutic approach to control fibrosis. It is important to note that TGF-β-induced collagen gene expression in skin fibroblasts is abrogated by inhibition of acetyltransferase activity using Anacardic acid indicating the suppression of acetyltransferase activity of p300 is a feasible approach to prevent or delay the progress of fibrogenesis. 102 Furthermore, inhibition of p300 acetyltransferase activity using Curcumin, a natural inhibitor of acetyltransferase activity, blunts hypertension-induced cardiomyocyte hypertrophy and significantly inhibits the perivascular fibrosis in salt-sensitive Dahl rats. Furthermore, Curcumin normalizes wall thickness in myocardial-infarcted (MI) rat hearts and improves left ventricular systolic function. 187 Together, it is reasonable to propose that selective pharmacological inhibition of p300-specific acetyltransferase activity or disruption of the Smad-p300 interaction using a safe and effective small molecule inhibitor will be an ideal approach to attenuate fibrogenesis.
Histone deacetylases and fibrosis
The transcription rate of a message from a gene promoter is tightly controlled by an accurate balance of acetyltransferase and deacetylases which activate and repress transcription respectively. Histone deacetylases deacetylate histones and transcription factors and play a significant role in epigenetic regulation of numerous genes involved in different biological processes. The HDAC family consists of three classes with eighteen members including eleven HDACs and seven Sirtuins. 188 While elevated acetyltransferase p300 activates collagen gene transcription, excess HDAC1 abrogates p300-induced collagen gene transcription indicating an antagonistic action of HDAC1 on p300 acetyltransferase activity (Ref. 36; and Ghosh AK, unpublished data). Having that information, one can postulate that inhibition of HDAC activity will increase histone acetylation and elevate collagen synthesis. Paradoxically, suppression of HDAC activity using the HDAC inhibitor Trichostatin A (TSA), a specific inhibitor of HDAC, is associated with abrogation of TGF-β-induced collagen synthesis.189–191 Although TSA prevents elevated collagen, fibronectin and α-SMA synthesis in different cells derived from different organs, the molecular mechanisms of suppression may be cell-type and context-dependent. In rat skin fibroblasts, TSA abrogates TGF-β-induced collagen synthesis possibly via time-dependent activation of anti Smad7, TG box interacting factor (TGIF), a corepressor of TGF-β signaling pathway, and inhibition of Smad2/3 activation. 190 On the other hand, in human skin fibroblasts, TSA blunts TGF-β-induced collagen gene expression via suppression of Sp1 expression without affecting TGF-β-induced Smad activation pathway and Smad-dependent reporter gene activity. Furthermore, overexpressed Sp1 rescues TGF-β-induced collagen gene expression in the presence of TSA suggesting Sp1 inhibition may be one of the mechanisms of TSA-induced inhibition of profibrotic responses. 191 Consistent with these observations, Wang et al. 192 also demonstrate that TSA blocks TGF-β-induced Sp1 activity but not Smad2/3 activation in human mesenchymal stem cells. In contrast, Kaimori et al. 193 demonstrate that in hepatocytes, TSA disrupts TGF-β-induced Smad-p300 complex formation and activates antifibrotic Fli-1 expression during epithelial to mesenchymal transition (EMT) thus blocking EMT and its contribution to elevated collagen synthesis.
