
Abstract
Transcriptional factor paired box 6 (PAX6) is very important for the development of the eyes, central nervous system, and pancreas. PAX6 mutations are associated with a diabetic phenotype and abnormal glucose metabolism. Our previous study showed that PAX6 directly bound to and activated the prohormone convertase 1/3 (Pc1/3) gene promoter and subsequently regulated proinsulin processing. Prohormone convertase 2 (PC2) is the essential enzyme for pancreatic proinsulin processing. To study the regulation of PAX6 in Pc2 expression, we did research on the pancreas of Pax6 R266Stop mutant mice, where truncated mutations happened in the C-terminal of the PAX6 protein. Our studies showed that the mutant PAX6 protein was stable and regulated the activity of Pc2 promoter as shown by luciferase activity assays. We found that the wild-type PAX6 protein imparts a transcriptional effect, and the mutant PAX6 can also regulate the downstream molecules. The results provide new insights into the mechanism of truncated PAX6 in regulating the functions of the pancreas and endocrine system.
Introduction
Paired box 6 (PAX6) is a transcriptional factor and belongs to the paired-box gene family.1,2 It plays a crucial role in regulating the development of several organs, such as pancreas, central nervous system, and eyes.3–6A paired domain, a homeodomain, a linker region, and a transactivation region constitute the PAX6 protein.1,2 The paired domain, together with the homeodomain, binds to the promoter DNA, so that the transactivation region could regulate the expression of its target genes.7,8
Beings with Pax6 mutation showed some naturally occurring mutant phenotypes: aniridia in humans,9–15 small eye (Sey) in rodents,16–18 and lack of eyes in Drosophila. 19 Most intragenic mutations lead to truncated PAX6 protein. The mutations of PAX6 proteins happen at its C-terminal region; however, its result on the mutants is not clear.20–22 Singh et al. 23 reported that the mutant PAX6 proteins not only were stable or loss of function simply, but also the mutants obtained strong DNA-binding capacity and functioned as dominant-negative repressors because of the lack of most of the transactivation activity. We found that PAX6 mutants within intact DNA-binding domains could inhibit downstream molecules’ expression by inhibiting their promoter activity.
In our previous study, we showed that PAX6 regulated the expression of prohormone convertase 1/3 (Pc1/3) directly, which encoded the PC1/3 protein, a serine protease essential for proinsulin processing. The downregulation of PC1/3 resulted in defective proinsulin processing and impaired glucose metabolism in PAX6-deficient patients and in Pax6 mutant mice. 24 PC2 is also a crucial protease for proinsulin processing.25,26 In this study, we showed that Pc2 expression is repressed in islets of Pax6 mutant mice. We found that the PAX6-truncated mutation functioned as a repressor to inhibit Pc2 expression in the mouse pancreatic islets.
Materials and methods
Cell culture, plasmids, and transfection
The mouse pancreatic β-cell line NIT-1 cells 27 were maintained in 1640 PRIM with 10% fetal bovine serum (FBS). HEK293 cells were cultured in DMEM/high glucose with 10% FBS. All of the cell culture media were products of Gibco (Gaithersburg, MD, USA), whereas FBS was obtained from Biochrom AG (Biochrom AG, Berlin, Germany). siRNAs for Pax6 (siRNA ID: s71268/s71269) were obtained from Ambion (Austin, TX, USA), and all results about RNAi were repeated and confirmed using different siRNA IDs. Following the manufacturer’s protocols, cells were transfected with siRNA using lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) or lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Animals
The animals used were described previously. 24 Simply, an injection of the chemical mutagen N-ethyl-N-nitrosourea caused a stop-codon mutation at 266aa of Pax6. Then the mutation was transmitted to newborns by breeding the heterozygous Pax6 mutant mice with normal C57BL/6 J mice. The mice with heterozygous Pax6 gene mutation were selected based on the Sey phenotype. Five to six mice were housed per cage in a controlled environment, at a constant temperature (23.8℃), with 12-h light/dark cycle, and free access to water and regular mouse chow. The study on animals was strictly carried out according to the ‘Guide for the Care and Use of Laboratory Animals of the National Institutes of Health’ 28 . All animal protocols were approved by the Peking University Animal Ethics Committee. And we have made all efforts to minimize suffering. All relevant data without special explanation were obtained from the mice of six months after birth.
