
Abstract
Melittin, a major polypeptide in honeybee venom, have been used to treat inflammatory disease. Various studies have demonstrated the anti-bacterial, anti-viral, anti-inflammatory and anticancer effects of bee venom and melittin. However, the precise mechanism of melittin in liver disease is not yet known. Apoptosis contributes to liver inflammation and fibrosis. Knowledge of the apoptotic mechanisms is important to develop new and effective therapies for treatment of cirrhosis. In the present study, we investigated the anti-apoptotic effect of melittin on tumor necrosis factor (TNF)-α/actinomycin (Act) D-induced apoptosis in hepatocytes. Our results show significant protection from DNA damage by melittin treatment compared with corresponding TNF-α/Act D-treated hepatocytes without melittin. Melittin inhibited TNF-α/Act D-induced activation of the caspase, bcl-2 family of proteins and poly ADP-ribose polymerase (PARP)-1. Our results also indicate that melittin decreased nuclear factor-kappa B (NF-κB) by degradation of phosphorylation of IκB kinase (p-IKK) and NF-κB DNA binding activity in TNF-α/Act D-treated hepatocytes. These results suggest that melittin possesses a potent suppressive effect on apoptotic responses in TNF-α/Act D-treated hepatocytes via the NF-κB pathway.
Introduction
Tumor necrosis factor (TNF)-α is a potent cytokine that can elicit a wide range of cellular effects, including proliferation, differentiation, and apoptosis, depending on the cell type. 1 Apoptosis is a cell suicide program characterized by a series of morphologic and biochemical changes, including the accumulation of sub-G1 cell population, fragmentation of the nucleus and DNA, and caspase activation in most cases. 2 In the liver, TNF-α-induced hepatocyte apoptosis has been implicated in many liver diseases. 3 TNF-α combined with Act D is known to induce hepatocyte apoptosis in vitro. 4 Apoptosis of hepatocyte is a major cause of liver injury during endotoxemia and liver ischemia. 5 Reduction/oxidation (redox) state is involved in the hepatocyte apoptosis induced by TNF-α/Act D and fas. 6 Recent study has suggested that the carbon monoxide protects hepatocytes from TNF-α/Act D by inhibition of the caspase-8-mediated apoptotic pathway. 7 TNF-α also mediates both cell survival and cell death signaling through TNF receptor-1 by activating nuclear factor-kappa B (NF-κB) and caspase-8, respectively. 8 NF-κB has been strongly implicated in the regulation of apoptosis induced by death factors, such as TNF-α. 9 However, despite the cell death signaling, hepatocytes are highly resistant to TNF-α-induced apoptosis unless the early activation of NF-κB is blocked. 10 Thus, transcriptional activation by NF-κB plays a pivotal role in the survival of hepatocytes after TNF-α stimulation. 11 Nevertheless, most cell lines are resistant to TNF-α-induced apoptosis, though exactly how this resistance is achieved is still unclear. 12
Bee venom is a natural toxin produced by the honeybee (Apis mellifera), and consists of several biologically active peptides, including melittin, apamin, adolapin, and mast cell degranulating peptide. 13 Recent study has suggested that bee venom protects the activation of the microglial response and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic cell apoptosis. 14 We have recently shown that bee venom protects transforming growth factor (TGF)-β1 - and ethanol-treated hepatocyte injury by inhibiting the activation of the bcl-2 family, caspase, and PARP-1 cleavage.15,16 The major component of bee venom, melittin, has anti-bacterial, anti-viral and anti-inflammatory, in various cell types and it can induce cell cycle arrest, growth inhibition. 17 Melittin inhibits platelet-derived growth factor (PDGF)-BB-induced aortic vascular smooth muscle cell (VSMC) proliferation, a major event in atherosclerosis and restenosis, by suppressing NF-κB, Akt activation, and the mitogen-activated protein kinase (MAPK) pathway. 18 However, the mechanisms of anti-apoptotic effect of melittin have not been fully elucidated in hepatocytes. The present study tested the hypothesis that melittin protects against TNF-α/Act D-induced hepatocyte death via inhibition of apoptosis signaling and NF-κB pathway. We focused our analysis on the precise molecular mechanism of melittin in TNF-α-induced apoptosis in hepatocytes.
Materials and methods
AML12 hepatocyte culture
A nontumorigenic mouse hepatocyte cell lines, AML12 (America Tissue Culture Collection, CRT-2254; ATCC, VA), were cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s Medium/Ham’s F-12 medium (Gibco, NY) containing 5 µg/mL ITS premix (Sigma-Aldrich, MO), 40 ng/mL dexamethasone (Sigma-Aldrich), and 10% fetal bovine serum (FBS, Gibco). Cell cultures were maintained at 37℃ in a humidified atmosphere of 5% CO2. For serum stimulation, cells at ∼80–90% confluence were made quiescent in serum-free media.
Induction of hepatocyte apoptosis with TNF-α/Act D and treatment with melittin
TNF-α/Act D treatment previously has been demonstrated to induce cell death, specifically apoptosis. 19 Therefore, we examined hepatocytes were pre-treated with melittin (0.5, 1, and 2 µg/mL, Sigma-Aldrich) for 30 min with additional administration of TNF-α (10 ng/mL, R&D system, MN)/Act D (200 ng/mL, Sigma-Aldrich) for 9 h. Melittin was dissolved in distilled water and lyophilized by freeze dryer and stored in 4℃.
Cytotoxicity assays
To determine the effects of melittin in hepatocyte apoptosis, cell viability was evaluated by the 3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenylterazolium bromide (MTT) reduction assay. In brief, cells (1 × 105 cells/mL) were seeded in 96-well plates and treated with melittin and TNF-α/Act D. The control was added to saline of equal volume. Cells were incubated with 0.5 mg/mL MTT solution. After being incubated for 4 h at 37℃ and 5% CO2, the supernatant was removed, and dimethyl sulfoxide (DMSO) with ethanol was added. Cells were incubated at 37℃ for 30 min and measured using a microplate reader at 540 mm. The percentage of cell survival was calculated by taking the optical density of cells given a particular treatment, dividing that number by the optical density for the untreated control cells, and then multiplying by 100.
Protein isolation and immunoblot analysis
Mitochondrial and cytosolic protein fractions were obtained as described. 15 Protein concentration was determined with a Bio-Rad Bradford kit (Bio-Rad Laboratories, CA). The mitochondrial and cytosolic lysates were subjected to immunoblot analysis as described below. Samples were boiled for 5 min and equal volumes (30 µL per lane) were loaded on a sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After separation the proteins were transferred to a nitrocellulose membrane during 1 h at 4℃ and blocked overnight with PBS-T (0.1% [v/v] Tween-20, 5% [w/v] powdered milk in 137 nm NaCl, 2.7 mmol/L KCl, 1.5 mmol/L KH2PO4, pH 7.4) at 4℃. Immune complexes were detected with a horseradish peroxidase (HRP)-conjugated secondary antibody and were visualized by enhanced chemiluminescence (ECL) detection system (Amersham, NJ). The primary antibodies used in this study were probed with anti-caspase-3, -8, -9, anti-bid, anti-bax, anti-VDAC, anti-cytochrome c, anti-PARP-1, p-IKK, p-IκB and NF-κB (Cell signaling, MA), anti-bcl-2 and anti-β-actin (Santa Cruz, CA). Signal intensity was quantified by image analyzer (Las 3000, Fuji, Japan).
