
Abstract
We demonstrate for the first time the selective cytotoxicity of Bacillus thuringiensis subsp. israelensis Cry4B toxin mediated by BT-R3 using a cell-based system, which employs High Five insect cells stably expressing BT-R3. Discovery and validation of BT-R3 as the Cry4B receptor was accomplished using a web-based computational pipeline platform that facilitates high-throughput insecticidal target identification utilizing the Anopheles gambiae genome. Once the Cry4B toxin binds to the BT-R3 receptor, a cell death pathway is manifested by sequential cytological changes that include membrane blebbing, cell swelling and lysis. Cry4B toxin associates with cell membrane in both oligomeric and monomeric forms. Monomeric toxin binds specifically to BT-R3 whereas oligomer interacts with cell membrane non-specifically. Cytotoxicity and cell death are the direct result of binding of toxin monomer to BT-R3. The oligomeric form of Cry4B toxin is not involved in cell death. Both the location of the toxin-binding region within BT-R3 and its structural motif are critical to the binding affinity and specificity of the toxin. The toxin-binding region of BT-R3 appears to be located in EC11, the most membrane proximal EC module within the extracellular domain. It is characterized by the presence of two highly conserved amino acid sequences within their N- and C-termini that flank EC11. These sequences represent signature motifs that mark the toxin-binding function in BT-R3. The two sequences form two adjacent β-strands within the β-barrel of EC11, the positioning of which is a hallmark of all Cry toxin receptors thus far reported.
Introduction
The Cry toxins produced by Bacillus thuringiensis exert their insecticidal activity by binding with high-affinity to their cognate cadherin receptors located on the surface of epithelial cells that line the midgut of susceptible insects. Binding of toxin, in turn, triggers an internal signalling event that stimulates heterotrimeric G protein and adenylyl cyclase, resulting in production of cyclic adenosine monophosphate and activation of protein kinase A (PKA). Activation of PKA is associated with an array of cellular changes such as cytoskeletal rearrangement and ion fluxing.1,2 Together, these events lead to oncotic-like cell death. A typical insect cadherin receptor consists of (i) an extracellular (EC) domain with a number of β-barrel cadherin repeats (referred to as EC modules), all of which are connected by interdomain linkers, (ii) a membrane proximal EC domain (MPED), (iii) a transmembrane domain (TM) and (iv) a cytoplasmic domain (CYTO). 3 Cry toxins bind to the most membrane proximal EC module within the EC domain, 4 blocking of which inhibits toxic action.1,3,5
B. thuringiensis subsp. israelensis (Bti) produces a complex of parasporal crystals during the sporulation phase of its life cycle. 6 Mosquitocidal activity resides within the parasporal crystals, which are oligomers composed of polypeptide protoxin subunits. Upon proteolytic activation of the various protoxins, multifarious insecticidal Cry toxins are generated.7,8 They include Cry4A, Cry4B, Cry10A and Cry11A. Each toxin has specific insecticidal activity against different mosquitoes. For example, Cry4A is more toxic to Culex species whereas Cry11A is more effective against Aedes species. Cry4B is primarily insecticidal to Anopheles gambiae, the principal vector of malaria. 6 The insecticidal activity of native Cry10A has not been established because it is unstable under experimental conditions. 6
Previously, we reported a web-based computational pipeline platform for high-throughput insecticidal target identification using A. gambiae as a model system.
9
Ninety-one genes, each of which represents a cell-surface protein, were identified and cloned from a complementary DNA (cDNA) library of A. gambiae. These genes encode cell surface molecules, including cadherins and integrins, apoptosis-related proteins and G protein-coupled receptors, among others. Among the cadherins identified in our A. gambiae library, some share sequence homology with cadherin ESTs in the A. gambiae genome
10
and VectorBase database (www.vectorbase.org/Anopheles_gambiae). To identify the Cry4B cadherin receptor among these cadherins, amino acid sequence alignments were done using cadherins similar to those identified in our library as well as orthologs in moths and beetles recognized as receptors for various Cry toxins.11–19 Sequence comparison revealed clustering of A. gambiae cadherins in three groups (Figure 1). The largest group, group I, includes a cadherin, designated BT-R3 (circled in red), and several homologs as well as orthologs known to be receptors for a number of Cry toxins. Groups II and III include other distantly related cadherins distinctive to A. gambiae. Within group I, BT-R3 was most similar to the Cry toxin cadherin receptors typical of other insects susceptible to various Cry toxins.