Isolation and selection of mice pancreatic islets
Pancreatic islets of male mice were isolated following Hori’s method. 29 Briefly, using type V collagenase (Sigma-Aldrich, St Louis, MO, USA), mice pancreases were digested and then centrifuged (2700 × g) in cold PBS. Finally, individual and complete islets were handpicked under a dissecting microscope, without any attached vascular, acinar, or ductal tissues.
Quantitative RT-PCR
The RNA of mice islets or cultured cells was isolated using an RNeasy Micro Kit (Qiagen, Valencia, CA, USA) and reversed to cDNA. Quantitative RT-PCR (qRT-PCR) was performed using forward and reverse primers to amplify Pax6 mRNA (5′-ACCCGGCAGAAGATCGTAG-3′, 5′-TTTGCATCTGCATGGGTCT-3′), Pc2 (5′-AGCTGGAGAGGTGGATTTTGA-3′, 5′-CTCATCCTGGGAGGACAGA-3′), or Gapdh (5′-CGTGCCGCCTGGAGAAACCTG-3′, 5′-AGAGTGGGAGTTGCTGTTGAAGTCG-3′). The results were calculated using the comparative threshold method with Gapdh as an endogenous control. All reactions were conducted in triplicate.
Western blotting analysis
Protein samples were obtained by lysing isolated islets or cells using lysis buffer (20 mmol/L Tris-HCl, pH 7.5, 1% Nonidet P-40, 150 mmol/L NaCl), sonicating on ice, centrifuging (10,000 × g) for 15 min. Supernatants were collected, and then 60 µg of protein was fractionated using 10% SDS-PAGE. Protein was probed with antibodies after transferring to polyvinylidene difluoride membranes. Rabbit PC2-specific antibody (Abcam), rabbit PAX6-specific antibody (Abcam, Cambridge, UK), and mouse β-actin-specific monoclonal antibody (Sigma-Aldrich, St. Louis, MO, USA) were used in this experiment.
Immunofluorescence
The mouse pancreases were fixed using 4% paraformaldehyde in PBS at 4℃ for about 10 h, and then prepared as 5-mm-thick cryosections. The cryosections were immunostained using rabbit antibody to PC2 (Abcam, Cambridge, UK) overnight at 4℃, incubated first with anti-rabbit IgG, and then with the mounting medium (DAPI, Vector Laboratories, Inc., Burlingame, CA, USA).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assay was performed on the NIT-1 cells. Simply, the antibody specific to PAX6 (Chemicon, Rosemont, IL, USA) was used to immunoprecipitate the chemical-linked complexes of PAX6 protein and genomic DNA. The antibody–protein–DNA complexes were collected. After releasing the crosslinks, purified DNA was analyzed with PCR by using the following primers: 5′-CAGTCCGGACTCCCGCTGGACT-3′ and 5′-TCCCTGTTGCTCTTCTTTGCTA-3′ to amplify the −815 to −525 region, 5′-TAGACCAGGATGGCCTCAAACTAAAA-3′ and 5′-AGCCCTAAACCCTTACTTCTGACCT-3′ to amplify the −4116 to −3526 region of PC2 promoter.
Electrophoretic mobility shift assay
Using LightShift Chemiluminescent EMSA Kit (Pierce, Rockford, USA) electrophoretic mobility shift assay (EMSA) was performed according to the manufacturer’s protocols. Briefly, 5 µg of nuclear extract from NIT-1 cells was added to a reaction mixture. The following oligonucleotides were used: 5′-TTTGCCCAAAGTGGAGGGGGCGGGAAGG-3′. 32 P was used to label the 5′ terminal of the probes at. We preincubated the nuclear extracts with antibody on ice for 10 min, then added the DNA probe labeled by 32 P, subsequently incubated the mixture at room temperature for 10 min. A probe unlabeled at 100-fold excess was used as the competitor. All reaction mixtures were separated with a 6% nondenaturing polyacrylamide gel, and then the gel was exposed for 1 min to X-ray film.