Flow cytometric cell cycle analysis
Hepatocytes were plated at a density of 1 × 105 cells/mL in a total volume of 10 mL and exposed for 24 h at 37℃ to melittin or TNF-α/Act D. After treatment, cells were collected by centrifugation for 5 min at 4000g, resuspended in 300 µL of ice-cold phosphate-buffered saline (PBS), fixed in ice-cold 95% ethanol and stored at −20℃ for at least 30 min. Fixed cells were collected by centrifugation and resuspended in PBS containing RNase A (500 UI/mL). Samples were kept at room temperature for 30 min. To determine cellular DNA content, cells were stained with propidium iodide (50 µg/mL), incubated for 20 min on ice, and then analyzed in duplicate by flow cytometry using a Coulter EPICS XL apparatus (Beckman-Coulter, FL). The percentage of cells in G0/G1, S, and G2/M phases of the cell cycle and the percentage of cells in the sub-G0/G1 peak were calculated using MultiCycle AV Software (Phonenix Flow Systems, CA) that eliminated the debris effect.
Quantitation of apoptosis and assessment of mitochondrial membrane potential
For the analysis of apoptotic cell death, we performed nuclear staining with the DNA-binding dye Hoechst 33342 (Sigma-Aldrich). Hoechst 33342 is cell membrane-permeable nuclear binding dye used for the evaluation of apoptosis. 20 Also, to measure the mitochondrial membrane potential, JC-1 dye (Stratagene, CA), a sensitive fluorescent probe, was used. 21 Hepatocytes were collected by centrifugation at 200 g for 5 min, washed with ice-cold PBS and then fixed with 2% paraformaldehyde in PBS for 10 min at 4℃. Fixed cells were washed with PBS, incubated with Hoechst 33342 and JC-1 for 30 min in the dark. Blue fluorescence was visualized by using fluorescence microscopy (excitation/emission = 330–380 nm/460 nm, Nikon ECLIPSE 80i). Fluorescence microscopy with a 488 nm filter was used for the excitation of JC-1. Emission filters of 535 and 595 nm were used to quantify the population of mitochondria with green (JC-1 monomers) and red (JC-1 aggregates) fluorescence, respectively.
Electrophoretic mobility shift analysis (EMSA)
AML12 cells were lysed and nuclei were extracted as reported previously. 22 In brief, 5 µg of nuclear protein were incubated at room temperature for 30 min with a Dig gel shift kit (Roche, Mannheim, Germany). The DNA–protein complexes were separated by electrophoresis in 6% nondenaturing polyacrylamide gels using 0.25× Tris-borate-EDTA as a running buffer. After electrophoresis, gels were transferred to nylon membranes and detected chemiluminescently. Complementary standard DNA oligos containing transcription factors NF-κB binding site, respectively, were annealed and DiG labeled. The sequence was as follows: NF-κB: 5′-AGT TGA GGG GAC TTT CCC AGG C-3′.
NF-κB promoter activity