Phylogenetic relatedness of selected cadherin proteins from A. gambiae and other insects. The neighbour-joining method of Saitou and Nei
29
was used to construct the phylogenetic tree based on alignments of the deduced amino acid sequences of selected cadherin proteins in A. gambiae and Cry protein-specific cadherin receptors from several moths and a beetle by using the CLUSTALW program (www.genom.jp/tools/clustalw) as described in ‘Materials and methods’ section. The deduced amino acid sequences for selected cadherins, accession numbers are shown, were obtained from the databases of the National Center for Biotechnology Information (NCBI). The phylogram illustrates clustering of the cadherins that serve as receptors for Cry toxins in one group (group I), which includes Mse, Manduca sexta (tobacco hornworm); Bmo, Bombyx mori (silkworm); Har, Helicoverpa armigera (cotton bollworm); Hvi, Heliothis virescens (tobacco budworm); Pxy, Plutella xylostella (diamondback moth); and Tmo, Tenebrio molitor (mealworm). BT-R3, which closely resembles the Cry toxin receptors in these insects, falls in this group, circled in red. Branch lengths are proportional to genetic distances between sequences. The three cadherin proteins selected for structural analyses are circled in blue (see Figure 5). Numbers at nodes represent the percentage bootstrap values of 100 replicates. The 0.1 value in the scale bar below the figure indicates 10% amino acid dissimilarity (10% amino acid substitutions per 100 amino acids)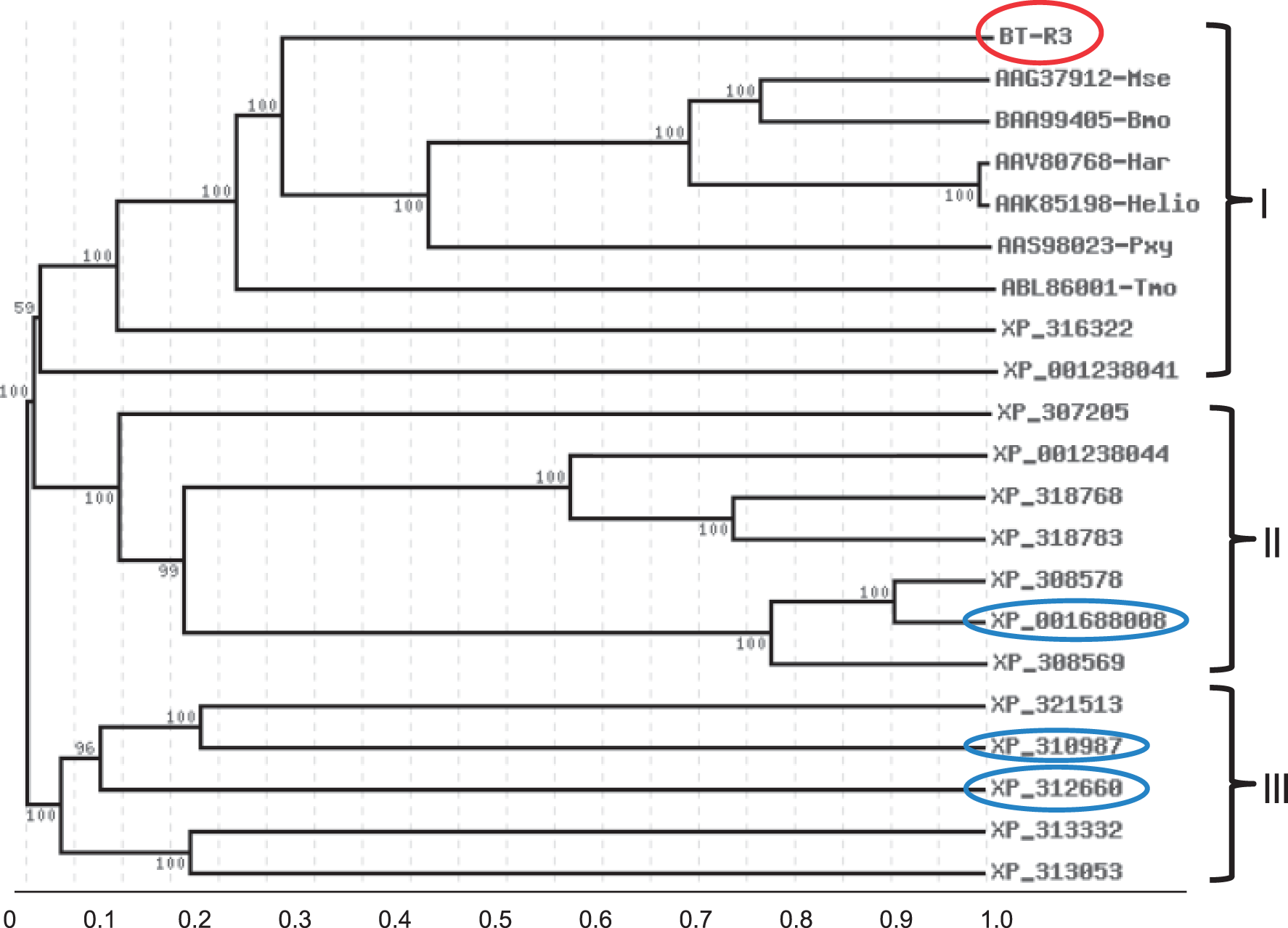
As do all Cry toxins, mosquitocidal Cry4B contains three structural domains, 20 of which domains II and III are involved in receptor binding. The structural features of the receptor-binding region of the Cry4B toxin and the toxin-binding region (TBR) of its commensurate receptor, BT-R3, together confer specificity, selectivity and affinity of binding. 6 The cadherin receptor BT-R3 contains 11 EC modules in its EC domain.6,21 In the present study, the TBR of BT-R3, which apparently lies within EC11, was found to be discernible by two signature motifs near its N- and C-termini. Indeed, comparative sequence analysis of the TBR of BT-R3 and the corresponding region in several of the cadherins representative of A. gambiae, together with secondary and three-dimensional structure predictions, revealed the presence of signature motifs that mark the toxin-binding function in BT-R3 only. To determine whether BT-R3 mediates Cry4B toxicity, we cloned and expressed its coding sequence in High Five insect cells, a model system established in our laboratory, 1 and found that binding of Cry4B toxin to BT-R3 triggers cytotoxic events which lead to cell death. We report here for the first time the cytotoxicity of Cry4B in living cells stably expressing BT-R3.
Materials and methods
Recombinant Cry4B toxin purification
Recombinant Cry4B was obtained by cloning the full-length cry4B protoxin gene in pDEST-17 and expressing it in Escherichia coli BL21 (DE3) as previously described. 6 The protoxin, produced as inclusion bodies, was solubilized in 50 mmol/L NaOH (pH 12) also as previously described 6 and dialysed against 20 mmol/L (4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid) (HEPES) buffer (pH 7.5). Cry4B toxin was generated by trypsin activation of Cry4B protoxin.
Cloning and sequence analysis of BT-R3
Total RNA was purified from third-instar A. gambiae larvae using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). First-strand DNA (cDNA) was synthesized as follows: 2 µg of total RNA was reverse-transcribed with oligo (dT)21 using MonsterScript™ reverse transcriptase (Epicenter Biotechnologies, Madison, WI, USA) at 60℃ for 40 min according to the manufacturer’s protocol. The cDNA was converted to double-stranded DNA using the two specific primers BT-R3-F0 (5′-CACCAGTTGGTTGCACTTGGGTTGG-3′) and BT-R3-R5 (5′-CTAGAACCGGTGGGACAGCTCGTCGTCAGTTTCGTGAAG-3′) in a second polymerase chain reaction (PCR) reaction.
BT-R3-F0 and BT-R3-R5 were designed based on in silico annotation of a 40-kb genomic DNA of A. gambiae that includes the bt-r3 cadherin gene (chromosome 2R: 28,040 k-28,080 k). The full-length coding sequence of bt-r3 (∼5.2 kb) was amplified and cloned in the Gateway Entry vector pENTR/D-TOPO (Invitrogen, Carlsbad, CA, USA). The complete nucleotide sequence was determined using a 3730XL Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA). Sequence analysis and alignments were performed using BLAST 2.0 (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Structural features of BT-R3 were determined based on motif searches using different algorithms such as the Motif Scan (http://myhits.isb-sib.ch/cgi-bin/motif_scan), HAMAP profiles, Pfam HMMs local and global models and PROSITE profiles. Signal peptide (SP) and transmembrane predictions were accomplished using the web-based algorithms SignalP 4.0 (http://www.cbs.dtu.dk/services/SignalP) and dense alignment surface, respectively.