Luciferase reporter assay
Human PC2 promoter (−4.5 kb to −1 bp) luciferase reporter vector was kindly provided by Theodore C. Friedman, University of California. Wild-type Pax6 and mutated Pax6 were cloned and inserted into pcDNA3.1 (−) vectors. HEK293 cells were cultured in 24-well plates and 24 h later the cells were transfected with mouse Pc2 promoter or pGL3-promoter vector along with pcDNA3.1 (−) vector, full length or truncated Pax6 cDNA at varying doses, while the truncated Pax6 cDNA expressed the moiety as in the Pax6 R266Stop mutant. After transfection for 48 h, cells were collected and assayed in triplicate with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Statistical analysis
Data are presented as mean ± SEM and analyzed by independent samples t-test. P < 0.05 was considered statistically significant.
Results
Our previous study showed that PAX6 regulated glucose metabolism through proinsulin processing mediated by PC1. To detect whether PC2, another crucial enzyme for proinsulin processing, is altered in Pax6 mutated mice, a heterozygous Pax6 R266Stop mutated mouse model
24
was used to analyze the production of Pc2. The mRNA and protein levels of PC2 were significantly decreased in Pax6 mutated pancreatic islets, detected using qRT-PCR and Western blotting analysis, respectively (Figure 1(a) and (b)). NIT-1, a β-cell line,30,31 was used as a protein control for Western blotting analysis. Immunofluorescent results also showed a decreased PC2 staining in islets of Pax6 mutants compared with wild-type islets (Figure 1(c)).