Along with these in vitro data, the significance of HDAC inhibitors as antifibrotic agents has also been evidenced by numerous in vivo studies. For example, TSA ameliorates bleomycin-induced increased mouse dermal thickness. 194 In a UUO-induced murine model, renal injury is associated with increased expression of HDAC1 and HDAC2, and decreased histone acetylation. 195 Administration of TSA blocks UUO-induced inflammation, α-SMA expression and renal fibrosis via suppression of UUO-induced CSF-1, an inflammatory chemokine that mediates macrophage infiltration, 195 or via inhibition of renal cell apoptosis. 196 TSA also abrogates atrial fibrosis and atrial arrhythmia in homeodomain-only protein (HOP)-induced murine model of cardiac hypertrophy. 197 Furthermore, HDAC inhibitor administration blunts pressure overload-induced cardiac hypertrophy and myocardial fibrosis and improves cardiac functions. 198 However, the effect of an HDAC-inhibitor on pulmonary artery banding (PAB)-induced right ventricular hypertrophy, matrix remodeling and right ventricular function are controversial. While Cho et al. 199 reported that the HDAC inhibitor Valproate blocks PAB-induced right ventricular hypertrophy and fibrosis in rats, Boggard et al. 200 reported that the HDAC inhibitors TSA or Valproate worsen PAB-induced right ventricular hypertrophy and fibrosis, and that this is associated with decreased expression of VEGF and Angiopoietin1. These differences in experimental outcome may be due to difference in the age of the rat, the method of artery banding, or in post-surgery treatment time frame with TSA. Taken together, the results of most of these studies provide sufficient experimental evidence to support the antifibrotic activity of HDAC inhibitors and thus the selective inhibition of HDAC using non-toxic HDAC inhibitors may be a potential therapeutic approach to control fibrosis in different organs including lung, heart, kidney, liver and skin.
Methyltransferases and fibrosis
Like acetyltransferase-mediated acetylation of histones, methylation of histones, in particular lysine residues, play a pivotal role in epigenetic control of eukaryotic gene expression. Numerous histone methyltransferases have been identified which are involved in controlling different biological processes. 201 During myofibroblast differentiation of hepatic stellate cells, a wide variety of histone methyltransferases are upregulated including MLL1, MLL5, Set1 and ASH1 compared to quiescent cells. Methylation of H3K4 by these methyltransferases is associated with transcriptional activation of target genes. While Ash1 methyltransferase activates α-SMA, TIMP1 and TGF-β1 profibrotic genes in hepatic stellate cells (HSC) via direct interaction with the promoter of these genes, depletion of Ash1 abrogates this stimulation. Along with these profibrotic genes, Ash1 also stimulates methylated CpG binding protein 2 (MeCP2) gene expression. 202 MeCP2 controls myofibroblast differentiation of HSC. In addition, MeCP2 increases H3K9 methylation on the PPAR-γ gene promoter, a potent endogenous antifibrotic factor, recruits the transcriptional repressor heterochromatin protein-1 (HP1) and represses PPAR-γ transcription. Deficiency of MeCP2 activity blunts myofibroblast differentiation and ameliorates CCl4-induced liver fibrosis in a rat model. Therefore, pharmacologic blocking of histone methyltransferases may blunt liver and lung fibrosis 184 via suppression of profibrotic genes including α-SMA, Timp1 and TGF-β, and releasing the repression of antifibrotic gene PPAR-γ. Additionally, Hu et al. 203 reported that MeCP2 is essential for bleomycin-induced myofibroblast differentiation, collagen synthesis and lung fibrosis because MeCP2-knockout mice show less inflammation, reduced myofibroblast differentiation and lung fibrosis in response to bleomycin instillation. This further implicates the positive role of MeCP2 in fibrogenesis. 203 The angiostatic chemokine IP-10 (IFN-γ-inducible 10 KDa protein) is a potent antifibrotic factor and the expression of IP-10 is significantly decreased in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis (IPF). Importantly, suppression of IP-10 in IPF fibroblasts is due to formation of an inhibitory transcriptional complex containing methyltransferase G9a, Suv 39 H, HDAC and HP-1 on the IP-10 gene promoter. Treatment of IPF fibroblasts with a HDAC inhibitor and a methyltransferase G9a inhibitor rescue IP-10 expression indicating the complex epigenetic regulation of profibrotic and antifibrotic genes controls fibrogenesis. 204 Therefore, these results collectively suggest that selective pharmacological inhibition of specific histone methyltransferase may be a viable therapeutic approach to control fibrogenesis via suppression of profibrotic factors and the simultaneous induction of antifibrotic factors like PPAR-γ and IP-10 proteins.