Reporter gene activity was evaluated by cell-based analysis methods for assaying NF-κB activity. NF-κB promoter-luciferase construct was transiently transfected by using transfection reagent Lipofectamine 2000 (Invitrogen, CA). After harvest cells were lysed in reporter lysis buffer (Promega, WI). Cell extract was mixed with 100 µL of luciferase assay reagent and the emitted light intensity was measured using luminometer FLUOstar OPIMA (BMG Labtech, Germany). The luciferase activity is represented as the fold induction compared with normal control cell.
Statistical analysis
The data are presented as means values ± standard error (SE) of the mean of at least three separate experiments. Comparisons were made using Student’s t-test. For all analysis, a two-sided P value < 0.05 was considered to indicate statistical significance.
Results
Inhibitory effects of melittin on TNF-α/Act D-treated AML12 cells
TNF-α/Act D treatment has previously been demonstrated to induce cell death, specifically apoptosis.
19
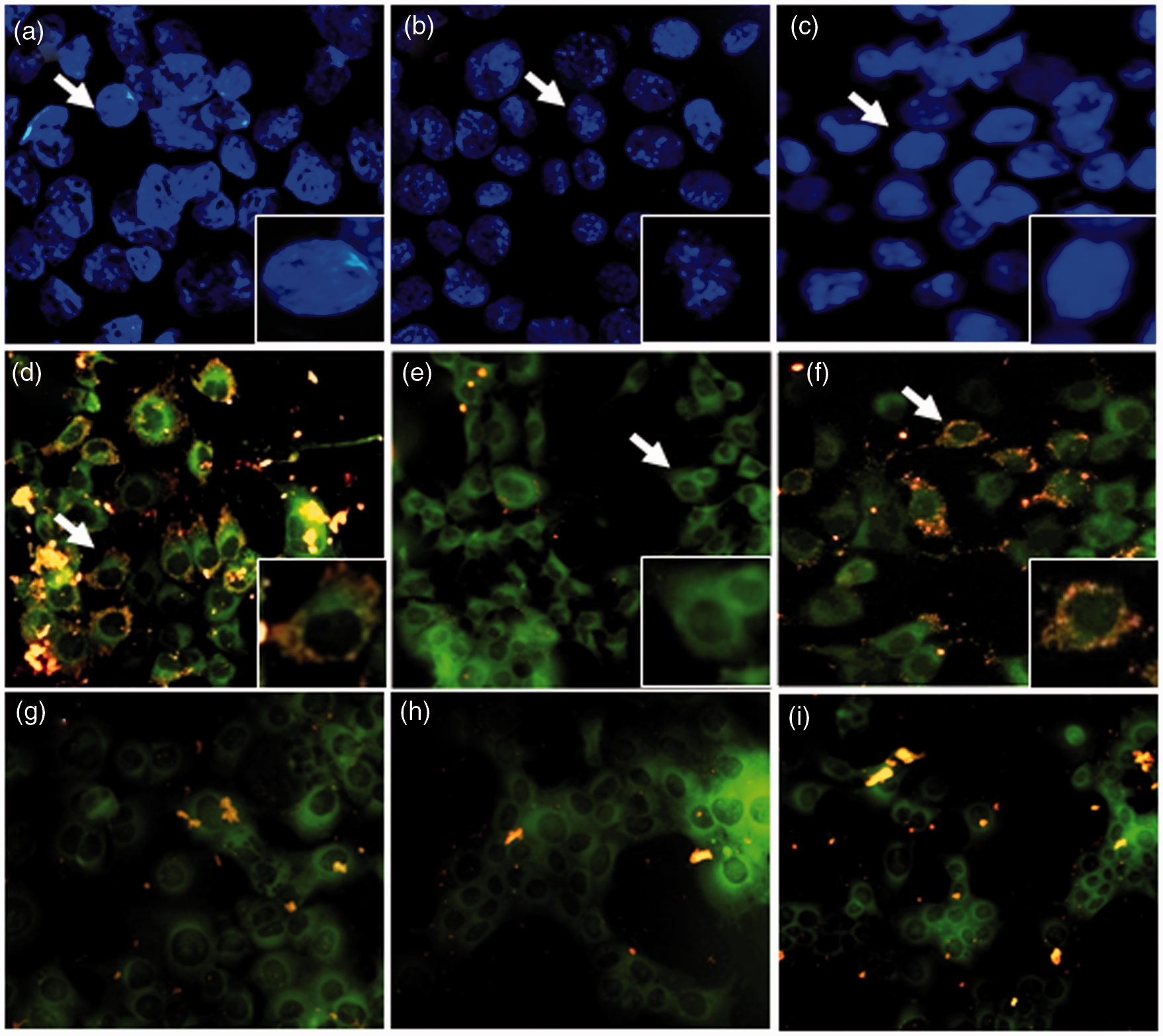
In order to determine the optimal dose of melittin for studying its effects on AML12 cells after TNF-α/Act D treatment, an MTT assay was carried out. We tested the viability of cells treated with TNF-α/Act D for 9 h. Viability was decreased by about 45% as compared to normal control cells. In addition, AML12 cells were treated with melittin (0.5, 1, and 2 µg/mL) for 30 min with additional administration of TNF-α/Act D for 9 h (Figure 1a). There were no significant changes in cell viability when the AML12 cells were incubated with melittin alone. Thus, the effects of melittin on normal AML12 cells are minimal. Cells treated with 1 µg/mL of melittin showed a significant increase in cell viability compared with TNF-α/Act D-treated AML12 cells without addition of melittin. These results showed the melittin treatment chosen as optimal for use in subsequent experiments on injured AML12 cells was 1 µg/mL for 9 h. We next used flow cytometry to investigate whether melittin treatment was also involved in cell cycle distribution (Figure 1b–c). When AML12 cells were treated with TNF-α/Act D for 24 h, the G0 phase increased from 1.2% of the normal control to 71.1%. About 17.5% of the TNF-α/Act D-treated AML12 cells appeared as apoptotic cells at 24 h after treatment (Figure 1c). In contrast, the G1 phase was increased 45.9% by melittin treatment compared with TNF-α/Act D (Figure 1d). These results suggest that melittin arrests cells in the G1 phase, confirming that melittin protects against TNF-α/Act D-induced apoptosis. We examined the nuclear morphology, cell cycle distribution, and mitochondrial membrane potential of TNF-α/Act D-treated and melittin-treated AML12 cells. AML12 cells were observed through fluorescence microscopy following treatment with Hoechst33342 stain for nuclear morphology (Figure 2a–c). The assay