Pax6 mutation led to the decrease of PC2 production in pancreatic islets of Pax6 R266Stop mutant mice. The mRNA and protein levels of PC2 were significantly decreased in Pax6 mutated pancreatic islets compared with the wild type detected with qRT-PCR (a) and Western blotting analysis (b). (**P < 0.01, n = the number of the animal from which the sample was obtained to produce the blot shown). (c), Pax6 mutant mice exhibited lowered production of PC2 detected using immunofluorescence. A representative experiment is shown; the experiment was repeated three times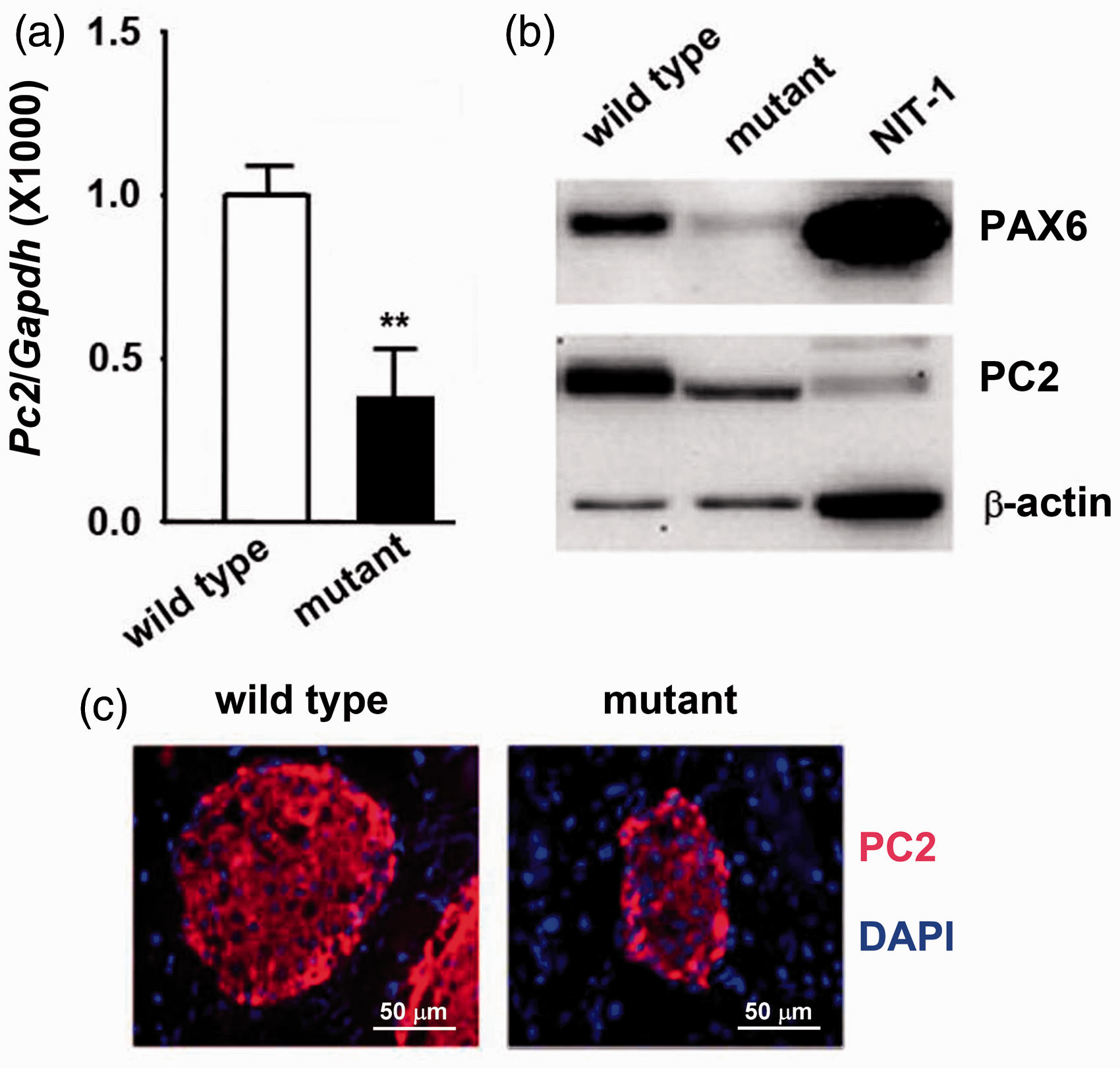
To verify the production of the wild-type and mutant PAX6 protein, we transfected the wild-type and truncated Pax6 expressive plasmid into 293 A cells to perform Western blotting analysis by using a polyclonal antibody which could directly against the N-terminal of PAX6. The mutant PAX6 showed smaller molecular weight and similar protein levels, compared with the wild-type PAX6 (Figure 2(a)). To determine whether the truncated PAX6 affects the expression of Pc2, we overexpressed the mutated PAX6 in NIT-1 cells based on overexpression of wild-type PAX6, and then performed the Western blotting analysis to detect the protein level of PC2. As shown in Figure 2(b), when an equimolar amount of mutant PAX6 was transfected, the production of PC2 was observed to be significantly lower than that in the wild type (Figure 2(b), lane 3). The reduction suggested that mutant PAX6 downregulated Pc2 expression.