MicroRNAs and fibrosis
MicroRNAs are short ∼22 nucleotide non-coding RNA that regulate the expression of numerous genes at the post-transcriptional level by degrading mRNA and preventing protein synthesis. Generally, these small non-coding RNAs target to 3”UTR of specific mRNA and induce RNaseIII Drosha and Dicer-mediated degradation. Expressions of numerous microRNAs are regulated by the profibrotic cytokine TGF-β. Recent studies demonstrate the pivotal roles of miRNAs in profibrotic pathways in different tissues. 185 While several miRNAs driving profibrogenic responses including miR-1, miR-21, and miR-155 are elevated in fibrotic tissues, the decreased expression of several antifibrotic miRNAs including mIR-19b, miR-29, miR-30, and miR-133a are also identified in multiple fibrotic organs including skin, heart, lung, liver and kidney. 185 Thum et al. 205 demonstrate that the level of miR-21 is significantly elevated in failing hearts and in a pressure overload-induced animal model of cardiac fibrosis. Treatment of mice with miR-21 antagomir ameliorates pressure-overload-induced ventricular hypertrophy, and cardiac fibrosis. In fibrotic hearts, elevated miR-21 targets Sprouty, an endogenous repressor of profibrotic ERK-1/2 MAP kinase, and in the absence of Sprouty, elevated ERK-1/2MAP kinase contributes to pressure-overload-induced collagen synthesis and myocardial fibrosis. In a separate report, Roy et al. 206 also demonstrate that miR-21 is elevated in a myocardial ischemia/reperfusion (I/R) murine model and miR-21-mediated matrix remodeling in the I/R murine model is possibly via suppression of PTEN (phosphatase and tensin homologue), a direct target of miR-21 and associated increased MMP-2 activity. Elevated levels of miR-21 have also been reported in the UUO-induced murine model of kidney fibrosis, and suppression of miR-21 using anti-miR-21 ameliorates UUO-induced kidney fibrosis indicating the significant role of miR-21 in fibrogenesis. 207 On the other hand, miR-133 and miR-30 are downregulated in fibrotic hearts. While overexpressed miR-30 and miR-133 downregulate CTGF, a potent profibrogenic regulator, in cardiomyocytes and fibroblasts and thus block profibrotic responses, downregulation of these miRNAs stimulate CTGF expression and increased profibrotic responses suggesting miR-133 and miR-30 are cardioprotective. Based on these findings, profibrotic and antifibrotic microRNAs may be novel therapeutic targets for the treatment of abnormal matrix remodeling and fibrogenesis.
EndMT and fibrosis
EndMT is a biological process by which vascular endothelial cells gradually lose endothelial traits including loss of CD31, VE cadherin and simultaneously acquire mesenchymal traits such as α-SMA and collagen expression. Although EndMT is a normal biological process pivotal for heart valve and pulmonary artery formation during embryonic development,208–213 almost quarter a century ago, the possible existence of such endothelial plasticity and its significance in vascular remodeling during dermal wound healing in adult rats was indicated (Ref. 214, reviewed in Ref. 212). The possible role of EndMT-derived myofibroblast-like cells in atherosclerosis and hypoxia-induced pulmonary vascular remodeling has also been reported (reviewed in Ref. 212). Using cell lineage analysis of fibroblasts in double transgenic mice, Zeisberg et al. 215 elegantly demonstrate that EndMT-derived myofibroblast-like cells contribute to cardiac fibrosis in a pressure overload-induced murine model and 27–35% of total collagen producing myofibroblasts are EndMT-derived in this model of cardiac fibrosis. These results indicate clearly the significance of EndMT and its contribution to pathological wound healing in adults. Interestingly, endothelial cell plasticity during EndMT is controlled by the profibrotic cytokine TGF-β,14,23,210,212,213,215,216 a master regulator of matrix remodeling and fibrogenesis.12,25
The TGF-β-induced EndMT is driven by multiple transcription factors including activated Smad2/3, Snail and β-Catenin.2,23,215–218 As the levels of acetyltransferase p300, a potent profibrotic epigenetic regulator, 15 and p53, a negative modulator of TGF-β-induced profibrotic response,38,160,161 are significantly elevated and downregulated respectively during EndMT, elevated p300 and p53 deficiency may also contribute to EndMT. 216 Interestingly, a small molecule inhibitor of acetyltransferase p300 blunts TGF-β-induced EndMT in vitro (Ghosh AK unpublished data). In addition, a recent study also demonstrates that a subset of microRNAs are differentially expressed during EndMT including microRNAs let-7c, let-7 g, miR-21, miR-125b and miR-195 etc., 216 few of which are known to be deregulated in fibrotic tissues. Furthermore, miRNA-21 contributes partially to EndMT possibly via downregulation of inhibitory Smad7, a direct target of miR-21, 219 suggesting that other miRNAs may also contribute to EndMT and the pathogenesis of fibrosis.