The TBR of BT-R3, which lies within EC modules 7–11 plus the MPED (residues: 874–1569; designated EC7–11:MPED) was amplified by PCR using the two primers Bt-R3-CR7: 5′-CACCGAGAAAATACCCTCCAATACGG-3′ and BT-R3-R4: 5′-CTACTGCAGCGTTTCGTCCGCCTCGGTCAG-3′. A stop codon was introduced at the 5′-end of BT-R3-R4. Amplicons were purified, cloned in pET102/D-TOPO and transformed into E. coli BL21 (DE3) for expression.
A bootstrapped neighbour-joining tree of selected cadherin proteins in A. gambiae and Cry protein-specific cadherin receptors from several insects were generated based on alignments of their deduced amino acid sequences available in the GenBank database. Multiple sequence alignments were performed using CLUSTALW (www.genom.jp/tools/clustalw), which was set to the following parameters: protein weigh matrix, BLOSUM; gap extension, 0.1; and gap penalty (DS units), 1.0. Presentation of the tree was accomplished using the web-based TreeTop-Phylogenetic Tree Prediction program (www.genebee.msu.su/services/phtree_reduced) using the output tree type PHYLIP (http://evolution.genetics.washington.edu/phylip/doc/distance.html).
Binding of Cry4B to the EC7-11:MPED of BT-R3
The CR7-11:MPED coding sequence of BT-R3 was expressed in E. coli and partially purified. EC7-11:MPED polypeptide was bound to a Ni+2 resin column through His-tag fusion and the unbound proteins were removed by washing with five column-volumes of HEPES buffer. Cry4B was added to the column and incubated at room temperature for 2 h. Unbound Cry4B was removed also by washing with five column-volumes of HEPES. Bound proteins were eluted with 2 × sodium dodecyl sulphate (SDS)-protein sample buffer and detected using Western blot analysis. A second Ni+2 resin column incubated with Cry4B only, not EC7-MPED, was used as a negative control and treated in the same manner to test for non-specific binding.
Cloning and expression of BT-R3 in High Five™ insect cells
The BT-R3 coding sequence was mobilized from the pENTR/D-TOPO vector to the Gateway destination vector pXINSECT-DEST38 using LR Clonase II according to the manufacturer’s instructions (Invitrogen, Grand Island, NY, USA). The resulting recombinant plasmids were transformed to the E. coli OmniMax2 T1 phage-resistant host and grown in LB plates containing ampicillin (100 µg/mL) and chloramphenicol (34 µg/mL). Colonies that grew in ampicillin but not chloramphenicol were chosen for further screening. The selected recombinant plasmids were purified and used to transfect High Five™ (H5) insect cells (Invitrogen) using the transfection reagent Cellfectin (Invitrogen, Carlsbad, CA, USA). H5 cells transfected with pXINSECT-DEST38-bt-r3 also were co-transfected with the neomycin-resistant plasmid, pBmA-neo. Transfected H5 cells were cultured in insect-Xpress medium (Cambrex, East Rutherford, NJ, USA) supplemented with G418 (geneticin, 800 µg/mL). Geneticin-resistant H5 cells were sub-cultured for several generations to obtain stably transfected cells. H5 cells stably expressing full-length BT-R3 are designated M5. Expression of bt-r3 in M5 cells was confirmed with reverse transcriptase-PCR (RT-PCR) using bt-r3-specific primers.
Cytotoxicity assays
Cells were seeded in 96-well plates (1 × 104 cells/well) and allowed to grow attached to the surface of the plate bottom. Growth medium was replaced with fresh medium containing Cry4B at various concentrations (37.5–375 nmol/L) and the cells were incubated at room temperature for 4 h. Cell death was determined by Trypan blue exclusion. Ten microlitres of Trypan blue (0.4%, wt/vol) were added directly to each well and incubated for 5 min. Stained cells were viewed immediately under a microscope (Nikon TE600) and photomicrographs were taken with an RTE/CCD-1300 camera (Roper Scientific, Planegg/Martinsried, Germany). Cells were not detached from the plate bottom within the time of observation and photographing. Microphotographs (×200 magnification) were analysed using MetaMorph 4 imaging software (Universal Imaging, Downingtown, PA, USA) to count the number of blue-stained dead cells (NB) and transparent viable cells (NT), respectively, and to record cytological changes such as cell swelling, membrane blebbing and ruffling and lysis of the cells treated with Cry4B toxin. Cytotoxicity was calculated by the ratio NB/(NB + NT).
Preparation of cell membrane fractions
M5 and H5 cells (1 × 106) were washed in phosphate-buffered saline (4℃) and lysed in CytoBuster protein extraction reagent (Novagen, Billerica, MA, USA). Cell lysates were centrifuged at 13,000 × g for 10 min at 4℃ and the supernatants were collected as soluble fractions. The membrane pellets were dissolved in membrane protein buffer (5 mol/L urea, 2 mol/L thiourea, 2% [w/v] CHAPS and 40 mmol/L Tris-HCl) and collected as membrane fractions. The M5 and H5 cells were incubated with Cry4B at a specified concentration and time before preparation of cell extracts for measurement of toxin incorporation. All cell extracts were freshly prepared for Western and immunoligand blotting analysis.
Immunoblot and overlay assays
For Western blot analysis, proteins were separated by 7.5–10% SDS-polyacrylamide gel electrophoresis (PAGE). Proteins from SDS gels were electrically transferred to polyvinylidene difluoride (PVDF) membranes (EMD Millipore Corporation, Billerica, MA, USA) using a 2051 Midget Multiblot Electrophoretic Transfer Unit (Hoefer Scientific Instruments, San Francisco, CA, USA). Membranes were blocked for 2 h at room temperature with Tris-HCl-buffered saline (pH 8) containing 5% non-fat dry milk powder, 5% glycerol and 0.1% Tween 20. For overlay assays, Cry4B toxin was added to the blocking buffer at a concentration of 5 nmol/L and incubated with the membrane for 2 h at room temperature. Cry4B binding to BT-R3 was detected using anti-Cry4B antibody and visualized with rabbit anti-goat antibody coupled with horseradish peroxidase (Pierce, Rockford, IL, USA) using an Amersham ECL Plus Western Blotting Detection System (GE Health-Care Bio-Sciences Corp., Piscataway, NJ, USA).