revealed the occurrence of nuclear condensation in TNF-α/Act D-treated AML12 cells (Figure 2b). Treatment with 1 µg/mL of melittin resulted in the cellular death of AML12 cells by involving apoptotic mechanisms, as demonstrated in both the detached and the adherent cell populations. Melittin significantly decreased apoptosis induced in AML12 cells by treatment with TNF-α/Act D for 9 h (Figure 2c). Also, we used JC-1 staining to measure mitochondrial membrane potential and investigate whether mitochondrial membrane integrity is affected by melittin (Figure 2d–i). Treatment with 1 µg/mL of melittin clearly decreased the amount of mitochondria with collapsed membrane potential in TNF-α/Act D-treated cells (Figure 2f). Taken together, these results indicate that melittin protects hepatocytes against mitochondrial injury caused by TNF-α/Act D.
Effects of melittin on TNF-α/Act D-induced AML12 cells apoptosis. (a) MTT reduction was calculated to indicate cellular proliferation for 9 h. Melittin was applied at concentrations of 0.5, 1, and 2 µg/mL for 9 h in TNF-α/Act D-induced hepatocytes apoptosis. The percentage of cell survival was defined relative to the number of surviving untreated AML12 cells. FACS analysis of AML12 cells stained with PI. cells appearing as a G0, G1, S, and G2/M pear were scored as apoptotic. Panels correspond to (b) normal control, (c) AML12 cells treated with TNF-α/Act D, (d) AML12 cells treated with 1 µg/mL melittin together with TNF-α/Act D. Data are means ± SE (n = 3). *P < 0.05 compared to normal control, #P < 0.05 compared to cells treated with TNF-α/Act D Fluorescent photographic assessment of apoptosis in AML12 cells. AML12 cells were evaluated by morphological criteria after Hoechst 33342 nuclear staining (a–c) and JC-1 mitochondria staining (red and green; d–i) by using fluorescent microscope. (a) Normal control showed round nuclei stained at blue color. (b) AML12 cells treated with TNF-α/Act D showed fragmented nuclei. (c) TNF-α/Act D-treated AML12 cells with addition of 1 µg/mL melittin had decreased numbers of fragmented nuclei compared with TNF-α/Act D-treated AML12 cells. (d) Normal control showed a mixture of red and green structures. (e) In TNF-α/Act D-treated AML12 cells most of the fluorescence was green, with a great diffuse distribution in the cytosol. (f) TNF-α/Act D-treated AML12 cells with addition of 1 µg/mL melittin had increased mixture of red and green structures. Mitochondria respiratory inhibitors: rotenone (g), antimycin (h), oligomycin (i). a–c magnifications × 400; arrows region × 600; d–i magnifications × 400. (A color version of this figure is available in the online journal.)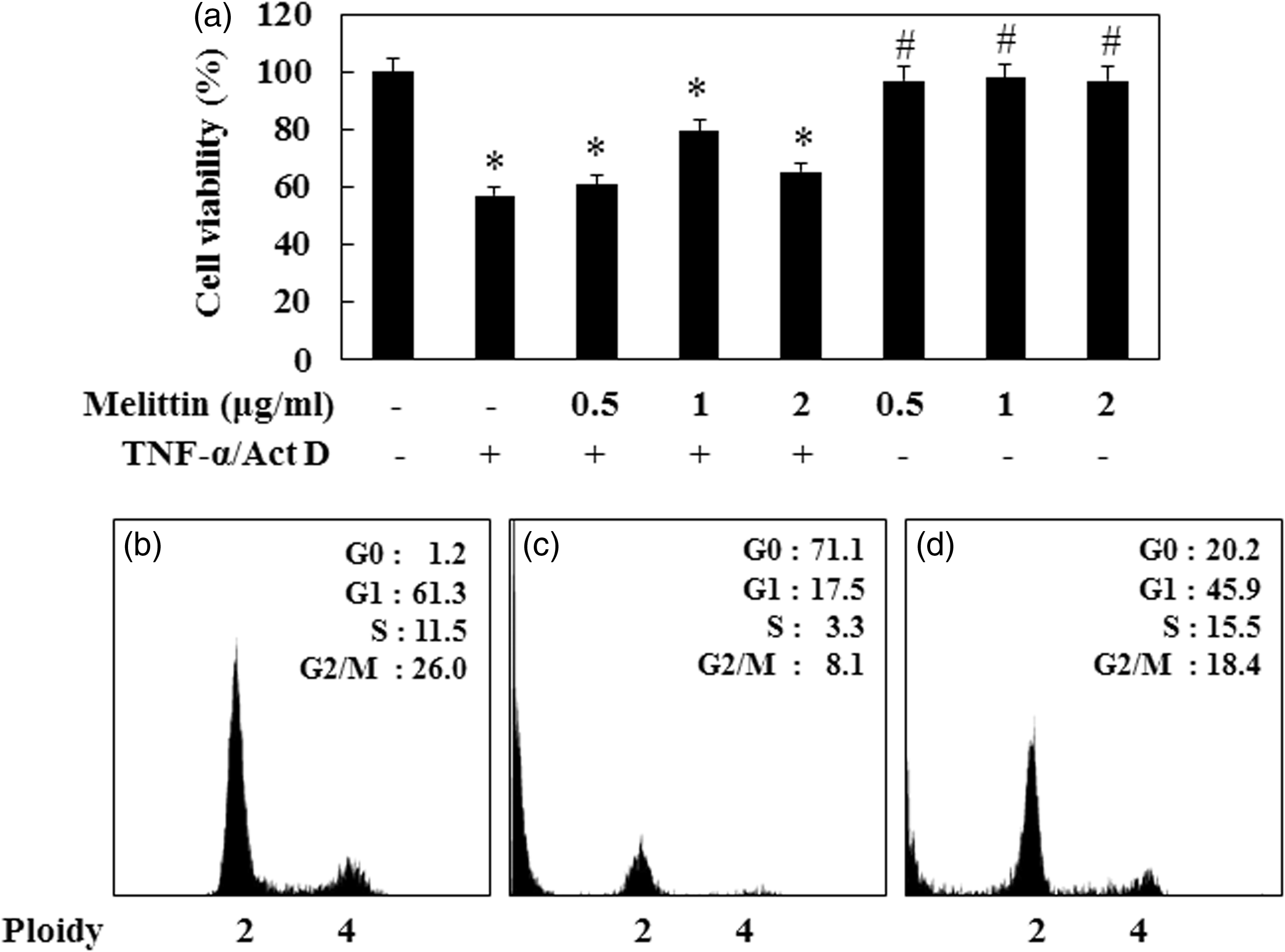
Melittin protects against caspase- and PARP-1-induced apoptosis in TNF-α/Act D-treated AML12 cells