Therefore, these results collectively indicate that targeting EndMT-inducing epigenetic regulators such as microRNAs or acetyltransferase p300 will be an attractive alternate approach to control EndMT in adult affected tissues and to develop a novel therapy to ameliorate EndMT contributed organ fibrosis. Recent in vivo studies support the notion that EndMT may be a novel target in amelioration of tissue fibrosis. For example, administration of recombinant human BMP-7 significantly blocks EndMT and ameliorates the pressure overload-induced cardiac fibrosis in mice. 215 Similarly, HGF blocks TGF-β-induced EndMT in human coronary artery endothelial cells and importantly, pressure-overload-induced cardiac fibrosis is significantly decreased in HGF expressing transgenic mice indicating activation of antifibrotic HGF may attenuate fibrosis via suppression of EndMT. 220 Additional supportive evidence comes from a very recent observation demonstrating that the Angiotensin II receptor blocker Irbesartan blocks high glucose-induced EndMT of human coronary artery cells in culture and attenuates EndMT and diabetic cardiomyopathy associated fibrosis in vivo. 221 Collectively these results are supporting the notion that EndMT may be a potential therapeutic target for treating fibrosis.
Senescence and fibrosis
During physiological wound healing, synthesis of collagen and other extracellular matrix proteins accelerates the repair process and maintains tissue homeostasis. 222 Deregulation or sustained activation of myofibroblasts and collagen synthesis leads to fibrosis. The obvious question is what determines or controls the physiological wound healing? One of the newly emerged concepts is that in response to injury, activated and differentiated myofibroblasts produce collagen and other ECM proteins required for proper wound healing. However, at a certain time point, collagen and ECM producing myofibroblasts undergo a senescence state where senescent myofibroblasts stop further synthesis of collagen and other ECM proteins and produce ECM protein degrading enzymes like MMPs preventing scar formation. Jun and Lau 223 elegantly demonstrated this new concept using both in vitro and in vivo studies. The metricellular protein CCN/Cyr61 plays a significant role in the conversion of ECM protein producing myofibroblasts to senescent myofibroblasts during physiological wound healing. CNN1/Cyr61-mediated induction of senescence in myofibroblasts involves a series of events: i) interaction of CCN-1/Cyr61 with adhesion receptor α6β1 and heparan sulphate proteoglycan, ii) activation of p53 and RAC1-Nox1, and iii) induction of ROS which in turn induces senescence via activation of the p16/Rb axis. Induction of senescence in myofibroblasts halts further ECM synthesis and controls wound healing without scar. The significance of CCN/Cyr61 in this senescent induction process is confirmed by the observations that CCN1/Cyr61 mutant mice show decreased senescence and increased fibrosis and topical application of CCN1/Cyr61 protein to a skin wound ameliorates fibrosis. These results strongly indicate that CCN-1/Cyr61 protein acts as an antifibrotic factor via induction of senescence in collagen producing myofibroblasts. The idea that cellular senescence is a counteracting biological process of fibrogenesis has been further evidenced by the observation that IL-22, which signals through IL-10R2 and IL-22R1 receptors, induces the expression of Stat3 in HSC and inhibits HSC apoptosis. In IL-22 transgenic mice, adenoviral overexpressed IL-22 abrogates CCl4-induced liver fibrosis which is associated with an increased number of senescent HSC and decreased myofibroblast differentiation and ECM protein synthesis. The IL-22-induced Stat3 and Stat3-induced p53 and p21 pathways play an important role in the induction of senescence and amelioration of liver fibrogenesis. 224 It is important to note that p53 itself is a repressor of induced collagen synthesis.38,160–162 These results strongly suggest that tissue-type or cell-type specific induction of the senescence pathway may be an attractive approach to control fibrogenesis in injured organs.