Results
Expression, purification and activation of recombinant Cry4B toxin
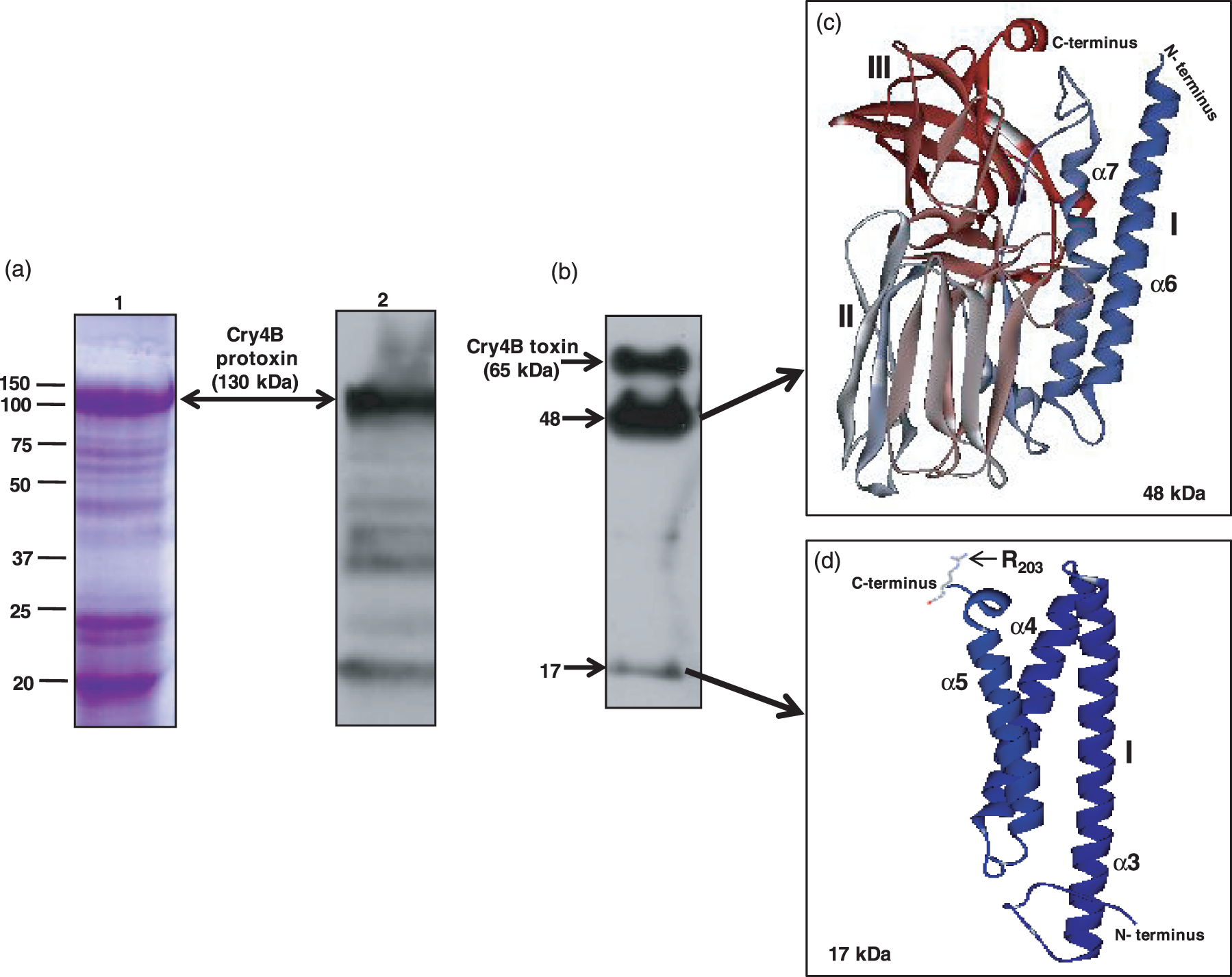
Expression of cloned cry4B protoxin in E. coli was confirmed by SDS-PAGE and Western blot analysis (Figure 2). As can be seen in Figure 2(a), the molecular weight of Coomassie-stained and immunoblotted protoxin was ∼130 kDa (double arrow, lanes 1 and 2, respectively). Recombinant protoxin was partially purified and activated to toxin (65 kDa) using trypsin. Figure 2(b) is a Western blot showing three major polypeptides (65, 48 and 17 kDa, arrows) produced by activation of the protoxin. The 48- and 17-kDa polpeptides are trypsin cleavage products that constitute the 65-kDa toxin. The 48-kDa polypeptide consists of α-helices 6 and 7 of domain I plus domains II and III (Figure 2c) and is not toxic. The 17-kDa polypeptide represents the remainder of domain I (Figure 2d), which also is not toxic. A trypsin cleavage site (R203) is located in the loop connecting α-helices 5 and 6 (Figure 2d, arrow).
Expression of Cry4B protein in Escherichia coli and its proteolytic activation by A. gambiae gut juice. (a) Inclusion bodies from recombinant E. coli were purified and solubilized at pH 12. Soluble fractions were analysed by 10% SDS-PAGE (lane 1) and detected by immunoblotting with anti-Cry4B goat antibody and horseraddish peroxidase conjugate (lane 2). (b) Cry4B protoxin (∼130 kDa) was converted to toxin (∼65 kDa) by subjecting protoxin to A. gambiae gut juice. Cry4B proteolytic fragments (48 and 17 kDa, arrows) were separated by 15% SDS-PAGE and transferred to PVDF membrane. Molecular weights (kDa) are indicated to the left of a and b. c and d show the predicted 3D structures of the 48-kDa and 17-kDa proteolytic fragments in ribbon format, respectively. The 3D structures were generated based on the known crystal structure of Cry4Ba (residues 84–641; PDB: 1W99)
20
and by using the SWISS-MODEL
30
protein modeling server as previously described.
6
Structural features of BT-R3
The nucleotide and deduced amino acid sequences of bt-r3 are deposited in GenBank under the accession number KC310451. Structural features of BT-R3 are based on different motif-search algorithms as described in ‘Materials and methods’ section. Using these algorithms, computational motif predictions were performed based on known and well characterized proteins in different databases. A schematic representation of predicted structural features of BT-R3 is presented in Figure 3. As portrayed in Figure 3(a), BT-R3 contains 1725 amino acids and consists of an N-terminal SP (amino acids 1–25), with putative peptidase recognition sequence between C20-Q21. Downstream of the SP is the EC, which consists of 11 EC modules (amino acids 166–1456), followed by the MPED (amino acids 1457–1569) and the TM (amino acids 1564–1587). The CYTO contains amino acid residues 1588–1725. Not shown are 11 putative calcium-binding motives—DRD, DPD or DYD—distributed throughout the EC domain as well as one DRD signature located in the CYTO. Two putative integrin recognition motifs, RGD and LDV, were identified in EC1 and EC5. Unlike the BT-R1 in moth
3
, no cadherin binding sequence (HAV) is present in BT-R3.