Mutated PAX6 protein was stable and repressed the production of PC2. (a) Truncated PAX6 protein was stable and analyzed using western blotting analysis. (b) Overexpression of mutated Pax6 reduced the level of PC2 production in NIT-1 cells detected using western blotting analysis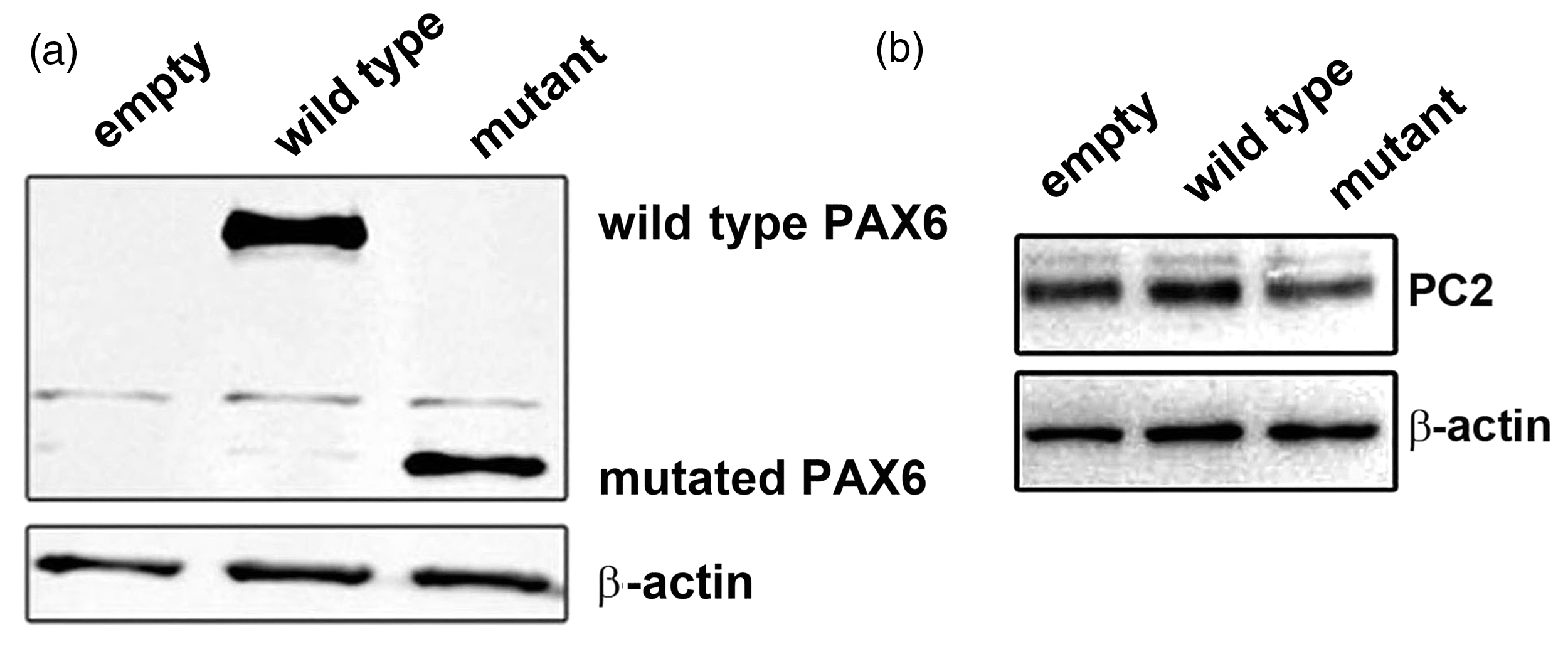
To investigate the method by which the PAX6 mutation led to PC2 deficiency, the binding of PAX6 to the Pc2 promoter was detected using ChIP assay in NIT-1 cells. As shown in Figure 3(a), PAX6 was found to bind to a region of the Pc2 promoter (−815 to −525 bp), which was specific because negligible binding was detected in the genomic sequence −2.5 kb upstream of the Pc2 gene transcription start site or with a control antiserum. This result suggests that PAX6 indeed physically associates with the Pc2 promoter.
PAX6 bound to Pc2 promoter and mutated PAX6 protein directly downregulated Pc2 expression. (a) PAX6 binding to the Pc2 promoter. Soluble chromatin from NIT-1 cells was immunoprecipitated with PAX6 antibody or rabbit normal IgG. The final DNA extractions were PCR amplified using primers that covered the proximal promoter region of the Pc2 gene. (b) Analysis of the binding site of PAX6 to the Pc2 promoter by EMSA. EMSA was performed using nuclear extracts of NIT-1 cells. Specific competitor (cold probe, lane 2) was used as the control. For supershift, antibody to PAX6 (lane 4) or rabbit normal IgG (lane 3) was incubated with nuclear extracts before being added to the reaction mixture. Arrows indicate the shifted or supershifted band and original band of protein–DNA complexes. (c) Wild-type PAX6 showing no effect on the activity of full-length promoter of Pc2 luciferase activities in HEK293 cells. (d) Mutant PAX6 downregulation of the activity of full-length promoter of Pc2 luciferase activities in HEK293 cells in a dose-dependent manner. The experiment shown is representative of three separate experiments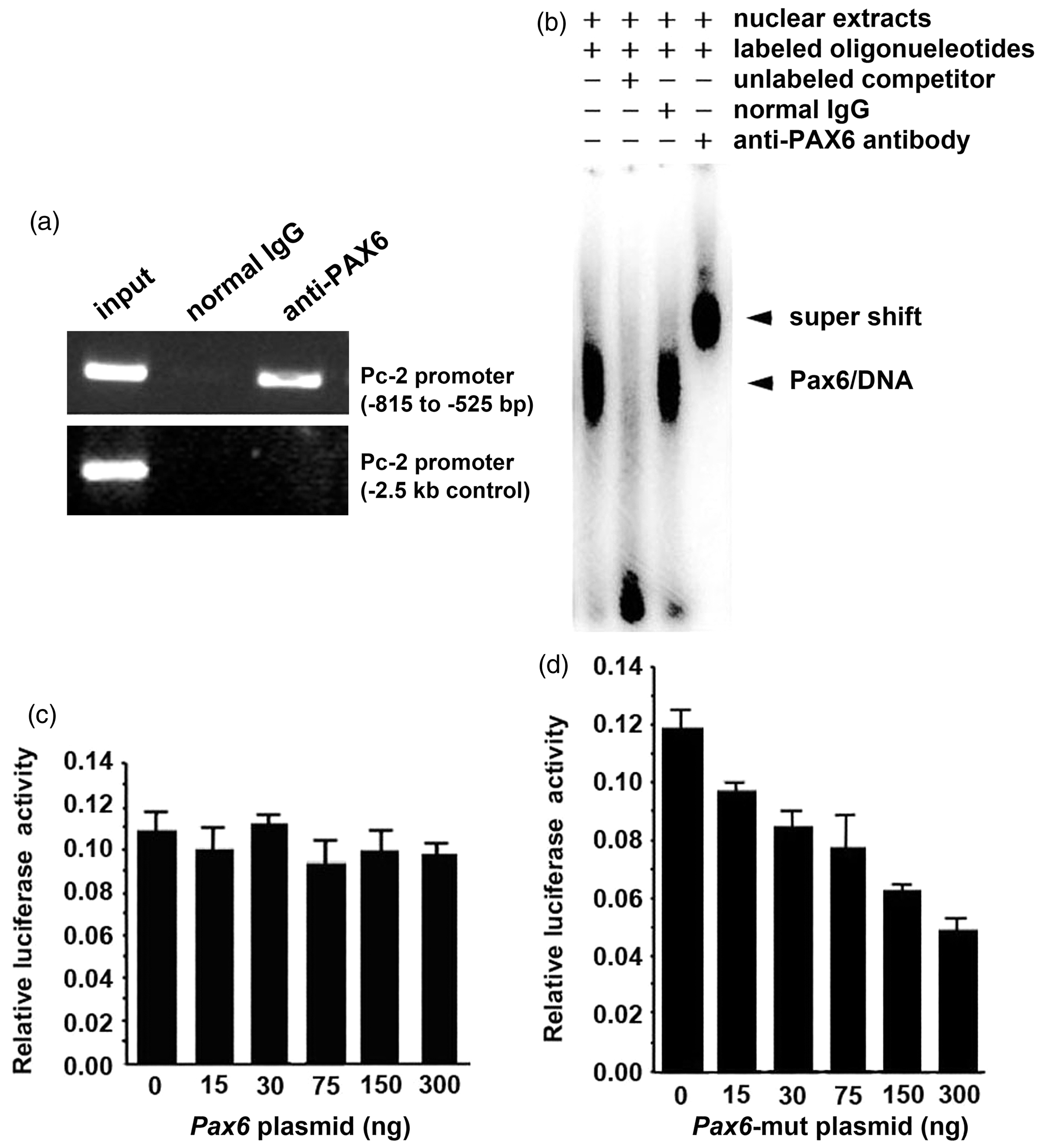