Most forms of apoptosis are mediated by the proteolytic action of caspase. Since caspase activation plays an important role in apoptosis and PARP-1 is a target of caspase activity associated with apoptosis, we investigated the activation of caspase 8 (18 kDa), caspase-9 (37 kDa), caspase-3 (17 kDa), and PARP-1 after the cells were treated with melittin at different concentrations (0.5, 1, and 2 µg/mL) for 9 h. TNF-α/Act D-treated AML12 cells showed increased cleavage of caspase-8, -9, and -3 (Figure 3). The addition of 1 µg/mL of melittin inhibited the proteolytic fragmentation of these caspases in TNF-α/Act D-treated AML12 cells. Moreover, the cleaved PARP-1 (89 kDa) appeared after 9 h of TNF-α/Act D treatment and its cleavage was reduced by the 1 µg/mL melittin treatment. Since PARP-1 cleavage coincides with pro-caspase processing, these results demonstrate that melittin protects TNF-α/Act D-induced apoptosis at a site upstream of caspase-3 activation.
Effect of melittin on TNF-α/Act D-induced caspase family and PARP-1 activation. (a) The total lysates from AML12 cells were examined by immunoblot. Treatment with 1 µg/mL of melittin attenuated caspase-8 (18 kDa), -9 (37 kDa), -3 (17 kDa), and PARP-1 (89 kDa) cleavage in TNF-α/Act D treated AML12 cells. β-Actin was used as loading controls. (b) caspase-8, (c) caspase-9, (d) caspase-3, and (e) PARP-1 followed by densitometric analysis. Data are means ± SE (n = 3). *P < 0.05 compared to normal control, #P < 0.05 compared to cells treated with TNF-α/Act D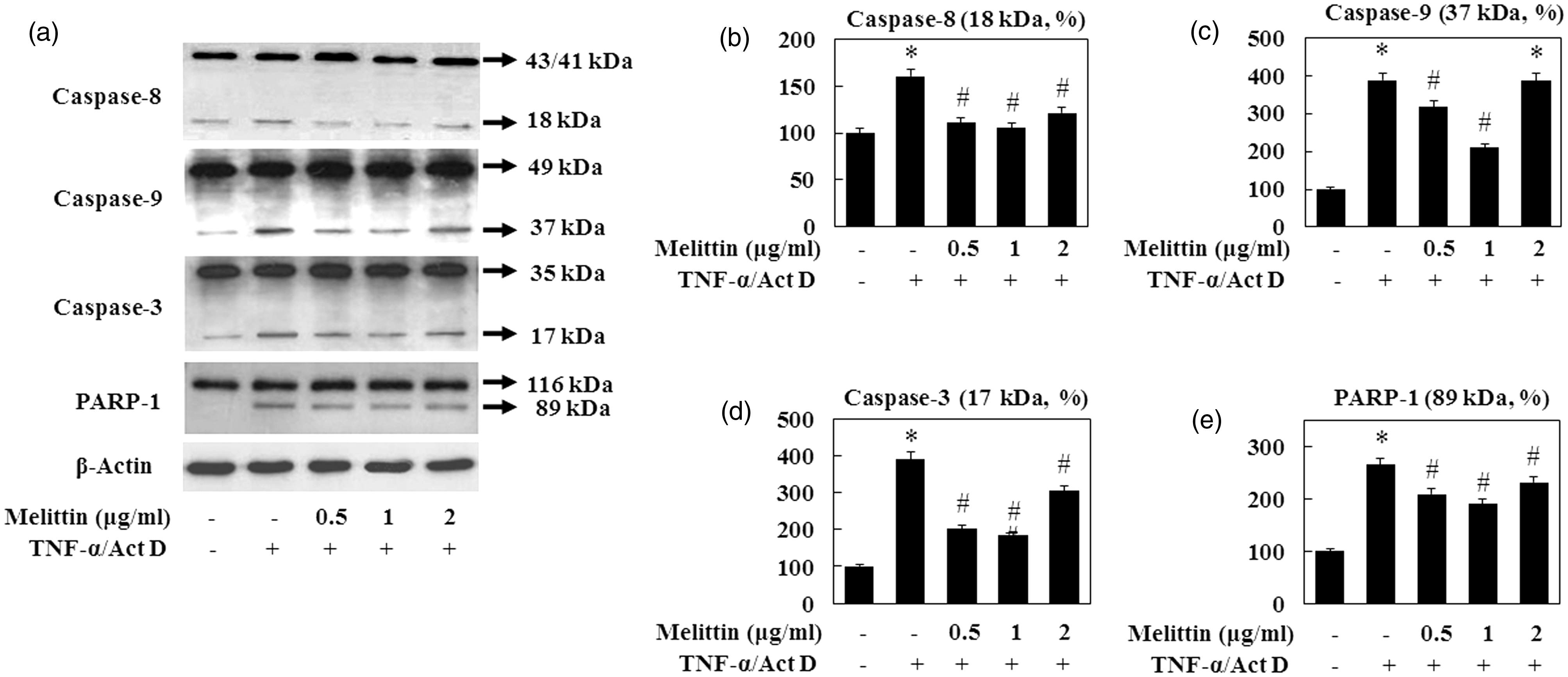
Inhibitory effects of melittin on activation of bcl-2 family during TNF-α/Act D-induced AML12 cells
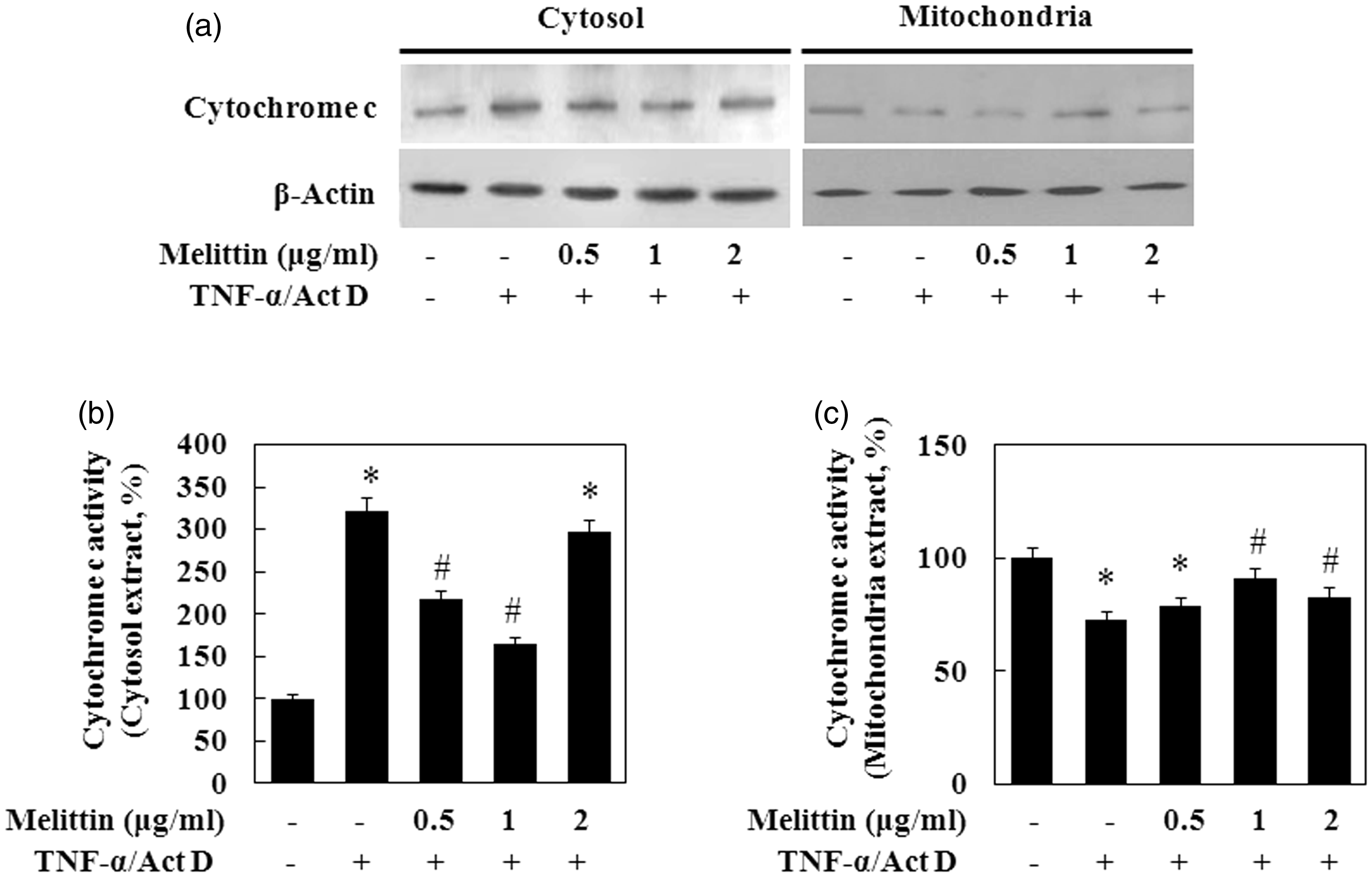
To evaluate the effects of TNF-α/Act D on the production of apoptotic proteins related to mitochondria, we measured bcl-2 family activation in TNF-α/Act D-treated AML12 cells by immunoblot analysis of mitochondrial proteins lysed from the cells (Figure 4). The anti-apoptotic protein bcl-2 was decreased and the pro-apoptotic protein bax was increased in the mitochondrial fraction of TNF-α/Act D-treated AML12 cells. The expression of bcl-2 in the TNF-α/Act D-treated AML12 cells with addition of 1 µg/mL of melittin treatment was significantly increased, while the expression of bax was decreased. In addition, TNF-α/Act D-treated AML12 cells showed increased tbid while the addition of 1 µg/mL of melittin inhibited this proteolytic fragmentation. In cells undergoing apoptosis, cytochrome c is released from the intermembrane space to the cytoplasm. Cytochrome c accumulated in the cytosol of cells exposed to TNF-α/Act D (Figure 5). This release of cytochrome c was significantly inhibited by treatment with 1 µg/mL melittin. These results are in agreement with the above findings that melittin suppresses TNF-α/Act D-induced cleavage of caspase-8.