Summary and concluding remarks
Wound healing in every organ is ignited by stress-induced vascular injury and inflammation. Inflammatory cells help to repair the wound via secretion of key cytokines and growth factors, stimulation of cell proliferation, migration, myofibroblast differentiation, and extracellular matrix biosynthesis by activated or differentiated myofibroblasts to replace damage tissues in the wound area. However, chronic inflammation, sustained myofibroblast activation and collagen synthesis lead to fibrosis, an end stage pathological manifestation of numerous human diseases that leads to organ failure. Every step of this pathway that leads to physiological wound healing is tightly controlled by the balanced activity of different cytokines (TGF-β, IFN-γ, IL-10), transcriptional regulators (Sp1, Egr1, Smads, Fli-1, PPAR-γ, p53, Klotho), and epigenetic regulators (p300, Methyltransferases, HDACs, microRNAs). Deregulation of these regulators leads to imbalanced synthesis of ECM proteins (collagen), ECM degrading proteases (uPA/tPA/Plasmin/MMPs) and their inhibitors (PAI-1/TIMPS) that causes fibrosis (Figure 2). Based on existing literature as described here, it is reasonable to claim that targeting these imbalanced transcriptional regulators or epigenetic regulators will be a viable approach to attenuate fibrogenesis by converting the activated ECM producing cells to either a quiescent or in senescent state while their production of ECM is normalized (Figure 3). The role of TGF-β signaling and its cross-talk with complex profibrogenic network in fibrogenesis are well-documented and widely endorsed (Figure 1). However, long term TGF-β neutralizing antibody therapy is not a viable approach based on the outcome of clinical trials as previously described. The role of IFN-γ as an antifibrotic cytokine and its downstream pathway is also very well-studied and holds some promise for fibrosis therapy. However, dose and duration of treatment has not yet been established for effective and safe clinical trials in humans. Additionally, the outcomes of past IFN-γ-based clinical trials for fibrosis therapy are mixed. The new in vivo data supporting the role of p53 as an antifibrotic agent is encouraging. However, animal studies using a safe pharmacological inducer of p53 are essential to evaluate its impact in amelioration of fibrogenesis. The significant contributions of Sp1, Egr-1, p300, Fli-1, PPAR-γ and Smad7 as profibrotic and antifibrotic factors in fibrogenesis and physiological wound healing are well documented. However, identification of drugable non-toxic small molecules for suppression of profibrotic factors or activation of antifibrotic factors is challenging. Although a large number of articles in the last two decades described the significance of several transcription factors in the induction or suppression of profibrogenic responses using in vitro and in vivo models of organ fibrosis and concluded that their chosen transcriptional regulators had potentiality as therapeutic target for fibrosis, almost no progress has been made to bring these potential regulators from bench to bedside. There are limited data on the identification small molecules that can specifically block the activity of a profibrotic activator or coactivator and specifically activate an antifibrotic repressor or corepressor. However, the efficacy of these discovered small molecules using a large cohort of animals has not been tested. What are the main causes that stalled this progress? The possibilities are: i) lack of a consortium effort for working with a specific profibrotic or antifibrotic factor in different organs, which is pivotal for such an endeavor; and ii) lack of adequate funding required to catalyse the initiative and enable the identification of drugable small molecules and the use of those molecules in preclinical studies using animal models of organ fibrosis, and if safe, for testing in humans. With the cumulative knowledge in genetics, and the cellular and molecular biology of many of these factors, fibrosis researchers working with the same molecule in different organs can work cooperatively to advance the field. A more coordinated effort will be helpful in accelerating the identification of candidate small molecules or biological agents that can be tested for efficacy and safety in animal studies and eventually in clinical trials.