To further confirm the binding of PAX6 to the Pc2 promoter in NIT-1 cells, nuclear extracts from NIT-1 cells were incubated with 32 P-labeled Pc2 promoter fragment, and an EMSA was performed. As shown in Figure 3(b), corresponding to the DNA–protein complex (Figure 3(b), lane 1), a supershift band was observed (Figure 3(b), lane 4) when anti-PAX6 antibodies were added. If the incubation was added with an excessive amount of unlabeled oligonucleotides containing the same region, the band (Figure 3(b), lane 2) completely disappeared (Figure 3(b), lane 2). Because PAX6 can bind to the Pc2 promoter, we next examined the effect of PAX6 on Pc2 promoter activity using Pc2 promoter-driven luciferase reporter constructs. Different from our expectations, the transcriptional activity of the Pc2 gene promoter was not activated by PAX6 (Figure 3(c)). When we detected the effect of truncated PAX6 on Pc2 promoter activity, these results showed that in NIT-1 cells the truncated PAX6 inhibited the Pc2 promoter activity in a dose-dependent manner (Figure 3(d)). These observations are consistent with the study of Singh et al., 23 which reported that the C-terminal-truncated PAX6 proteins are stable, and their DNA-binding function are kept. Their results suggest that these mutations might compete the target DNA-binding sites with wild-type PAX6 to decrease activity.