Binding of Cry4B toxin to the EC7-11:MPED region of BT-R3. (a) Schematic diagram portrays the domain composition of BT-R3 and the EC7-11:MPED that was cloned and tested for Cry4B toxin binding. (b) SDS-PAGE reveals the total cellular proteins of E. coli harbouring the recombinant plasmid pET102/EC7-11:MPED after IPTG induction (lane 1). Lane 2 represents total soluble proteins transferred to PVDF membrane, which was subsequently overlaid with Cry4B toxin. Binding of toxin to the EC7-11:MPED protein was detected using anti-Cry4B antibody and anti-goat HRP conjugate (arrow). Lane 3 is a negative control, which was not overlaid with Cry4B. (c) Total soluble proteins shown in lane 1 were loaded onto a column containing Ni+2-sepharose beads and allowed to bind to the beads through His-tag fusion. Cry4B was added to bound EC7-11:MPED. After extensive washing, all bound proteins were eluted with SDS-sample buffer, separated by SDS-PAGE, transferred to PVDF membrane and detected with anti-Cry4B antibody. Lane 1 represents Cry4B toxin binding to EC7-11:MPED. The monomeric form of Cry4B (∼65 kDa) is indicated by a white arrow in lane 1. Binding of the 48-kDa fragment (black arrow) also is evident. Lane 2 is a negative control in which EC7-11:MPED was excluded. The 48-kDa fragment of Cry4B bound non-specifically to the Ni+2-sepharose beads (lane 2, black arrow). Molecular weight markers are indicated on the left of the gels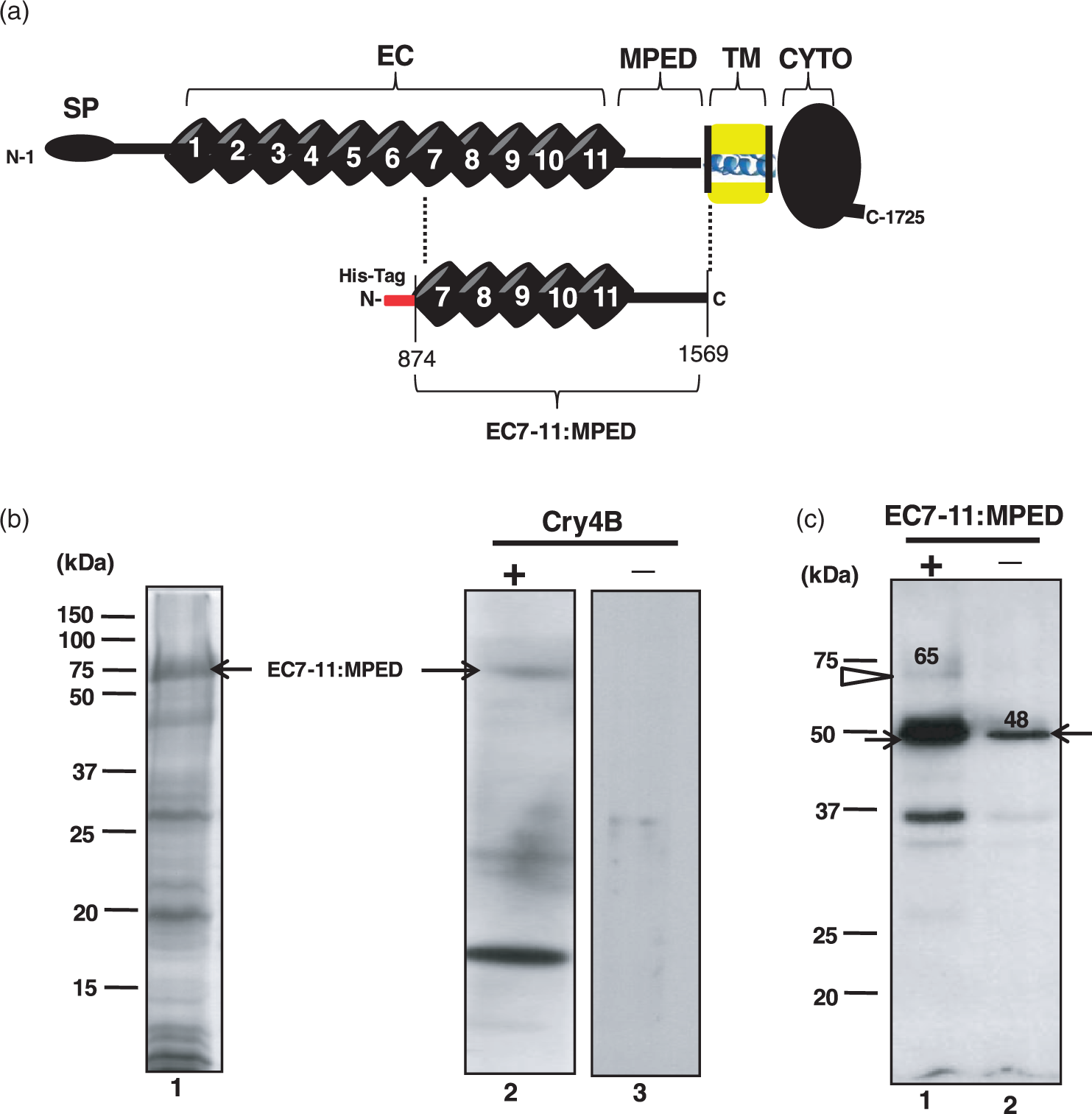
BT-R3 has 43% and 36.5% amino acid identity with two orthologous mosquito cadherins, XP_001652804 in Aedes aegypti and XP_001864657 in Culex pipiens var. quinquefasciatus, respectively. When compared to orthologs from moth and beetle representatives, BT-R3 shares 23% and 17% amino acid identity with BT-R1 of Manduca sexta (AAG37912) and TmCad1 of Tenebrio molitor (ABL86001), respectively. Despite the low sequence homology with these two cadherins, BT-R3, as well as the other mosquito cadherins, does share the same topology, in terms of domain organization, with BT-R1 and TmCad1. 21
Binding of Cry4B to BT-R3
The binding of recombinant Cry4B toxin to BT-R3 was accomplished by utilizing a segment of the receptor that contains the TBR. The segment extend from EC7 through the MPED and is designated EC7-11:MPED (amino acid residues 874–1569, Figure 3a). The part of bt-r3 that encodes for EC7-11:MPED (75 kDa) was cloned in pET102-D-TOPO and expressed in E. coli BL21-DE3 as a fusion protein linked to His-tag (Figure 3b, lane 1, arrow). The partially purified EC7-11:MPED was transferred to PVDF membrane, overlaid with Cry4B toxin and the membrane probed with anti-Cry4B antibody (Figure 3b, lane 2). As seen in Figure 3(c), EC7-11:MPED bound Cry4B toxin (lane 2, arrow) whereas no binding was detected in the absence of the toxin (lane 3).