Constellation of biological events during wound healing process: Involvement of cytokines, transcriptional regulators and epigenetic factors. Injury/stress-induced inflammation is essential for wound healing. Biological processes including cell proliferation, migration and differentiation are involved in wound healing and fibrogenesis. Activated myofibrobalst-synthesized ECM and PAI-1 accelerate wound healing. Imbalanced activities of transcriptional activators, repressors and epigenetic regulators are responsible for sustained activation of myofibroblasts and fibrogenesis. (A color version of this figure is available in the online journal) Schematic model showing the involvement of myofibroblasts in physiological and pathological wound healing. Cellular senescence may play an important role in physiological wound healing. (A color version of this figure is available in the online journal)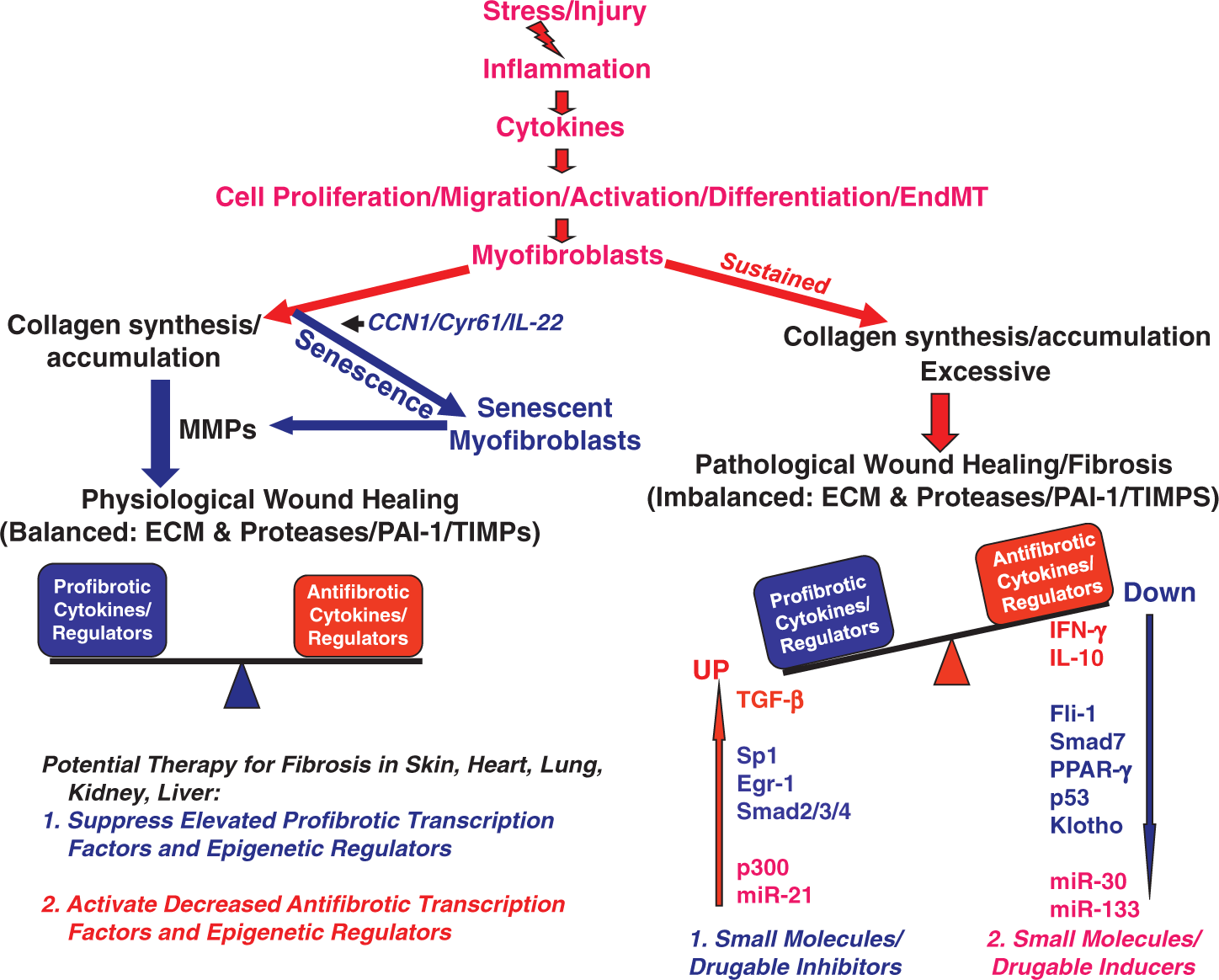
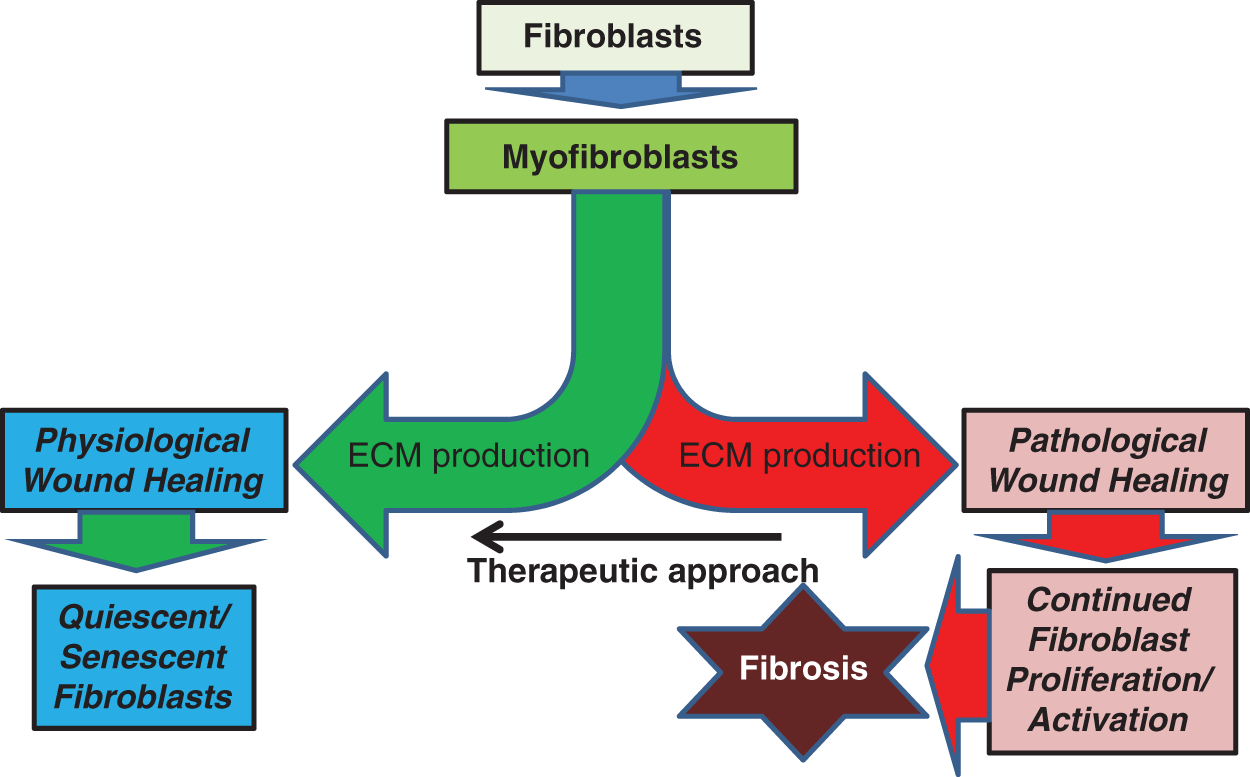
Footnotes
Author contribution
AKG wrote the article and drew the illustrations. SEQ and DEV provided insights and edited the manuscript.
Acknowledgements
The authors thank Sheila B Murphy for critical reading of the manuscript. This work was supported by grants from NIH-NHLBI (HL051387 and 1P01HL108795-01).