Melittin inhibits mitochondrial translocation of apoptotic proteins. (a) Immunoblot of total lysates of bid and bax, mitochondria fractions of bcl-2 were examined. Treatment with 1 µg/mL of melittin treatment increased bcl-2 expression, decreased tbid and bax expression in TNF-α/Act D-treated AML12 cells. VDAC and β-actin were used as loading controls. (b) bcl-2, (c) bax, and (d) t-bid followed by densitometric analysis. Data are means ± SE (n = 3). *P < 0.05 compared to normal control, #P < 0.05 compared to cells treated with TNF-α/Act D Effect of melittin on TNF-α/Act D-induced mitochondrial cytochrome c release. (a) Immunoblot of cytosol and mitochondria fractions of cytochrome c was examined. VDAC and β-actin were used as loading controls. (b) Cytosol fraction of cytochrome c and (c) mitochondria fraction of cytochrome c followed by densitometric analysis. Data are means ± SE (n = 3). *P < 0.05 compared to normal control, #P < 0.05 compared to cells treated with TNF-α/Act D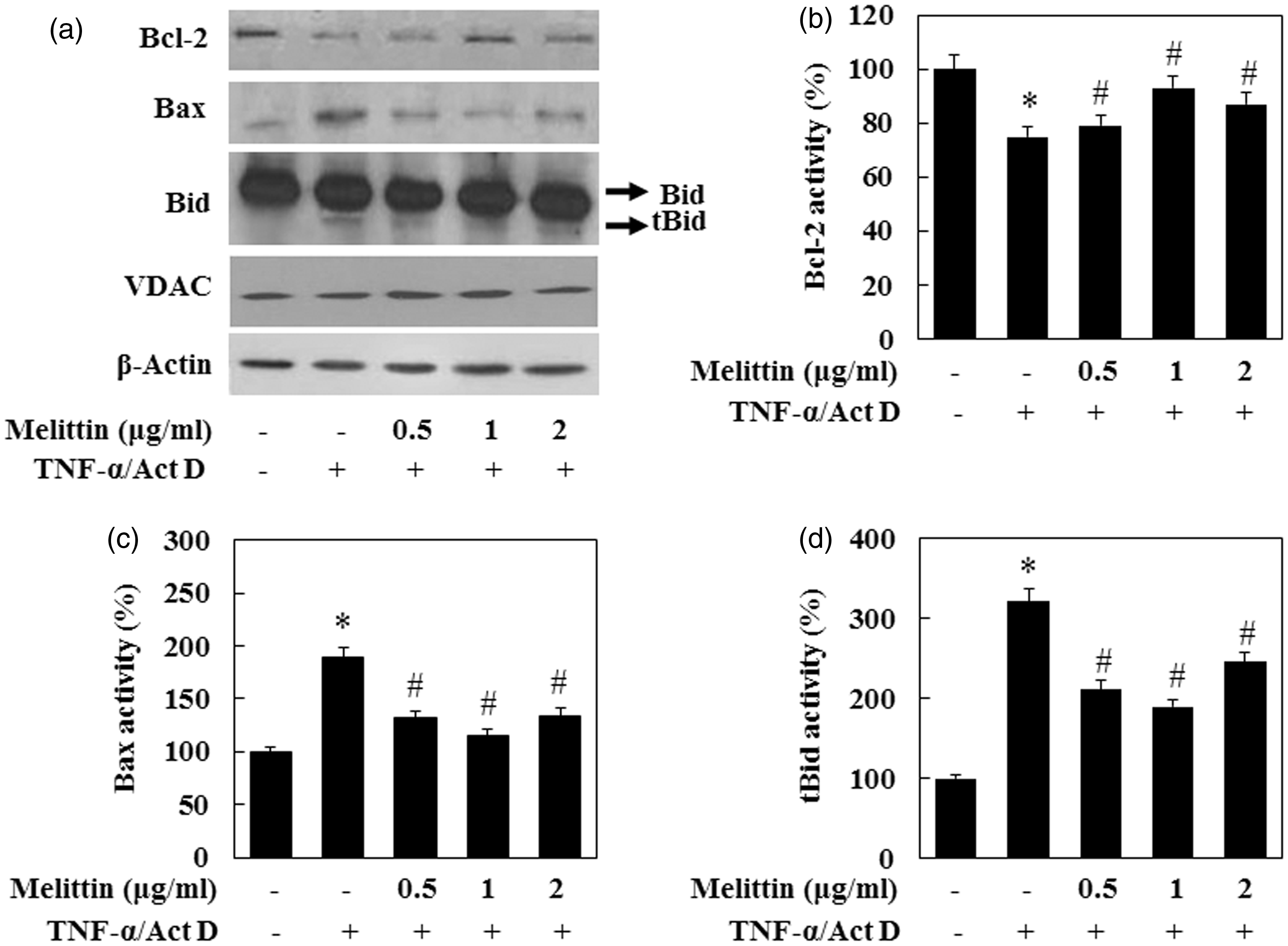
Effects of melittin on TNF-α/Act D induced the activity of NF-κB
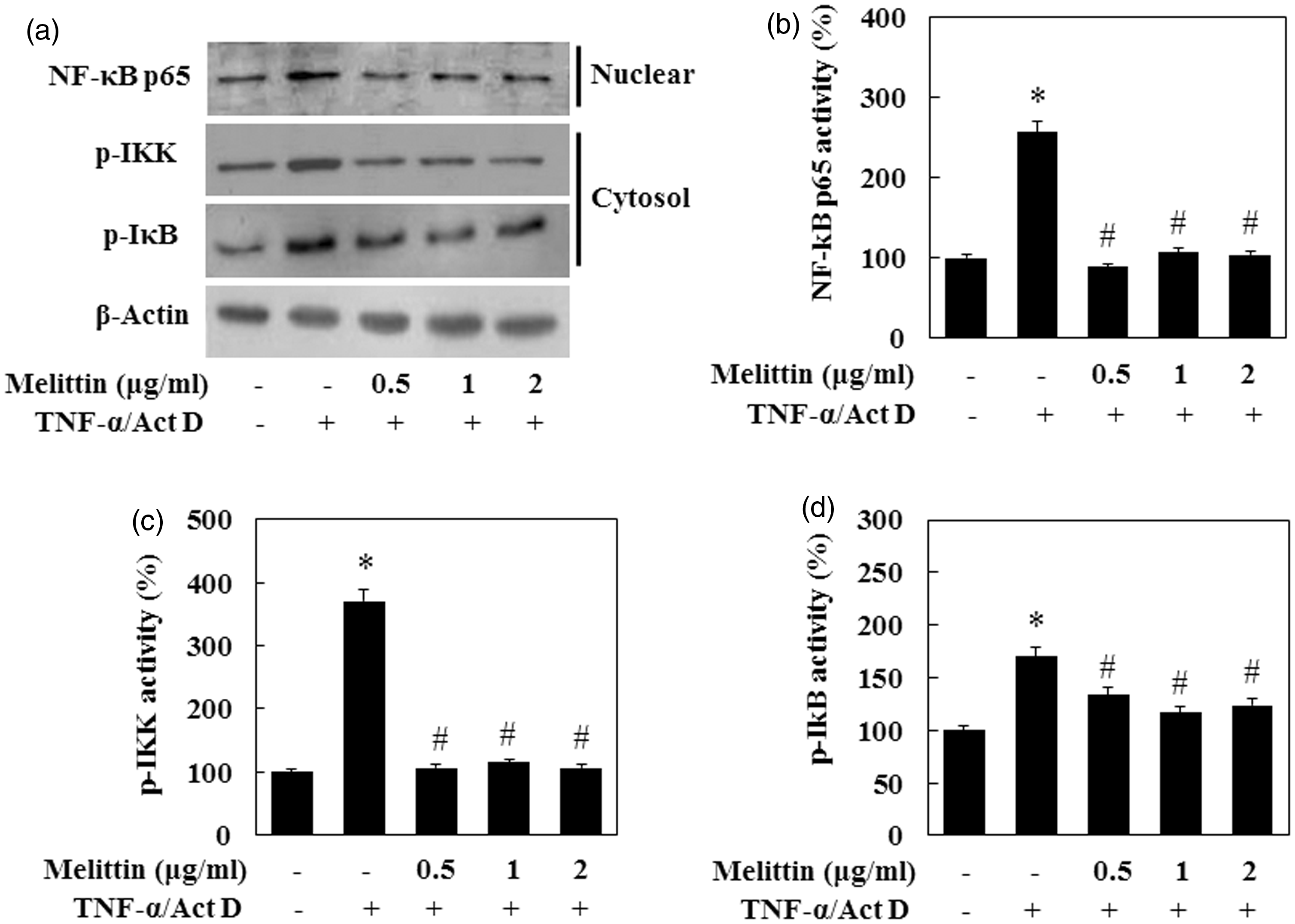
NF-κB has a role in the liver as part of a pro-survival signaling pathway. The activation of IκB kinase (IKK) is a key step in the activation of NF-κB. The effectiveness of melittin was investigated by EMSA for NF-κB nuclear binding and immune blot analysis. To investigate the mechanism for suppression of NF-κB transcription by melittin, we examined the effect of melittin on NF-κB promoter activity by transiently transfected luciferase reporter plasmid containing the NF-κB promoter sequence. As shown in Figure 6a, melittin significantly inhibited NF-κB promoter activity induced by TNF-α/Act D. Melittin suppressed NF-κB expression to the basal level at 1 µg/mL. To investigate the effect of melittin on NF-κB DNA binding activity in TNF-α/Act D-treated AML12 cells, nuclear extracts prepared from isolated AML12 cells were analyzed for NF-κB DNA binding activity (Figure 6b). TNF-α/Act D-treated AML12 cells readily responded to melittin treatment with the activation and translocation of NF-κB. NF-κB activity in the TNF-α/Act D-treated AML12 cells was strongly up-regulated. Increased NF-κB DNA binding activity was suppressed significantly in the TNF-α/Act D-treated AML12 cells with melittin treatment. We also examined whether adding melittin to TNF-α/Act D-treated AML12 cells has an effect on the activation of the NF-κB signaling pathway (Figure 7). Melittin inhibited the expression of p-IKK and NF-κB in TNF-α/Act D-treated AML12 cells. The expression of p-IκB decreased on TNF-α/Act D-treated AML12 cells with melittin treatment, but the difference of this expression was less than that of p-IKK. Taken together, these results suggest that melittin treatment reduces apoptotic protein expression via inhibition of NF-κB pathways.