Binding of Cry4B to BT-R3 in solution using affinity-tagged recombinant protein immobilized on Ni+2-sepharose beads revealed interaction not only with the 65-kDa toxin monomer (Figure 3c, lane 1, white arrow) but also with its 48-kDa cleavage product (black arrow). The toxin monomer was not detected in the absence of EC7-11:MPED although the 48-kDa fragment appeared to bind to the sepharose beads non-specifically (Figure 3c, lane 2, black arrow). Certainly, binding to this toxin fragment was greater in the presence of EC7-11:MPED than without, as evidenced by the more intense staining of the fragment in lane 1 (Figure 3c).
Cytotoxicity of Cry4B
H5 cells do not contain Cry toxin receptors and, as such, are not susceptible to any of the Cry toxins. Therefore, we routinely use these cells as surrogates for heterologous expression of receptor cDNA. In this study, H5 cells were transfected with the bt-r3 coding sequence using the pXINSECT-DEST38/pBmA-neo system, which includes the silk moth (Bombyx mori) cytoplasmic actin gene promoter, Pactin, upstream of the bt-r3 coding sequence and the plasmid vector pBmA-neo for antibiotic selection. Stably transfected H5 cells, referred to as M5, expressed bt-r3 as evidenced by RT-PCR (results not shown).
As anticipated, Cry4B was not toxic to H5 cells. They exhibited normal cell growth and cell division when exposed to 187.5 nmol/L Cry4B toxin in the culture medium (Figure 4a, lower left panel). In contrast, most of the M5 cells (>80% of the entire population) were killed by 187.5 nmol/L Cry4B toxin as demonstrated by Trypan blue exclusion (data not shown). Indeed, they underwent sequential cytological changes, including (i) membrane blebbing and ruffling within 40 min after exposure, (ii) cell swelling within 60 min after toxin exposure and (iii) cell lysis 90 min after exposure. Figure 4(a) (lower right panel) displays cells undergoing blebbing and swelling (arrows). A time-course of morphological events associated with M5 cells in the presence of Cry4B toxin is recorded micrographically in Figure 4(b). The lethal concentration that kills 50% of toxin-treated larvae (LC50) of Cry4B for M5 cells was 75 nmol/L.
Selective cytotoxicity of Cry4B to M5 cells. (a) Cytological changes in M5 cells associated with the progression of Cry4B toxin-induced cell death (187.5 nmol/L Cry4B) include membrane blebbing and cell swelling (arrows, lower right panel) and lysis. No such effects occurred in toxin-treated non-transfected H5 cells (lower left panel). Upper left panel shows normal H5 cells and upper right panel shows mock-transfected M5 cells, both in the absence of toxin. Bar = 10 µm. (b) The sequence of cytological changes during the course of toxin-induced cell death was captured by phase-contrast microscopy. The long arrow beneath the photographs indicates the relative time for each stage of cell death, i.e. toxin binding, membrane blebbing and cellular swelling. (c) Interaction of Cry4B toxin with cell membrane of H5 cells (lane 1) and M5 cells (lane 2) was assessed by immunoblotting using anti-Cry4B antibody. Lane 1 contains membrane fraction of H5 cells and lane 2 contains that of M5 cells. The black arrow points to a tetrameric form of Cry4B (∼240 kDa), which can be seen in both H5 and M5 cells. The white arrow points to the monomeric form of Cry4B (∼65 kDa), which is unique to M5 cells. The grey arrow points to the 48-kDa fragment of Cry4B. Molecular weight markers are indicated on the left side of the gel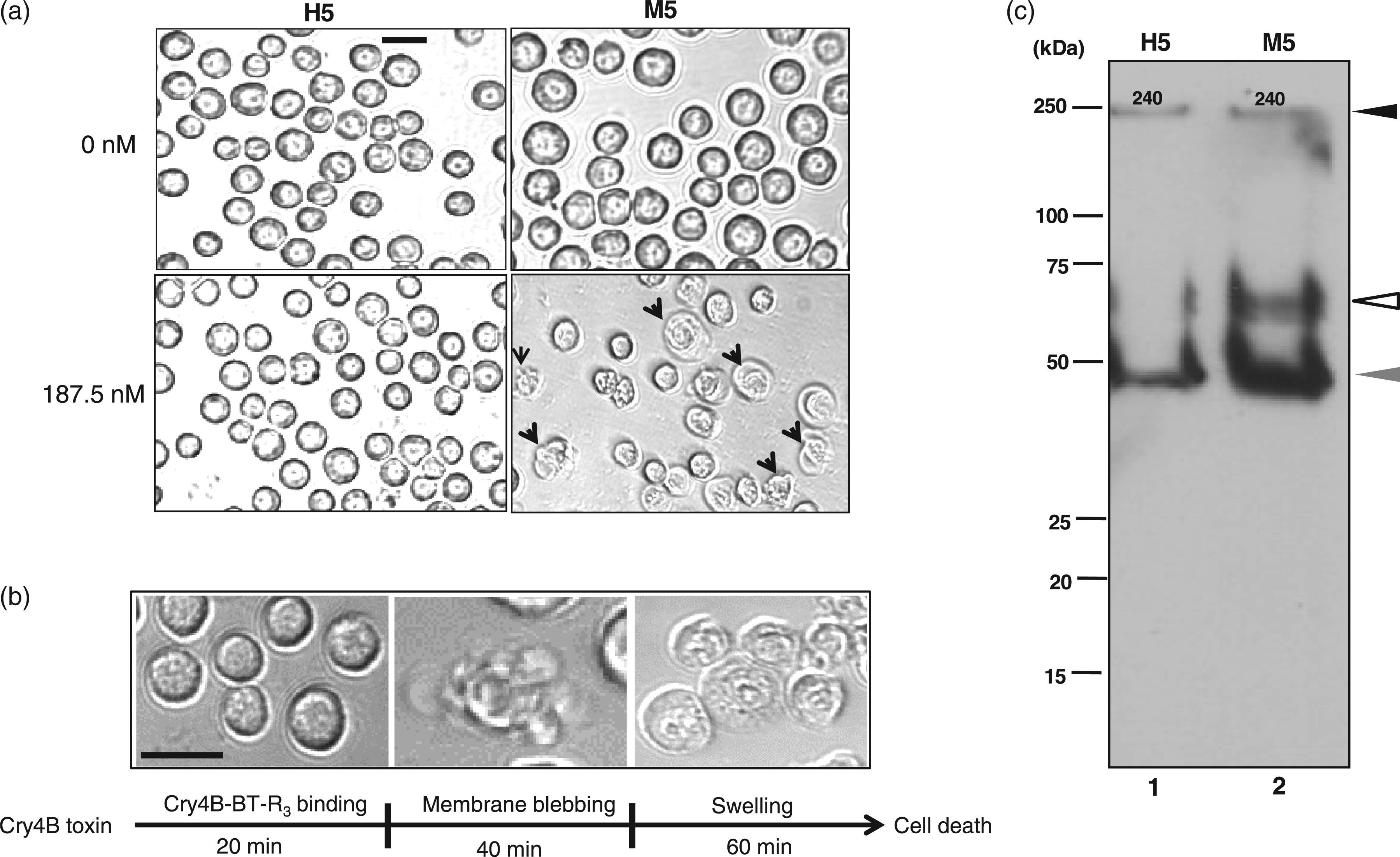
Interaction of Cry4B with cell membrane
In the tobacco hornworm, the first step in Cry1Ab toxin action is binding of toxin to the BT-R1 receptor located in the cell membrane.1,2 Importantly, it is the monomeric, not the oligomeric, form of Cry1Ab toxin that binds receptor.1,2 Oligomers of Cry1Ab can associate non-specifically with the surface of H5 cells without effect. We were interested in determining whether Cry4B interacts with the cytoplasmic membrane of H5 and M5 cells in a similar manner. Therefore, we analysed toxin-membrane interaction in both H5 and M5 cells by examining protein fractions prepared from cell membrane after treatment of the cells with Cry4B (Figure 4c). Antibody against Cry4B toxin was used to detect the toxin in protein fractions.