Taking these data together, we concluded that PAX6 directly bound to the Pc2 promoter region from −681 to −660 bp and that the PAX6 mutation is stable and can reduce the Pc2 promoter activity.
Discussion
Our previous study shows the significance of PAX6 as a crucial regulator of proinsulin processing through transcriptional regulation by directly binding to the Pc1 promoter. 24 Using the mouse model of Pax6 heterozygous R266Stop mutation, we report that mutant PAX6 protein regulates the transcription of Pc2 gene in the pancreas, providing new evidence for the important role of PAX6 in the differentiation and functioning of the endocrine system.
By using MRI assessments, Song et al. 32 reported that similar to the mouse model with the Pax6 mutation, most PAX6 mutation patients showed brain abnormalities. The abnormalities of neural development induced by Pax6 mutations in the embryo have been understood for a long time. These abnormal changes in adult brain should be due to dose accumulation of mutant PAX6 proteins with aging. This study confirmed that mutant PAX6 protein is stable and can downregulate PC2 expression by inhibiting the activity of Pc2 promoter. Furthermore, we characterized functional PAX6 binding site in the Pc2 gene promoter by both ChIP and EMSA assays (Figure 3(a) and (b)). This conclusion is supported by Tang et al., 33 who reported that the stability of mutant PAX6 proteins is not changed, and the binding activity of these mutant protein to promoter DNA remains unaffected. Overall, previously reported study results and our present data indicate that PAX6 is critical for Pc1 and Pc2 gene transcriptions through direct and indirect mechanisms involving mutated PAX6. We found that wild-type PAX6 has no direct role on the regulation of Pc2 expression, which is consistent with the observation of Katz et al., 34 that PAX6 regulates Pc2 expression through cMaf and Beta2/NeuroD. However, their study did not discuss the role of mutant PAX6 protein on Pc2 expression.
Although we found a novel regulation of Pc2 expression by PAX6 deficiency, the mechanism is still unknown. It is possible that PAX6 mutant protein inhibits Pc2 transcription through repressing the binding of a transcriptional-promoting complex at the Pc2 promoter-binding site. Further studies of the definition of this transcriptional complexes and the promoter-binding site are required.
PAX6 not only regulates proinsulin processing but also affects the transcription of glucagon gene and other factors which are important for α-cell function.24,35 All issues, including glucagon concentration in Pax6 mutants and the function of α- and β-cells in aging mice, require further research.
In conclusion, we showed that mutant PAX6 plays critical roles in the pancreatic endocrine system by acting as a repressor of Pc2 expression. We describe the transcriptional network in which PAX6 is a considerable factor for glucose metabolism and endocrine system functioning.
Footnotes
Author contributions
All authors contributed to the design, interpretation of the studies, analysis of the data and review of the manuscript; CYY and CWW (co-first authors) conducted the experiments; ZSX and SL supplied critical reagents; WJH designed the experiments and wrote and reviewed the manuscript.
Acknowledgments
We thank Dr. Theodore C. Friedman (University of California) for kindly providing us human PC2 promoter (−4.5 kb to −1 bp) luciferase reporter vector. This study was supported by Grants 973 Program (2011CB966203 and 2011CBA01102) and International S&T Cooperation Program (2011DFA31040) from the Ministry of Science and Technology of China. It was also supported by the National Natural Science Foundation of China (31171417) and Specialized Research Fund for the Doctoral Program of Higher Education (20103234120003).