Melittin suppresses NF-κB DNA binding activity in TNF-α/Act D-treated AML12 cells. (a) AML12 cells were transfected with NF-κB promoter-containing reporter vector and incubated with melittin in the absence or presence TNF-α/Act D. Luciferase activity was measured 9 h after transfection. (b) Nuclear extracts were subjected to NF-κB DNA binding assay by EMSA. (c) NF-κB DNA binding activity followed by densitometric analysis. Data are means ± SE (n = 3). *P < 0.05 compared to normal control, #P < 0.05 compared to cells treated with TNF-α/Act D Melittin suppresses NF-κB signal pathway in TNF-α/Act D-treated AML12 cells. (a) Immunoblot of cytosol fraction of p-IKK and p-IkB, and nuclear fraction of NF-κB p65 were examined. Treatment with 1 µg/mL of melittin inhibited expression of p-IKK, p-IkB, and translocation of NF-κB in TNF-α/Act D-treated AML12 cells. β-Actin was used as loading controls. (b) NF-κB p65, (c) p-IKK, and (d) p-IkB followed by densitometric analysis. Data are means ± SE (n = 3). *P < 0.05 compared to normal control, #P < 0.05 compared to cells treated with TNF-α/Act D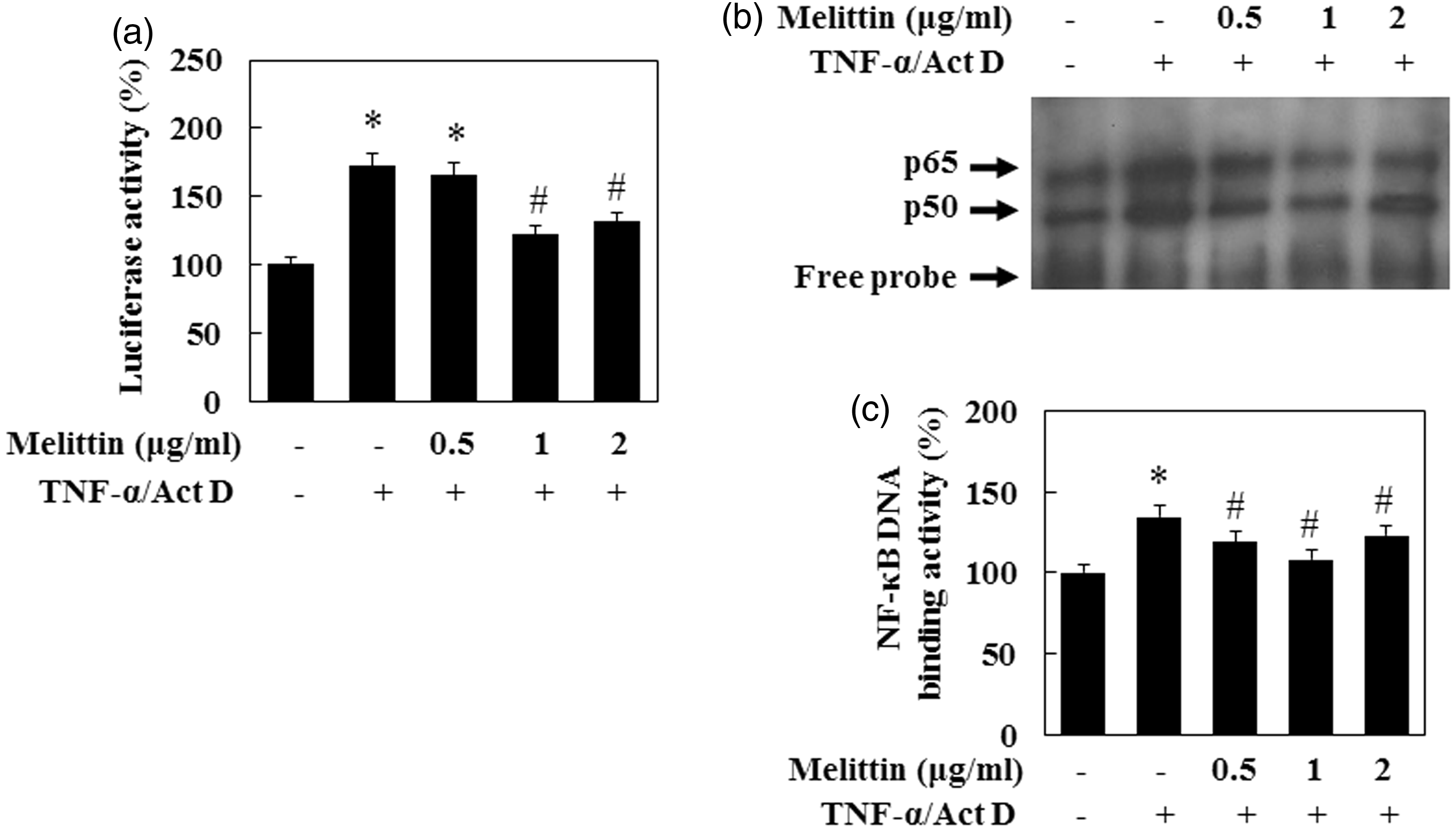
Discussion
Bee venom and melittin as potent inhibitors of NF-κB would support the many claims that bee venom and/or melittin can indeed be used as an effective anti-inflammatory remedy.23,24 Melittin has been studied extensively due to its lytic effects on biological and model membranes when inserted into phospholipid layer in high concentration.25,26 In contrast, some papers reported that concentrations of melittin lower than 2 µmol/L do not disrupt cell membranes of leukocytes. 13 Also, melittin exhibits an anti-inflammatory effect by inhibiting the lipopolysaccharide (LPS)-induced expression of cyclooxygenase (COX)-2, cytosolic phospholipase A2, inducible nitric oxide synthase (iNOS), generation of prostaglandin E2, nitric oxide (NO), intracellular calcium levels, and necrotic cytotoxicity in gastrointestinal cells.13,27,28 In one study, after 70% of the rat liver volume have been resected, certain stimulating effects of a single administration of bee venom on hepatocyte proliferation were revealed. 29 We have recently shown that bee venom protects TGF-β1 - and ethanol-treated hepatocyte injury by inhibiting the activation of the bcl-2 family of proteins, caspase, and PARP-1 cleavage.15,30 In addition, we have recently revealed that melittin protects TGF-β1-treated hepatocyte apoptosis. 31 However, the mechanisms of anti-apoptotic effect of melittin have not been fully elucidated in hepatocytes. The present study tested the hypothesis that melittin protects against TNF-α/Act D-induced hepatocyte death via inhibition of apoptosis signaling and NF-κB pathway.