Upon treatment with Cry4B, the toxin was detected as a monomer (∼65 kDa) in the membrane fraction of M5 cells (Figure 4c, lane 2, white arrow), not H5 cells (Figure 4c, lane 1). A tetrameric form of Cry4B (∼240 kDa) was detected in membrane fractions of both H5 and M5 cells (Figure 4c, lanes 1 and 2, respectively, black arrow), demonstrating that toxin oligomer was associated with membrane regardless of receptor. Also, the 48-kDa cleavage product of Cry4B was detected in both H5 and M5 membrane fractions (Figure 4c, lanes 1 and 2, respectively, grey arrow). Significantly, the toxin tetramer was SDS-resistant. It remained intact irrespective of sample treatment prior to gel electrophoresis and Western blotting. Heat treatment (including boiling) in the presence of β-mercaptoethanol and SDS did not dissociate toxin tetramer. These results indicate that the specificity of toxin interaction, which lead to cytotoxicity, correlated directly to the binding of monomeric toxin with the M5 cells expressing BT-R3.
Structural uniqueness of BT-R3 among other A. gambiae cadherins
In general, the TBR of all Cry toxin receptors identified so far, including BT-R3, were characterized by the presence of two highly conserved stretches of amino acid residues (highlighted in black boxes) within their N- and C-termini that flank the membrane-proximal EC module (Figure 5). These sequences represent signature motifs that fold close to each other and mark the toxin-binding function in these receptors.4,21 We were interested in comparing the amino acid sequence of the TBR in BT-R3 (amino acid residues 1337–1452) to corresponding regions in selected cadherins contained in A. gambiae (refer to Figure 1). Based on the phylogenetic relationships presented in Figure 1, we selected cadherins that represent the three distinct phylogenetic groups I, II and III—XP_310987 (residues 1516–1635), XP_001688008 (residues 1074–1239) and XP_312660 (residues 143–332). Figure 5 shows amino acid sequence comparisons and secondary structure approximations (Figure 5a–c) together with three-dimensional folding predictions (Figure 5d–g).
Comparative structural analysis of the Cry4B toxin-binding region (TBR) of BT-R3 and homologous regions in selected cadherins representing three phylogenetic groups (I, II and III; Figure 1) of A. gambiae. a, b and c are multiple sequence alignments and secondary structure predictions showing the putative TBR of BT-R3 (residues 1337–1452) and corresponding regions in XP_310987 (group I, residues 1516–1635), XP_001688008 (group II, residues 1074–1239) and XP_312660 (group III, residues 143–332). The predicted secondary structures, β-strands and loops, are shown above the amino acid sequence in BT-R3 and below the amino acid sequence in XP_310987, XP_001688008 and XP_312660. Gold arrows and black solid lines highlight predicted β-strands and loops, respectively, for BT-R3, whereas red arrows and black solid lines highlight predicted β-strands and loops, respectively, for XP_310987, XP_001688008 and XP_312660. The black boxes denote signature sequences in BT-R3. The red cylinder in XP_310987 signifies predicted α-helices. Multiple sequence alignments were performed using CLUSTALW and secondary structure prediction was accomplished using JPred 3.
31
d, e, f and g are proposed three-dimensional (3D) models in ribbon format for the TBR of BT-R3, and equivalent regions in XP_310987, XP_001688008 and XP_312660, respectively. The 3D structure predictions for BT-R3, XP_310987 and XP_312660 were generated based on the 3D structure of the EC1-EC2 of cadherin 23 (PDB code 3mvs).
32
The 3D prediction for XP_001688008 was generated based on the 3D structure of the LG1-3 region of the laminin α2 chain (PDB code 2wjs).
33
All 3D models were generated and viewed using the SWISS-MODEL
29
protein modeling server as previously described.
6
β-strands of all 3D are represented in blue and α-helices of XP_310987 is highlighted in red. Loops and coiled coils are shown in yellow. N and C denote N- and C-termini, respectively
The two BT-R3 signature sequences form two adjacent β-strands within the β-barrel fold of EC11 which is immediately adjacent to the MPED (Figure 5a, gold arrows: β-strands; black solid lines: loops; black boxes: signature sequences). The precise positioning of the two β-strands is a hallmark of all Cry toxin receptors thus far reported. This area provides an interface for toxin binding. The three-dimensional structure predictions revealed that the corresponding region in the other three cadherins (Figure 5e–g, red arrows: β-strands; black solid lines: loops; red cylinders: α-helices) is different from that in BT-R3 (Figure 5d). For example, no BT-R3 signature motifs exist in XP_310987 (group I, Figure 1) and XP_001688008 (group II, Figure 1). Furthermore, the C-terminal end of XP_001688008 consisted mainly of loops and coils and lacks β-strand. The equivalent regions of XP_310987 and XP_001688008 are not in close proximity to each other (Figure 5a and e and Figure 5b and f, respectively). XP_312660 (group III, Figure 1), which has sequence similarity in the equivalent region, is located in the distal area of the EC domain (structure not shown), not adjacent to the MPED as is the case for BT-R3. These findings point up the unique features of BT-R3 and, most likely, why the signalling events which lead to cell death depend on binding of Cry4B toxin to the conserved signature motifs in EC11 of this particular cadherin.
Discussion
It is well established that conserved regions in all Cry toxins studied so far are responsible for receptor binding. Likewise, there are highly conserved structural motifs in the cognate Cry toxin receptors, which are responsible for toxin binding. Inspection of some insects known to be susceptible to Cry toxins reveals that their cadherin receptors are related phylogenetically, clustering in a single group (group I, Figure 1). Foremost among this group is BT-R3 in A. gambiae, which binds Cry4B toxin (Figure 3), the primary mosquitocidal toxin produced by Bti in a membrane-encased protein packet alongside the endospore. 6 Those cadherins in A. gambiae not implicated in Cry toxin binding are more distantly related phylogenetically (groups II and III, Figure 1).
Unlike anthrax toxin, a three-protein exotoxin that is secreted by B. anthracis and the emetic toxin and enterotoxins produced by B. cereus, all of which make holes or channels in membranes,22,23 the Cry4B toxin binds to a cell surface cadherin, activating a death pathway. Indeed, binding of Cry4B toxin to BT-R3 in High Five insect cells brought about rather dramatic cytological changes (Figure 4). The time required for toxin to render dead cells was relatively short (about 60 min after exposure), which compares with the time required for the moth toxin Cry1Ab to kill High Five cells containing the BT-R1 cadherin receptor. 2 The sequential changes resemble those associated with oncosis, which involves membrane blebbing and ruffling along with the appearance of ghost nuclei followed by cell swelling and lysis. 24
As is true for all known Cry toxin cadherin receptors, it is the membrane-proximal EC module that is responsible for toxin binding and, ultimately, toxin action. We believe that the region of BT-R3 to which Cry4B binds (TBR) is located within the EC11, which is adjacent to the MPED (Figures 3a and 5d). Apparently, the location of the TBR is critical to the binding affinity and specificity of not only the Cry4B toxin but all other Cry toxins as well.
Just as the position of the TBR within the receptor is important, so is its structural motif. Whether the selective action of Cry4B for A. gambiae is dependent on conserved signature sequences, which characterize the toxin-binding function of BT-R3, remains to be determined. Two highly conserved amino acid sequences lie within the N- and C-termini of the TBR of BT-R3 (Figure 5, black boxes) and form two β-strands within the β-barrel fold of EC11. Not unlike other Cry toxin receptors, their exact arrangement is a distinguishing feature that establishes binding of Cry4B to BT-R3. The lack of BT-R3 signature motifs, or the displacement of sequences similar to such motifs in other A. gambiae cadherins not closely related phylogenetically, most likely preclude their ability to bind Cry4B. Remarkably, mosquitocidal toxin does not bind BT-R1 and does not kill the moth M. sexta. 11
As previously noted, Cry4B is unique among the Cry toxins in that it represents the key biological agent produced by Bti that kills A. gambiae, the principal mosquito vector of malaria. 6 Upon ingestion, the Cry4B toxin is activated by enzymes in the gut juice of mosquito larvae. Mosquito larval gut juice is rich in proteolytic enzymes—most notably trypsin. One of the main features of Cry toxins is their resistance to trypsin action. However, Cry4B is not resistant to trypsin and is cleaved by the enzyme into two fragments (48 and 17 kDa, Figure 2). A similar finding was previously reported. 25 Neither fragment is insecticidal 26 even though the 48-kDa cleavage product is associated with the plasma membrane of M5 expressing the BT-R3 receptor (Figure 4c). The transient nature of the 65-kDa Cry4B toxin, i.e. its instability, may explain why its incorporation into M5 membrane is less than that of Cry1Ab into S5 cell membrane containing the BT-R1 receptor. 1 The LC50 value for Cry1Ab against third-instar M. sexta larvae is 200 ng/cm 2 whereas the LC50 value for Cry4B against A. gambiae is 2 µg/mL.27,6 Less Cry4B than Cry1Ab available for incorporation into larval midgut cell membrane may account for the 10-fold difference in Cry4B toxicity compared to Cry1Ab.
As is true for Cry1Ab binding to BT-R1, 1 binding of the monomeric form of Cry4B to BT-R3 is an important condition for cytotoxicity. M5 cell membranes harvested from cells after toxin exposure contained Cry4B monomer as well as oligomer (240-kDa tetramer, lane 2, Figure 4c) whereas no monomer, only oligomer, was associated with H5 cell membranes lacking the receptor (lane 1, Figure 4c). As was the case for Cry1Ab, 1 the two forms of the Cry4B toxin appear to be mutually exclusive relative to toxin action because it is quite apparent that Cry4B tetramer has no obvious effect on H5 cells deficient in BT-R3 (Figure 4a, lower left panel). On the other hand, only those cells harbouring BT-R3 and displaying Cry4B monomer on their surface were susceptible to the toxin (Figure 4a, lower right panel). A homolog of BT-R3 from A. gambiae has been reported and shown to bind Cry4B in brush border membrane vesicles. 28 However, Drosophila cells transiently expressing the homolog did not bind Cry4B nor were they killed by the toxin. 28
Validation of BT-R3 as a functional receptor for Cry4B exemplifies the power of bioinformatics, genomics and proteomics to identify target proteins, enabling the design of specific primers or probes for subsequent cDNA screening. 9 The process of target selection starts with data mining of archived protein sequences available in various genome and proteome databases, and results in the selection and annotation of candidate proteins based on their potential to mediate insecticidal action. It brings together genome- and proteome-based target identification and target-directed screening for validating the action of insecticidal proteins such as Cry4B—engineered or otherwise. Targets selected for consideration can then be analysed in silico by docking calculations, molecular dynamics simulations and other techniques to characterize appropriate target interactions with chemically or genetically altered Cry toxins. We believe that this kind of strategy will facilitate protein design for creation of new customized Cry proteins and peptide mimics that might be more effective than the natural toxins themselves and, hopefully, less able to bring about insect host resistance.
Obviously, new approaches are needed in the area of malaria control because no effective vaccine currently exists and no available antimalarial medications have been designed or discovered that impede parasite resistance with long-term use. Certainly, genetic modification of wild mosquito vector populations to reduce vectorial capacity is fraught with unknown and hidden trials and tribulations.
Footnotes
Author contributions
All authors participated in the design, interpretation of the studies, analysis of the data and writing and review of the manuscript. MAI conducted all cloning and transfection experiments and constructed all 3D models. NBG participated in protein purification, characterization and analysis as well as maintaining cell cultures.
Acknowledgement
This work was supported by a grant from the National Science Foundation (Award No. 0412257).