We evaluated the effect of melittin on TNF-α/Act D-induced apoptosis in hepatocytes to gain a better understanding of the molecular mechanisms involved in the melittin of anti-apoptotic effects. Our results showed that in cell treated with TNF-α/Act D, viability was decreased by about 40% compared with untreated control cells, and that melittin 1 µg/mL treatment dramatically inhibits this damaged cell viability by 20%. We also showed that melittin did not stimulate normal cells, but inhibited TNF-α/Act D-induced apoptosis. Additionally, we found that TNF-α/Act D caused nuclear damage, as assessed by Hoechst 33342 stain. These results suggest that melittin 1 µg/mL treatment contributes to decreased TNF-α/Act D-induced apoptosis.
Caspase-3 is a key and common protease in both mitochondria- and death receptor-dependent pathways. 32 In many studies, it has been determined that a variety of chemotherapeutic agents induced apoptosis through the activation of caspase and degradation of PARP-1. 33 PARP-1 is cleaved by activated caspase-3 from its intact form of 116 kDa into 85 kDa fragments. It is cleaved before or concomitant with the degradation of nuclear DNA into nucleosomal fragments, and the presence of PARP inhibitors delays or prevents apoptosis. PARP cleavage has also been widely used as a characteristic phenotype of cell apoptosis and as indirect evidence of the activation of caspase-3 or caspase-7. 34
Various apoptotic stimuli activate the pro-apoptotic machinery in which the initiator caspase-8 is activated by a death receptor, while caspase-9 is activated by cytochrome c released from mitochondria. It has been reported that caspase-8 stimulates directly, as well as indirectly the activity of caspase-3. 35 On the other hand, caspase-8 has been shown to interact with bid, a bcl-2 family protein, which in turn mediates the release of cytochrome c from mitochondrial membranes, leading to the activation of caspase-3 subsequent to activation of caspase-9. 36 We therefore investigated whether melittin inhibits expression of caspases in TNF-α/Act D-treated AML12 cells. In this study, nuclear fragmentation induced by TNF-α/Act D increased the level of cytochrome c released from the mitochondria into the cytosol, enhanced the caspase-3 and -9 activities and the cleavage of PARP-1. Conversely, melittin 1 µg/mL treatment inhibited apoptosis in TNF-α/Act D-treated AML12 cells by suppressing caspase activation. Therefore, we conclude that melittin suppresses apoptotic signaling at the early stage by inhibiting caspase-induced proteolytic activation and cytochrome c release.
The bcl-2 family is a well-characterized regulator of cytochrome c release from the mitochondria into the cytosol, which it does by regulating the mitochondrial permeability transition pore that is composed of voltage-dependent anion channel (VDAC) in outer membrane, adenosine nucleotide translocated (ANT) in the inner membrane, and cyclophilin (Cyp)-D in the matrix assemblies. The bcl-2 family is classified into anti-apoptotic proteins such as bcl-2 and bcl-xL, which reduce the level of cytochrome c release 37 and pro-apoptotic proteins such as bax and bak, which induce the release of cytochrome c and loss of mitochondrial membrane potential. 38 Therefore, the ratio of pro-apoptotic and anti-apoptotic activities of the bcl-2 family may be pivotal in the release of cytochrome c from the mitochondria into the cytosol. In the present study, bcl-2 was shown to be up-regulated, whereas bax was down-regulated by melittin in TNF-α/Act D-treated AML12 cells.
The BH3 domain-only protein, bid, a death agonist member of the bcl-2/bcl-xL family, is localized in the cytosolic fraction of cells as an inactive precursor. 39 Its active form is generated upon proteolytic cleavage by caspase-8 in the fas signaling pathway. Cleaved bid (tbid) translocates to the mitochondria and induces cytochrome c release and mitochondrial damage. 40 Thus, bid relays an apoptotic signal from the cell surface to mitochondria. However, the precise molecular mechanisms for the translocation of the cleaved bid and for the subsequent release of cytochrome c during apoptosis are still unclear. Our data showed that melittin treatment decreased the expression of tbid in TNF-α/Act D-treated AML12 cells.
The role of NF-κB in the liver, as part of a pro-survival signaling pathway, has been studied. 41 Activation of the transcription factor NF-κB is a major effector of the inducible resistance to death receptor-mediated apoptosis by modulating anti-apoptotic genes. 7 Bee venom inhibits the DNA binding and transcriptional activity of NF-κB in Raw 265.7 cells, synoviocytes, and THP-1 cells, which are a monocytic cell line, in a dose-dependent manner. 13 The promoter region of the murine gene encoding iNOS and COX-2 contains NF-κB binding sites. 42 There, we investigated the effects of melittin on NF-κB because NF-κB is an important regulator of cell apoptosis. To investigate whether melittin inhibits NF-κB activation, we examined the effects on IKK and IκB, because these proteins are essential for the nuclear translocation and activation of NF-κB. 43 We found that melittin significantly inhibited p-IKK and p-IκB in TNF-α/Act D-treated AML12 cells. For further demonstration of the inhibitory effect of melittin in NF-κB activation, we examined NF-κB binding activity found that melittin potently attenuated NF-κB p65 subunit nuclear translocation. These results suggest that melittin suppresses NF-κB activation, leading to an inhibition of TNF-α/Act D-induced apoptosis in AML12 cells.
In conclusion, we have demonstrated that melittin protects hepatocytes against TNF-α/Act D-induced apoptosis. Melittin-mediated anti-apoptotic effects were associated with a decrease in proteolytic fragmentation activity of caspases and PARP-1. Melittin also suppressed the translocation of bid to mitochondria and inhibited the activation of bcl-2, and bax, resulting in inhibition of cytochrome c release and repressed NF-κB activation. Melittin possess a potent suppressive effect on apoptotic responses of TNF-α/Act D-treated AML12 cells via the NF-κB pathway, protecting cells and organs against TNF-α-mediated injury. These results indicate that an optimal dose of melittin exerts anti-apoptotic effects against TNF-α/Act D-induced injury to hepatocytes via the anti-apoptotic pathway.
Footnotes
Author contributions
J-HP, W-RL, and K-KP managed the overall experiment; J-HP, W-RL, S-MK, and H-SK carried out cell culture experiments (in vitro); J-HP, W-RL, S-MK, and H-SK performed Western blotting and EMSA; and Y-CC and K-KP performed flow cytometric cell cycle analysis.
Acknowledgment
This work was supported by a grant from the Next-Generation Biogreen 21 Program (No.PJ009519), Rural Development Administration, Republic of Korea.